
Cannabinoids Transmogrify Cancer Metabolic Phenotype via Epigenetic Reprogramming and a Novel CBD Biased G Protein-Coupled Receptor Signaling Platform



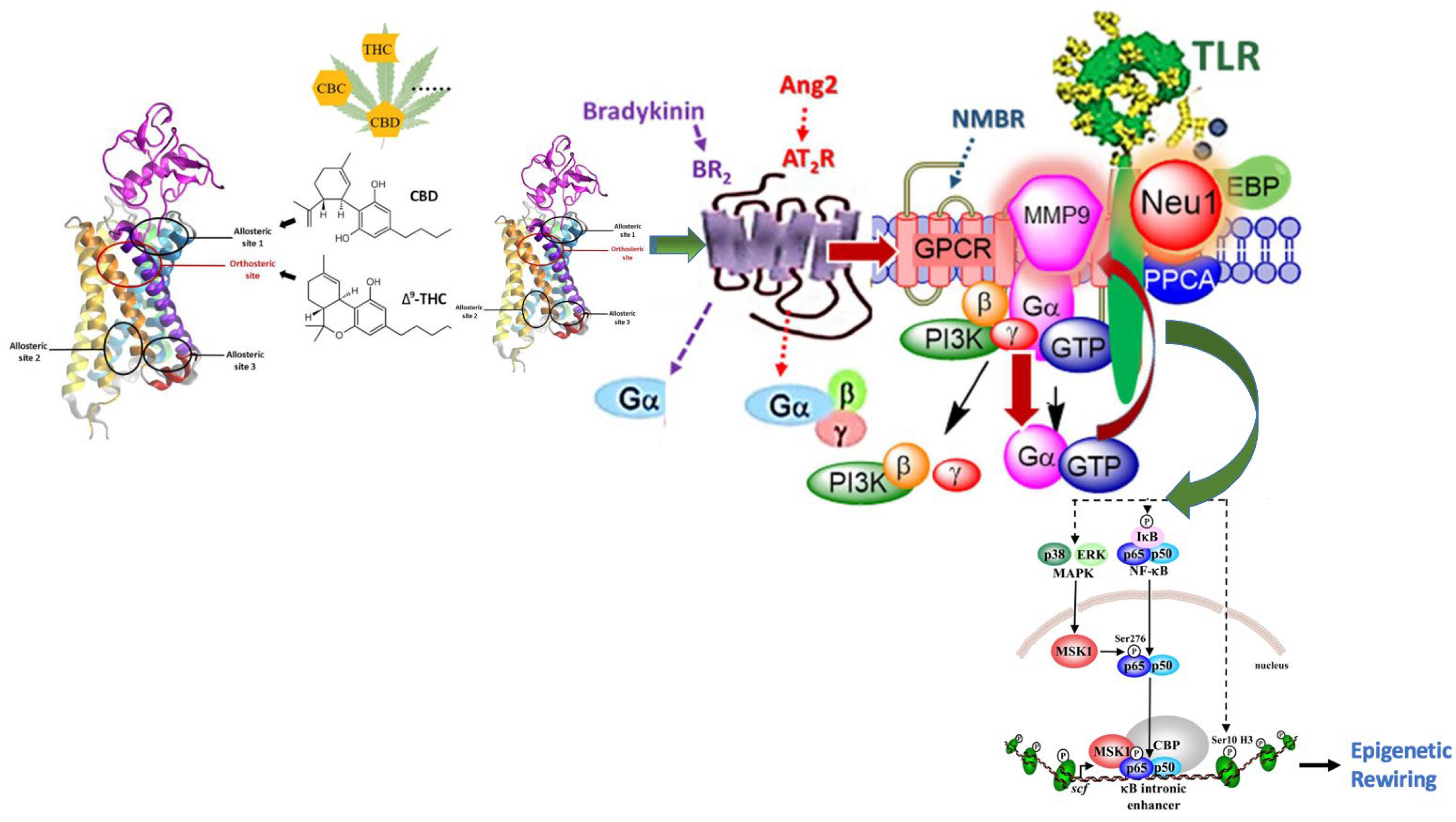{kind=link}
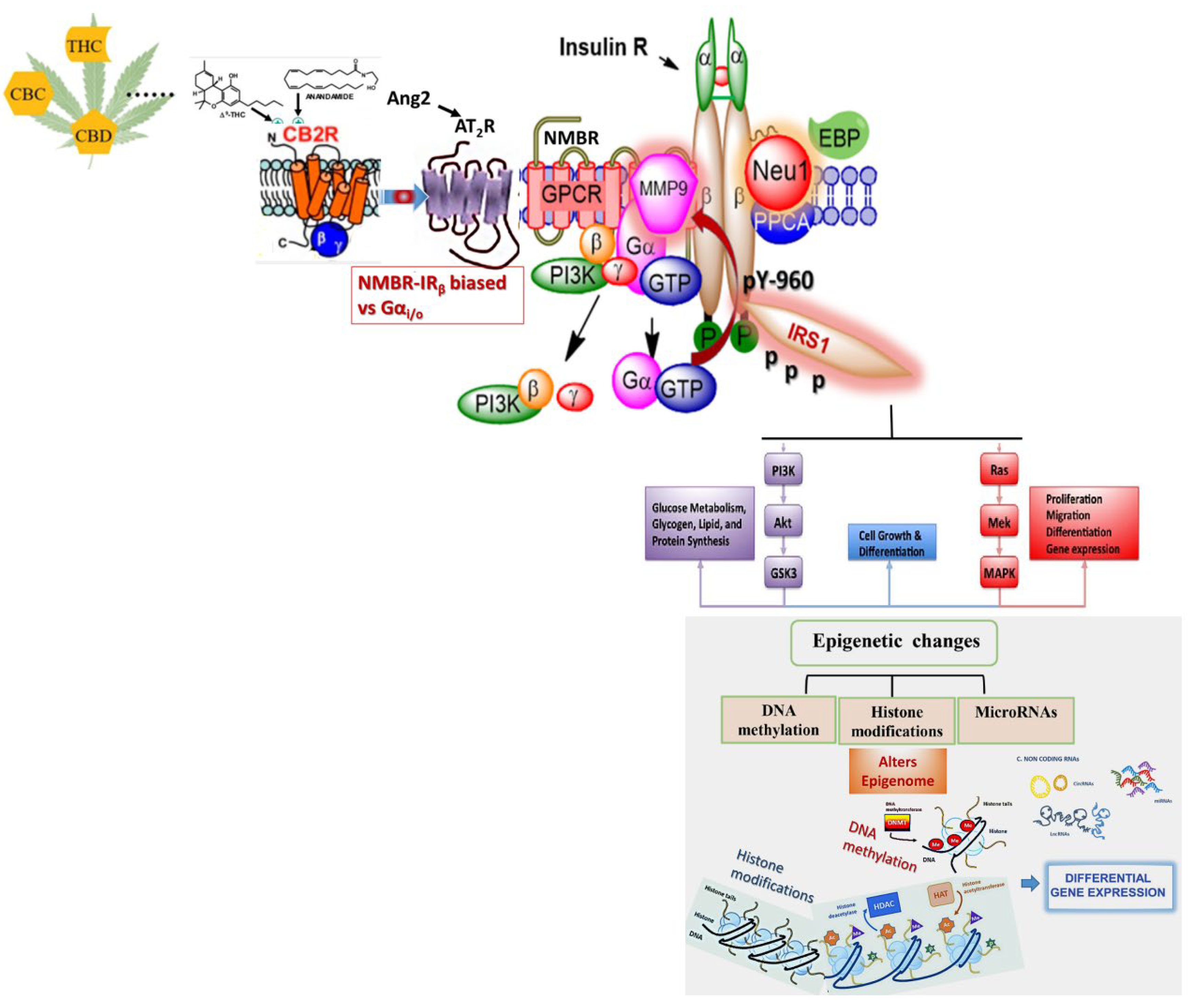{kind=link}
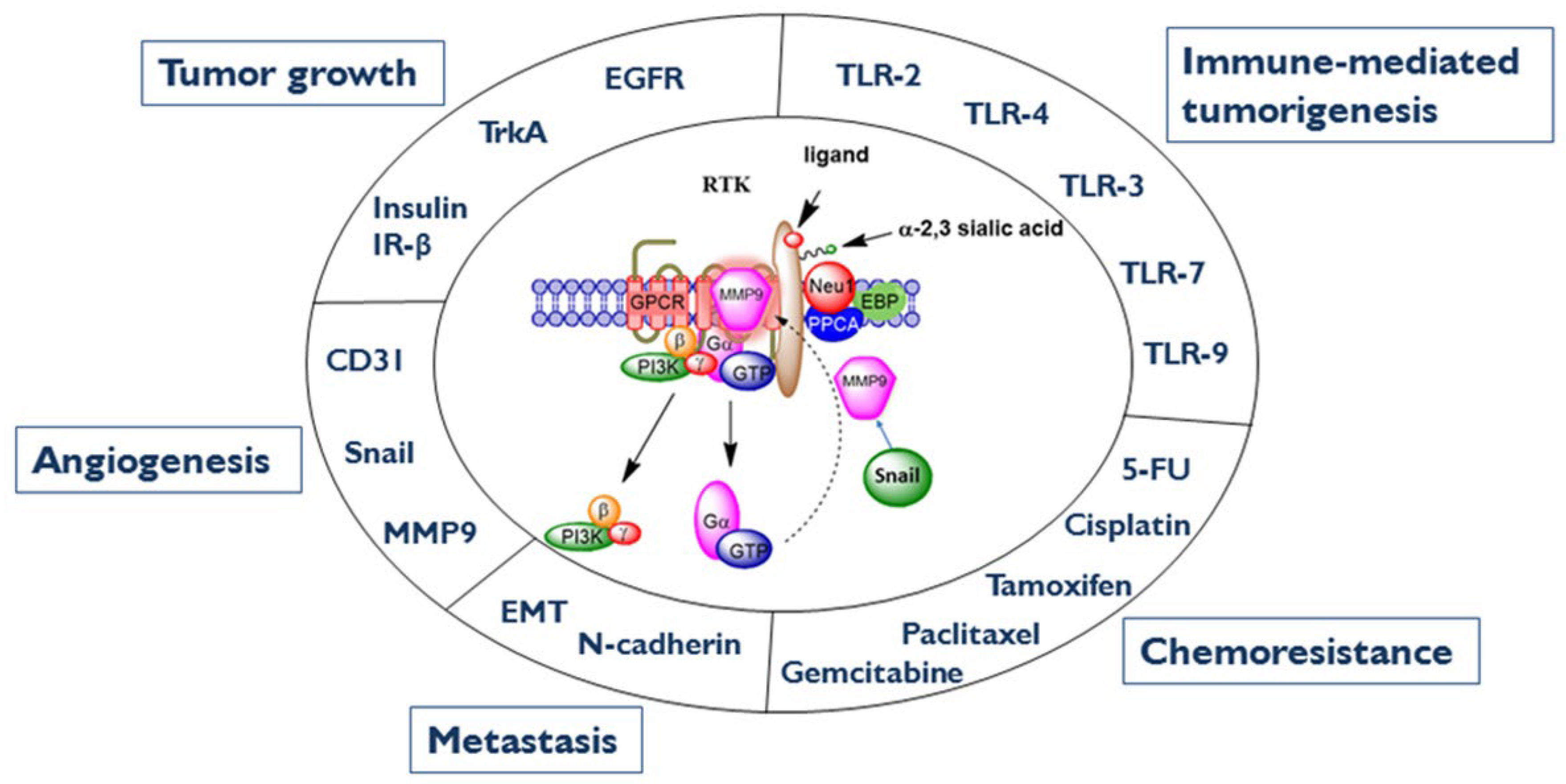{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Cannabis and the Endocannabinoid System
3. Cannabis and Cancer Metabolism
Cannabis and the Endocannabinoid System’s Potential as a Cancer Therapeutic
4. Cannabis and Epigenetics
4.1. Cannabis-Induced Epigenetic Effects
4.2. Molecular Insights into Cannabis-Induced Epigenetic Effects
4.3. Future Directions of Cannabis-Induced Epigenetic Rewiring Research
5. Neuraminidase-1
5.1. The Neuraminidase-1 Complex
5.2. Neuraminidase-1′s Impact on Cancer and Diabetes Progression
5.3. Inhibition of the Neu-1 Complex as a Potential Therapeutic to Cancer
6. MMP-9 as a Therapeutic Target of Cancer
6.1. MMP-9 and Its Interaction with the Neu-1 Complex
6.2. Inhibition of the NF-κB Pathway Results in Downregulation of MMP-9
6.3. Inhibition of the COX-2 Decreases MMP-9 Activity
7. Effects of Biased GPCR Agonists on the Insulin Receptor (IR)
Bias GPCR Agonism and the Insulin Receptor
8. Bias GPCR Agonism on the IR and Cancer
9. Conclusions
10. Future Research Directions and Limitations
- Exploring the structure-activity relationships of ligands that target the CB1 allosteric site or that behave as neutral CB1 and/or CB2 receptor antagonists.
- Assessing the therapeutic potential of CB1 and/or CB2 receptor allosteric modulators and neutral antagonists.
- CB1 and/or CB2 receptor allosteric modulators and neutral antagonists modulate for or against the presence of endocannabinoid mechanisms in mammalian cells.
- Validating and characterizing non-CB1 and non-CB2 targets such as RTK and TLR receptors for particular cannabinoids and developing compounds that can selectively activate or block such targets with reasonable potency for metabolic health and related diseases.
- Validating and characterizing that cannabinoid receptors may exist as homodimers or form heterodimers or oligomers with one or more classes of the non-cannabinoid receptor.
- Validating and characterizing the role played by the endocannabinoid system in ameliorating the symptoms and/or the underlying pathology of certain disorders.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Rock, E.M.; Parker, L.A. Constituents of Cannabis Sativa. Adv. Exp. Med. Biol. 2021, 1264, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Filipiuc, L.E.; Ababei, D.C.; Alexa-Stratulat, T.; Pricope, C.V.; Bild, V.; Stefanescu, R.; Stanciu, G.D.; Tamba, B.-I. Major Phytocannabinoids and Their Related Compounds: Should We Only Search for Drugs That Act on Cannabinoid Receptors? Pharmaceutics 2021, 13, 1823. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.; Williams, C.; Iversen, L.; Whalley, B. Molecular Pharmacology of Phytocannabinoids. Phytocannabinoids 2017, 103, 61–101. [Google Scholar]
- Wiley, J.L.; Owens, R.A.; Lichtman, A.H. Discriminative Stimulus Properties of Phytocannabinoids, Endocannabinoids, and Synthetic Cannabinoids. In The Behavioral Neuroscience of Drug Discrimination; Porter, J.H., Prus, A.J., Porter, J.H., Prus, A.J., Eds.; Springer: Cham, Switzerland, 2018; pp. 153–173. [Google Scholar]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef]
- Marzo, V.D.; Petrocellis, L.D. Plant, synthetic, and endogenous cannabinoids in medicine. Annu. Rev. Med. 2006, 57, 553–574. [Google Scholar] [CrossRef]
- Cohen, K.; Weinstein, A. The effects of cannabinoids on executive functions: Evidence from cannabis and synthetic cannabinoids—A systematic review. Brain Sci. 2018, 8, 40. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Pertwee, R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997, 74, 129–180. [Google Scholar] [CrossRef]
- Javid, F.A.; Phillips, R.M.; Afshinjavid, S.; Verde, R.; Ligresti, A. Cannabinoid pharmacology in cancer research: A new hope for cancer patients? Eur. J. Pharmacol. 2016, 775, 1–14. [Google Scholar] [CrossRef]
- Mielnik, C.A.; Lam, V.M.; Ross, R.A. CB1 allosteric modulators and their therapeutic potential in CNS disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 106, 110163. [Google Scholar] [CrossRef]
- Mackie, K. Distribution of Cannabinoid Receptors in the Central and Peripheral Nervous System. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2005; Volume 168, pp. 299–325. [Google Scholar] [CrossRef]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.-P.; Patel, S.; Perchuk, A.; Meozzi, P.A.; Myers, L.; Mora, Z.; Tagliaferro, P.; Gardner, E.; et al. Discovery of the Presence and Functional Expression of Cannabinoid CB2 Receptors in Brain. Ann. N. Y. Acad. Sci. 2006, 1074, 514–536. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef]
- Morales, P.; Reggio, P.H. An update on non-CB1, non-CB2 cannabinoid related G-protein-coupled receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef]
- Jett, J.; Stone, E.; Warren, G.; Cummings, K.M. Cannabis Use, Lung Cancer, and Related Issues. J. Thorac. Oncol. 2018, 13, 480–487. [Google Scholar] [CrossRef]
- Bogdanović, V.; Mrdjanović, J.; Borišev, I. A Review of the Therapeutic Antitumor Potential of Cannabinoids. J. Altern. Complement. Med. 2017, 23, 831–836. [Google Scholar] [CrossRef]
- Szutorisz, H.; Hurd, Y.L. High times for cannabis: Epigenetic imprint and its legacy on brain and behavior. Neurosci. Biobehav. Rev. 2018, 85, 93–101. [Google Scholar] [CrossRef]
- Montero-Oleas, N.; Arevalo-Rodriguez, I.; Nuñez-González, S.; Viteri-García, A.; Simancas-Racines, D. Therapeutic use of cannabis and cannabinoids: An evidence mapping and appraisal of systematic reviews. BMC Complement. Med. Ther. 2020, 20, 12. [Google Scholar] [CrossRef]
- Mouhamed, Y.; Vishnyakov, A.; Qorri, B.; Sambi, M.; Frank, S.S.; Nowierski, C.; Lamba, A.; Bhatti, U.; Szewczuk, M.R. Therapeutic potential of medicinal marijuana: An educational primer for health care professionals. Drug Healthc Patient Saf. 2018, 10, 45–66. [Google Scholar] [CrossRef]
- Peeri, H.; Koltai, H. Cannabis Biomolecule Effects on Cancer Cells and Cancer Stem Cells: Cytotoxic, Anti-Proliferative, and Anti-Migratory Activities. Biomolecules 2022, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Michalski, C.W.; Oti, F.E.; Erkan, M.; Sauliunaite, D.; Bergmann, F.; Pacher, P.; Batkai, S.; Müller, M.W.; Giese, N.A.; Friess, H.; et al. Cannabinoids in pancreatic cancer: Correlation with survival and pain. Int. J. Cancer 2008, 122, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yang, J.; Zhao, H.; Fang, X.; Li, H. Cannabinoid receptor 2 is upregulated in melanoma. J. Cancer Res. Ther. 2012, 8, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, B.; Ravi, J.; Ganju, R.K. Cannabinoids as therapeutic agents in cancer: Current status and future implications. Oncotarget 2014, 5, 5852–5872. [Google Scholar] [CrossRef]
- Pérez-Gómez, E.; Andradas, C.; Blasco-Benito, S.; Caffarel, M.M.; García-Taboada, E.; Villa-Morales, M.; Moreno, E.; Hamann, S.; Martín-Villar, E.; Flores, J.M.; et al. Role of cannabinoid receptor CB2 in HER2 pro-oncogenic signaling in breast cancer. J. Natl. Cancer Inst. 2015, 107, djv077. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, S23–S32. [Google Scholar] [CrossRef]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef]
- Ursini-Siegel, J.; Schade, B.; Cardiff, R.D.; Muller, W.J. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat. Rev. Cancer 2007, 7, 389–397. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Andradas, C.; Mira, E.; Pérez-Gómez, E.; Cerutti, C.; Moreno-Bueno, G.; Flores, J.M.; García-Real, I.; Palacios, J.; Mañes, S.; et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol. Cancer 2010, 9, 196. [Google Scholar] [CrossRef]
- Ramer, R.; Hinz, B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J. Natl. Cancer Inst. 2008, 100, 59–69. [Google Scholar] [CrossRef]
- Massi, P.; Solinas, M.; Cinquina, V.; Parolaro, D. Cannabidiol as potential anticancer drug. Br. J. Clin. Pharmacol. 2013, 75, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Vidinský, B.; Gál, P.; Pilátová, M.; Vidová, Z.; Solár, P.; Varinská, L.; Ivanová, L.; Mojžíš, J. Anti-proliferative and anti-angiogenic effects of CB2R agonist (JWH-133) in non-small lung cancer cells (A549) and human umbilical vein endothelial cells: An in vitro investigation. Folia Biol. 2012, 58, 75–80. [Google Scholar]
- Laezza, C.; D’Alessandro, A.; Paladino, S.; Maria Malfitano, A.; Chiara Proto, M.; Gazzerro, P.; Pisanti, S.; Santoro, A.; Ciaglia, E.; Bifulco, M.; et al. Anandamide inhibits the Wnt/β-catenin signalling pathway in human breast cancer MDA MB 231 cells. Eur. J. Cancer 2012, 48, 3112–3122. [Google Scholar] [CrossRef]
- Braile, M.; Marcella, S.; Marone, G.; Galdiero, M.R.; Varricchi, G.; Loffredo, S. The Interplay between the Immune and the Endocannabinoid Systems in Cancer. Cells 2021, 10, 1282. [Google Scholar] [CrossRef]
- Moreno, E.; Cavic, M.; Krivokuca, A.; Casadó, V.; Canela, E. The Endocannabinoid System as a Target in Cancer Diseases: Are We There Yet? Front. Pharmacol. 2019, 10, 339. [Google Scholar] [CrossRef]
- Zhu, L.X.; Sharma, S.; Stolina, M.; Gardner, B.; Roth, M.D.; Tashkin, D.P.; Dubinett, S.M. Δ-9-Tetrahydrocannabinol Inhibits Antitumor Immunity by a CB2 Receptor-Mediated, Cytokine-Dependent Pathway1. J. Immunol. 2000, 165, 373–380. [Google Scholar] [CrossRef]
- Hart, S.; Fischer, O.M.; Ullrich, A. Cannabinoids Induce Cancer Cell Proliferation via Tumor Necrosis Factor α-Converting Enzyme (TACE/ADAM17)-Mediated Transactivation of the Epidermal Growth Factor Receptor. Cancer Res. 2004, 64, 1943–1950. [Google Scholar] [CrossRef]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Δ-9-Tetrahydrocannabinol Enhances Breast Cancer Growth and Metastasis by Suppression of the Antitumor Immune Response1. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Ciaglia, E.; Gangemi, G.; Gazzerro, P.; Laezza, C.; Bifulco, M. Update on the endocannabinoid system as an anticancer target. Expert Opin. Ther. Targets 2011, 15, 297–308. [Google Scholar] [CrossRef]
- Sailler, S.; Schmitz, K.; Jäger, E.; Ferreiros, N.; Wicker, S.; Zschiebsch, K.; Pickert, G.; Geisslinger, G.; Walter, C.; Tegeder, I.; et al. Regulation of circulating endocannabinoids associated with cancer and metastases in mice and humans. Oncoscience 2014, 1, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic Activity of Cannabinoids2. JNCI J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef]
- Schwarz, R.; Ramer, R.; Hinz, B. Targeting the endocannabinoid system as a potential anticancer approach. Drug Metab. Rev. 2018, 50, 26–53. [Google Scholar] [CrossRef]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharmacol. 2019, 176, 1384–1394. [Google Scholar] [CrossRef]
- Kim, W.; Doyle, M.E.; Liu, Z.; Lao, Q.; Shin, Y.-K.; Carlson, O.D.; Kim, H.S.; Thomas, S.; Napora, J.K.; Lee, E.K.; et al. Cannabinoids Inhibit Insulin Receptor Signaling in Pancreatic β-Cells. Diabetes 2011, 60, 1198–1209. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, L.; Xiong, K.; Godlewski, G.; Mukhopadhyay, B.; Tam, J.; Yin, S.; Gao, P.; Shan, X.; Pickel, J.; et al. Hepatic Cannabinoid Receptor-1 Mediates Diet-Induced Insulin Resistance via Inhibition of Insulin Signaling and Clearance in Mice. Gastroenterology 2012, 142, 1218–1228.e1211. [Google Scholar] [CrossRef]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E.; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Kaufman, F.; Sandy, M.S.; Cardenas, A. Cannabis Exposure During Critical Windows of Development: Epigenetic and Molecular Pathways Implicated in Neuropsychiatric Disease. Curr. Environ. Health Rpt. 2020, 7, 325–342. [Google Scholar] [CrossRef]
- Schrott, R.; Acharya, K.; Itchon-Ramos, N.; Hawkey, A.B.; Pippen, E.; Mitchell, J.T.; Kollins, S.H.; Levin, E.D.; Murphy, S.K. Cannabis use is associated with potentially heritable widespread changes in autism candidate gene DLGAP2 DNA methylation in sperm. Epigenetics 2020, 15, 161–173. [Google Scholar] [CrossRef]
- Szutorisz, H.; DiNieri, J.A.; Sweet, E.; Egervari, G.; Michaelides, M.; Carter, J.M.; Ren, Y.; Miller, M.L.; Blitzer, R.D.; Hurd, Y.L. Parental THC Exposure Leads to Compulsive Heroin-Seeking and Altered Striatal Synaptic Plasticity in the Subsequent Generation. Neuropsychopharmacology 2014, 39, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Szutorisz, H.; Garg, P.; Martin, Q.; Landry, J.A.; Sharp, A.J.; Hurd, Y.L. Genome-Wide DNA Methylation Profiling Reveals Epigenetic Changes in the Rat Nucleus Accumbens Associated With Cross-Generational Effects of Adolescent THC Exposure. Neuropsychopharmacology 2015, 40, 2993–3005. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Fowler, J.S.; Wang, G.J.; Swanson, J.M. Dopamine in drug abuse and addiction: Results from imaging studies and treatment implications. Mol. Psychiatry 2004, 9, 557–569. [Google Scholar] [CrossRef] [PubMed]
- DiNieri, J.A.; Wang, X.; Szutorisz, H.; Spano, S.M.; Kaur, J.; Casaccia, P.; Dow-Edwards, D.; Hurd, Y.L. Maternal Cannabis Use Alters Ventral Striatal Dopamine D2 Gene Regulation in the Offspring. Biol. Psychiatry 2011, 70, 763–769. [Google Scholar] [CrossRef]
- Martínez-Peña, A.A.; Lee, K.; Pereira, M.; Ayyash, A.; Petrik, J.J.; Hardy, D.B.; Holloway, A.C. Prenatal Exposure to Delta-9-tetrahydrocannabinol (THC) Alters the Expression of miR-122-5p and Its Target Igf1r in the Adult Rat Ovary. Int. J. Mol. Sci. 2022, 23, 8000. [Google Scholar] [CrossRef]
- D’Addario, C.; Di Francesco, A.; Pucci, M.; Finazzi Agrò, A.; Maccarrone, M. Epigenetic mechanisms and endocannabinoid signalling. FEBS J. 2013, 280, 1905–1917. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Subbanna, S. Molecular Insights into Epigenetics and Cannabinoid Receptors. Biomolecules 2022, 12, 1560. [Google Scholar] [CrossRef]
- Khare, M.; Taylor, A.H.; Konje, J.C.; Bell, S.C. Δ9-Tetrahydrocannabinol inhibits cytotrophoblast cell proliferation and modulates gene transcription. Mol. Hum. Reprod. 2006, 12, 321–333. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lazar, M.A. Biochemical isolation and analysis of a nuclear receptor corepressor complex. Methods Enzymol. 2003, 364, 246–257. [Google Scholar]
- Pucci, M.; Rapino, C.; Di Francesco, A.; Dainese, E.; D’Addario, C.; Maccarrone, M. Epigenetic control of skin differentiation genes by phytocannabinoids. Br. J. Pharmacol. 2013, 170, 581–591. [Google Scholar] [CrossRef]
- Sido, J.M.; Yang, X.; Nagarkatti, P.S.; Nagarkatti, M. Δ9-Tetrahydrocannabinol-mediated epigenetic modifications elicit myeloid-derived suppressor cell activation via STAT3/S100A8. J. Leukoc. Biol. 2015, 97, 677–688. [Google Scholar] [CrossRef]
- Mondal, N.K.; Behera, J.; Kelly, K.E.; George, A.K.; Tyagi, P.K.; Tyagi, N. Tetrahydrocurcumin epigenetically mitigates mitochondrial dysfunction in brain vasculature during ischemic stroke. Neurochem. Int. 2019, 122, 120–138. [Google Scholar] [CrossRef]
- Sales, A.J.; Guimarães, F.S.; Joca, S.R.L. CBD modulates DNA methylation in the prefrontal cortex and hippocampus of mice exposed to forced swim. Behav. Brain Res. 2020, 388, 112627. [Google Scholar] [CrossRef]
- Fuchs Weizman, N.; Wyse, B.A.; Szaraz, P.; Defer, M.; Jahangiri, S.; Librach, C.L. Cannabis alters epigenetic integrity and endocannabinoid signalling in the human follicular niche. Hum. Reprod. 2021, 36, 1922–1931. [Google Scholar] [CrossRef]
- Paradisi, A.; Pasquariello, N.; Barcaroli, D.; Maccarrone, M. Anandamide Regulates Keratinocyte Differentiation by Inducing DNA Methylation in a CB1 Receptor-dependent Manner. J. Biol. Chem. 2008, 283, 6005–6012. [Google Scholar] [CrossRef]
- Hwang, Y.S.; Kim, Y.-J.; Kim, M.O.; Kang, M.; Oh, S.W.; Nho, Y.H.; Park, S.-H.; Lee, J. Cannabidiol upregulates melanogenesis through CB1 dependent pathway by activating p38 MAPK and p42/44 MAPK. Chem. Biol. Interact. 2017, 273, 107–114. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Zhang, L.; Xiao, X.; Arnold, P.R.; Li, X.C. Transcriptional and epigenetic regulation of immune tolerance: Roles of the NF-κB family members. Cell Mol. Immunol. 2019, 16, 315–323. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Verma, I.M. Nuclear factor (NF)-B proteins: Therapeutic targets. Ann. Rheum. Dis. 2004, 63, ii57–ii61. [Google Scholar] [CrossRef]
- Tagde, A.; Rajabi, H.; Stroopinsky, D.; Gali, R.; Alam, M.; Bouillez, A.; Kharbanda, S.; Stone, R.; Avigan, D.; Kufe, D. MUC1-C induces DNA methyltransferase 1 and represses tumor suppressor genes in acute myeloid leukemia. Oncotarget 2016, 7, 38974–38987. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Z.; Xie, Z.; Pang, J.; Yu, J.; Lehmann, E.; Huynh, L.; Vukosavljevic, T.; Takeki, M.; Klisovic, R.B.; et al. Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-κB–dependent DNA methyltransferase activity in acute myeloid leukemia. Blood 2008, 111, 2364–2373. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The Histone H3 Lysine-27 Demethylase Jmjd3 Links Inflammation to Inhibition of Polycomb-Mediated Gene Silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef]
- Federman, N.; de la Fuente, V.; Zalcman, G.; Corbi, N.; Onori, A.; Passananti, C.; Romano, A. Nuclear Factor B-Dependent Histone Acetylation is Specifically Involved in Persistent Forms of Memory. J. Neurosci. 2013, 33, 7603–7614. [Google Scholar] [CrossRef] [PubMed]
- Teferedegne, B.; Green, M.R.; Guo, Z.; Boss, J.M. Mechanism of Action of a Distal NF-κB-Dependent Enhancer. Mol. Cell Biol. 2006, 26, 5759–5770. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Vento-Tormo, R.; Rodríguez-Ubreva, J.; Lisio, L.D.; Islam, A.B.; Urquiza, J.M.; Hernando, H.; Lopez-Bigas, N.; Shannon-Lowe, C.; Martínez, N.; Montes-Moreno, S. NF-κB directly mediates epigenetic deregulation of common microRNAs in Epstein-Barr virus-mediated transformation of B-cells and in lymphomas. Nucleic Acids Res. 2014, 42, 11025–11039. [Google Scholar] [CrossRef]
- Kozela, E.; Pietr, M.; Juknat, A.; Rimmerman, N.; Levy, R.; Vogel, Z. Cannabinoids Δ9-Tetrahydrocannabinol and Cannabidiol Differentially Inhibit the Lipopolysaccharide-activated NF-κB and Interferon-β/STAT Proinflammatory Pathways in BV-2 Microglial Cells*. J. Biol. Chem. 2010, 285, 1616–1626. [Google Scholar] [CrossRef]
- Hassan, S. Effect of Cannabinoid Receptor Ligands on Microglial Cell Functions. Ph.D. Thesis, The University of Nottingham, Nottingham, UK, 2013. [Google Scholar]
- Huang, Y.; Wan, T.; Pang, N.; Zhou, Y.; Jiang, X.; Li, B.; Gu, Y.; Huang, Y.; Ye, X.; Lian, H.; et al. Cannabidiol protects livers against nonalcoholic steatohepatitis induced by high-fat high cholesterol diet via regulating NF-κB and NLRP3 inflammasome pathway. J. Cell Physiol. 2019, 234, 21224–21234. [Google Scholar] [CrossRef]
- González-Mariscal, I.; Carmona-Hidalgo, B.; Winkler, M.; Unciti-Broceta, J.D.; Escamilla, A.; Gómez-Cañas, M.; Fernández-Ruiz, J.; Fiebich, B.L.; Romero-Zerbo, S.-Y.; Bermúdez-Silva, F.J.; et al. (+)-trans-Cannabidiol-2-hydroxy pentyl is a dual CB1R antagonist/CB2R agonist that prevents diabetic nephropathy in mice. Pharmacol. Res. 2021, 169, 105492. [Google Scholar] [CrossRef]
- Krzyżewska, A.; Baranowska-Kuczko, M.; Jastrząb, A.; Kasacka, I.; Koz\lowska, H. Cannabidiol Improves Antioxidant Capacity and Reduces Inflammation in the Lungs of Rats with Monocrotaline-Induced Pulmonary Hypertension. Molecules 2022, 27, 3327. [Google Scholar] [CrossRef]
- Do, Y.; McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Activation through cannabinoid receptors 1 and 2 on dendritic cells triggers NF-κB-dependent apoptosis: Novel role for endogenous and exogenous cannabinoids in immunoregulation. J. Immunol. 2004, 173, 2373–2382. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, J.; Fan, N.; Teng, Z.-q.; Wu, Y.; Yang, H.; Tang, Y.-p.; Sun, H.; Song, Y.; Chen, C. Δ9-THC-Caused Synaptic and Memory Impairments Are Mediated through COX-2 Signaling. Cell 2013, 155, 1154–1165. [Google Scholar] [CrossRef]
- Jouroukhin, Y.; Zhu, X.; Shevelkin, A.V.; Hasegawa, Y.; Abazyan, B.; Saito, A.; Pevsner, J.; Kamiya, A.; Pletnikov, M.V. Adolescent Δ9-THC exposure and astrocyte-specific genetic vulnerability converge on NF-κB-COX-2 signaling to impair memory in adulthood. Biol. Psychiatry 2019, 85, 891–903. [Google Scholar] [CrossRef]
- Wei, T.-T.; Chandy, M.; Nishiga, M.; Zhang, A.; Kumar, K.K.; Thomas, D.; Manhas, A.; Rhee, S.; Justesen, J.M.; Chen, I.Y.; et al. Cannabinoid receptor 1 antagonist genistein attenuates marijuana-induced vascular inflammation. Cell 2022, 185, 1676–1693.e1623. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- Baltoumas, F.A.; Theodoropoulou, M.C.; Hamodrakas, S.J. Interactions of the α-subunits of heterotrimeric G-proteins with GPCRs, effectors and RGS proteins: A critical review and analysis of interacting surfaces, conformational shifts, structural diversity and electrostatic potentials. J. Struct. Biol. 2013, 182, 209–218. [Google Scholar] [CrossRef]
- Khajehali, E.; Malone, D.T.; Glass, M.; Sexton, P.M.; Christopoulos, A.; Leach, K. Biased agonism and biased allosteric modulation at the CB1 cannabinoid receptor. Mol. Pharmacol. 2015, 88, 368–379. [Google Scholar] [CrossRef]
- Olianas, M.C.; Dedoni, S.; Onali, P. Cannabinoid CB1 and CB2 receptors differentially regulate TNF-α-induced apoptosis and LPA1-mediated pro-survival signaling in HT22 hippocampal cells. Life Sci. 2021, 276, 119407. [Google Scholar] [CrossRef]
- Yang, H.; Wang, Z.; Capó-Aponte, J.E.; Zhang, F.; Pan, Z.; Reinach, P.S. Epidermal growth factor receptor transactivation by the cannabinoid receptor (CB1) and transient receptor potential vanilloid 1 (TRPV1) induces differential responses in corneal epithelial cells. Exp. Eye Res. 2010, 91, 462–471. [Google Scholar] [CrossRef]
- Fitzpatrick, J.-M.; Hackett, B.; Costelloe, L.; Hind, W.; Downer, E.J. Botanically-Derived Δ9-Tetrahydrocannabinol and Cannabidiol, and Their 1: 1 Combination, Modulate Toll-like Receptor 3 and 4 Signalling in Immune Cells from People with Multiple Sclerosis. Molecules 2022, 27, 1763. [Google Scholar] [CrossRef] [PubMed]
- Keresztes, A.; Streicher, J.M. Synergistic interaction of the cannabinoid and death receptor systems—A potential target for future cancer therapies? FEBS Lett. 2017, 591, 3235–3251. [Google Scholar] [CrossRef] [PubMed]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef] [PubMed]
- Sabatucci, A.; Tortolani, D.; Dainese, E.; Maccarrone, M. In silico mapping of allosteric ligand binding sites in type-1 cannabinoid receptor. Biotechnol. Appl. Biochem. 2018, 65, 21–28. [Google Scholar] [CrossRef]
- Qorri, B.; Kalaydina, R.V.; Velickovic, A.; Kaplya, Y.; Decarlo, A.; Szewczuk, M.R. Agonist-Biased Signaling via Matrix Metalloproteinase-9 Promotes Extracellular Matrix Remodeling. Cells 2018, 7, 117. [Google Scholar] [CrossRef]
- Jakowiecki, J.; Abel, R.; Orzeł, U.; Pasznik, P.; Preissner, R.; Filipek, S. Allosteric Modulation of the CB1 Cannabinoid Receptor by Cannabidiol—A Molecular Modeling Study of the N-Terminal Domain and the Allosteric-Orthosteric Coupling. Molecules 2021, 26, 2456. [Google Scholar] [CrossRef]
- Reber, L.; Vermeulen, L.; Haegeman, G.; Frossard, N. Ser276 phosphorylation of NF-κBp65 by MSK1 controls SCF expression in inflammation. PLoS ONE 2009, 4, e4393. [Google Scholar] [CrossRef]
- Verstrepen, L.; Bekaert, T.; Chau, T.L.; Tavernier, J.; Chariot, A.; Beyaert, R. TLR-4, IL-1R and TNF-R signaling to NF-κB: Variations on a common theme. Cell. Mol. Life Sci. 2008, 65, 2964–2978. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.-C. NF-κB in inflammation and renal diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef]
- Hua, F.; Hao, W.; Wang, L.; Li, S. Linear Ubiquitination Mediates EGFR-Induced NF-κB Pathway and Tumor Development. Int. J. Mol. Sci. 2021, 22, 11875. [Google Scholar] [CrossRef]
- Asimaki, O.; Sakellaridis, N.; Mangoura, D. Transactivation of epidermal growth factor receptor EGFR by CB1 cannabinoid receptor agonists. Ep. Klin. Farmakol. Kai Farmakokinet. 2007, 25, 19–21. [Google Scholar]
- Haxho, F.; Haq, S.; Szewczuk, M.R. Biased G protein-coupled receptor agonism mediates Neu1 sialidase and matrix metalloproteinase-9 crosstalk to induce transactivation of insulin receptor signaling. Cell. Signal. 2018, 43, 71–84. [Google Scholar] [CrossRef]
- Jayanth, P.; Amith, S.R.; Gee, K.; Szewczuk, M.R. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for neurotrophin activation of Trk receptors and cellular signaling. Cell. Signal. 2010, 22, 1193–1205. [Google Scholar] [CrossRef]
- Abdulkhalek, S.; Guo, M.; Amith, S.R.; Jayanth, P.; Szewczuk, M.R. G-protein coupled receptor agonists mediate Neu1 sialidase and matrix metalloproteinase-9 cross-talk to induce transactivation of TOLL-like receptors and cellular signaling. Cell. Signal. 2012, 24, 2035–2042. [Google Scholar] [CrossRef]
- Erica, Z.; Cristina, M.; Marina, G.; Tiziana, R.; Daniela, P. Chapter 6—Correlates and consequences of cannabinoid exposure on adolescent brain remodeling: Focus on glial cells and epigenetics. In Cannabis and the Developing Brain; Melis, M., Manzoni, O.J.J., Melis, M., Manzoni, O.J.J., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 95–106. [Google Scholar]
- Costa, B.; Siniscalco, D.; Trovato, A.E.; Comelli, F.; Sotgiu, M.L.; Colleoni, M.; Maione, S.; Rossi, F.; Giagnoni, G. AM404, an inhibitor of anandamide uptake, prevents pain behaviour and modulates cytokine and apoptotic pathways in a rat model of neuropathic pain. Br. J. Pharmacol. 2006, 148, 1022–1032. [Google Scholar] [CrossRef]
- Paszcuk, A.F.; Dutra, R.C.; da Silva, K.A.B.S.; Quintão, N.L.M.; Campos, M.M.; Calixto, J.B. Cannabinoid Agonists Inhibit Neuropathic Pain Induced by Brachial Plexus Avulsion in Mice by Affecting Glial Cells and MAP Kinases. PLoS ONE 2011, 6, e24034. [Google Scholar] [CrossRef]
- Rozenfeld, R.; Gupta, A.; Gagnidze, K.; Lim, M.P.; Gomes, I.; Lee-Ramos, D.; Nieto, N.; Devi, L.A. AT1R-CB₁R heteromerization reveals a new mechanism for the pathogenic properties of angiotensin II. EMBO J. 2011, 30, 2350–2363. [Google Scholar] [CrossRef]
- Alghamdi, F.; Guo, M.; Abdulkhalek, S.; Crawford, N.; Amith, S.R.; Szewczuk, M.R. A novel insulin receptor-signaling platform and its link to insulin resistance and type 2 diabetes. Cell. Signal. 2014, 26, 1355–1368. [Google Scholar] [CrossRef]
- Bordoni, L.; Gabbianelli, R. Primers on nutrigenetics and nutri(epi)genomics: Origins and development of precision nutrition. Biochimie 2019, 160, 156–171. [Google Scholar] [CrossRef]
- Hinek, A.; Bodnaruk, T.D.; Bunda, S.; Wang, Y.; Liu, K. Neuraminidase-1, a subunit of the cell surface elastin receptor, desialylates and functionally inactivates adjacent receptors interacting with the mitogenic growth factors PDGF-BB and IGF-2. Am. J. Pathol. 2008, 173, 1042–1056. [Google Scholar] [CrossRef]
- Abdulkhalek, S.; Amith, S.R.; Franchuk, S.L.; Jayanth, P.; Guo, M.; Finlay, T.; Gilmour, A.; Guzzo, C.; Gee, K.; Beyaert, R. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for Toll-like receptor activation and cellular signaling. J. Biol. Chem. 2011, 286, 36532–36549. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, A.M.; Abdulkhalek, S.; Cheng, T.S.W.; Alghamdi, F.; Jayanth, P.; O’Shea, L.K.; Geen, O.; Arvizu, L.A.; Szewczuk, M.R. A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell. Signal. 2013, 25, 2587–2603. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Seyrantepe, V.; Landry, K.; Ahmad, R.; Ahmad, A.; Stamatos, N.M.; Pshezhetsky, A.V. Monocyte differentiation up-regulates the expression of the lysosomal sialidase, Neu1, and triggers its targeting to the plasma membrane via major histocompatibility complex class II-positive compartments*. J. Biol. Chem. 2006, 281, 27526–27538. [Google Scholar] [CrossRef] [PubMed]
- Abdulkhalek, S.; Hrynyk, M.; Szewczuk, M.R. A novel G-protein-coupled receptor-signaling platform and its targeted translation in human disease. Res. Rep. Biochem. 2013, 3, 17–30. [Google Scholar]
- Vajaria, B.N.; Patel, P.S. Glycosylation: A hallmark of cancer? Glycoconj. J. 2017, 34, 147–156. [Google Scholar] [CrossRef]
- Sparrow, L.G.; Lawrence, M.C.; Gorman, J.J.; Strike, P.M.; Robinson, C.P.; McKern, N.M.; Ward, C.W. N-linked glycans of the human insulin receptor and their distribution over the crystal structure. Proteins Struct. Funct. Bioinform. 2008, 71, 426–439. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E. Insulin resistance: A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 1991, 14, 173–194. [Google Scholar] [CrossRef]
- Kahn, S.E. The importance of the β-cell in the pathogenesis of type 2 diabetes mellitus. Am. J. Med. 2000, 108, 2–8. [Google Scholar] [CrossRef]
- Hirabara, S.M.; Gorjão, R.; Vinolo, M.A.; Rodrigues, A.C.; Nachbar, R.T.; Curi, R. Molecular targets related to inflammation and insulin resistance and potential interventions. J. Biomed. Biotechnol. 2012, 2012, 379024. [Google Scholar] [CrossRef]
- Arabkhari, M.; Bunda, S.; Wang, Y.; Wang, A.; Pshezhetsky, A.V.; Hinek, A. Desialylation of insulin receptors and IGF-1 receptors by neuraminidase-1 controls the net proliferative response of L6 myoblasts to insulin. Glycobiology 2010, 20, 603–616. [Google Scholar] [CrossRef]
- Fischoeder, A.; Meyborg, H.; Stibenz, D.; Fleck, E.; Graf, K.; Stawowy, P. Insulin augments matrix metalloproteinase-9 expression in monocytes. Cardiovasc. Res. 2007, 73, 841–848. [Google Scholar] [CrossRef]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1, 2005-0010. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Bonten, E.J.; Annunziata, I.; d’Azzo, A. Lysosomal multienzyme complex: Pros and cons of working together. Cell. Mol. Life Sci. 2014, 71, 2017–2032. [Google Scholar] [CrossRef]
- Ono, M.; Kuwano, M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef]
- Gazdar, A.F. Personalized medicine and inhibition of EGFR signaling in lung cancer. N. Engl. J. Med. 2009, 361, 1018. [Google Scholar] [CrossRef]
- Haxho, F.; Neufeld, R.J.; Szewczuk, M.R. Neuraminidase-1: A novel therapeutic target in multistage tumorigenesis. Oncotarget 2016, 7, 40860–40881. [Google Scholar] [CrossRef]
- O’Shea, L.K.; Abdulkhalek, S.; Allison, S.; Neufeld, R.J.; Szewczuk, M.R. Therapeutic targeting of Neu1 sialidase with oseltamivir phosphate (Tamiflu®) disables cancer cell survival in human pancreatic cancer with acquired chemoresistance. Onco Targets Ther. 2014, 7, 117–134. [Google Scholar] [CrossRef]
- Abdulkhalek, S.; Geen, O.D.; Brodhagen, L.; Haxho, F.; Alghamdi, F.; Allison, S.; Simmons, D.J.; O’Shea, L.K.; Neufeld, R.J.; Szewczuk, M.R. Transcriptional factor snail controls tumor neovascularization, growth and metastasis in mouse model of human ovarian carcinoma. Clin. Transl. Med. 2014, 3, 28. [Google Scholar] [CrossRef]
- Wong, H.H.; Lemoine, N.R. Pancreatic cancer: Molecular pathogenesis and new therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Sarmento-Ribeiro, A.B.; Scorilas, A.; Gonçalves, A.C.; Efferth, T.; Trougakos, I.P. The emergence of drug resistance to targeted cancer therapies: Clinical evidence. Drug Resist. Updates 2019, 47, 100646. [Google Scholar] [CrossRef] [PubMed]
- Sambi, M.; Szewczuk, M.R. Introduction to the Acquisition of Resistance to Targeted Therapy. In Current Applications for Overcoming Resistance to Targeted Therapies; Springer Nature Switzerland AG: Cham, Switzerland, 2019; pp. 1–33. [Google Scholar]
- Al-Zeheimi, N.; Adham, S.A. Therapies to Overcome Multidrug-Resistant Receptors. In Current Applications for Overcoming Resistance to Targeted Therapies; Springer Nature Switzerland AG: Cham, Switzerland, 2019; pp. 131–159. [Google Scholar]
- Long, S.S.; Prober, C.G.; Fischer, M. Principles and Practice of Pediatric Infectious Diseases E-Book; Elsevier Health Sciences: Philadelphia, PA, USA, 2022. [Google Scholar]
- Haxho, F.; Allison, S.; Alghamdi, F.; Brodhagen, L.; Kuta, V.E.L.; Abdulkhalek, S.; Neufeld, R.J.; Szewczuk, M.R. Oseltamivir phosphate monotherapy ablates tumor neovascularization, growth, and metastasis in mouse model of human triple-negative breast adenocarcinoma. Breast Cancer Targets Ther. 2014, 6, 191. [Google Scholar]
- Amith, S.R.; Jayanth, P.; Franchuk, S.; Siddiqui, S.; Seyrantepe, V.; Gee, K.; Basta, S.; Beyaert, R.; Pshezhetsky, A.V.; Szewczuk, M.R. Dependence of pathogen molecule-induced Toll-like receptor activation and cell function on Neu1 sialidase. Glycoconj. J. 2009, 26, 1197. [Google Scholar] [CrossRef] [PubMed]
- Abdulkhalek, S.; Szewczuk, M.R. Neu1 sialidase and matrix metalloproteinase-9 cross-talk regulates nucleic acid-induced endosomal TOLL-like receptor-7 and-9 activation, cellular signaling and pro-inflammatory responses. Cell. Signal. 2013, 25, 2093–2105. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Fan, P.; Bauer, N.; Gladkich, J.; Ryschich, E.; Bazhin, A.V.; Giese, N.A.; Strobel, O.; Hackert, T.; et al. Aspirin counteracts cancer stem cell features, desmoplasia and gemcitabine resistance in pancreatic cancer. Oncotarget 2015, 6, 9999–10015. [Google Scholar] [CrossRef]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef]
- Yin, M.-J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase-β. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- Qorri, B.; Harless, W.; Szewczuk, M.R. Novel Molecular Mechanism of Aspirin and Celecoxib Targeting Mammalian Neuraminidase-1 Impedes Epidermal Growth Factor Receptor Signaling Axis and Induces Apoptosis in Pancreatic Cancer Cells. Drug Des. Devel. Ther. 2020, 14, 4149–4167. [Google Scholar] [CrossRef]
- Sambi, M.; Samuel, V.; Qorri, B.; Haq, S.; Burov, S.V.; Markvicheva, E.; Harless, W.; Szewczuk, M.R. A Triple Combination of Metformin, Acetylsalicylic Acid, and Oseltamivir Phosphate Impacts Tumour Spheroid Viability and Upends Chemoresistance in Triple-Negative Breast Cancer. Drug Des. Devel. Ther. 2020, 14, 1995–2019. [Google Scholar] [CrossRef]
- Qorri, B.; Mokhtari, R.B.; Harless, W.W.; Szewczuk, M.R. Next Generation of Cancer Drug Repurposing: Therapeutic Combination of Aspirin and Oseltamivir Phosphate Potentiates Gemcitabine to Disable Key Survival Pathways Critical for Pancreatic Cancer Progression. Cancers 2022, 14, 1374. [Google Scholar] [CrossRef]
- Qorri, B.; Mokhtari, R.B.; Harless, W.W.; Szewczuk, M.R. Repositioning of Old Drugs for Novel Cancer Therapies: Continuous Therapeutic Perfusion of Aspirin and Oseltamivir Phosphate with Gemcitabine Treatment Disables Tumor Progression, Chemoresistance, and Metastases. Cancers 2022, 14, 3595. [Google Scholar] [CrossRef]
- Blanco, M.J.; Moreno-Bueno, G.; Sarrio, D.; Locascio, A.; Cano, A.; Palacios, J.; Nieto, M.A. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002, 21, 3241–3246. [Google Scholar] [CrossRef]
- Jordà, M.; Olmeda, D.; Vinyals, A.; Valero, E.; Cubillo, E.; Llorens, A.; Cano, A.; Fabra, A. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J. Cell Sci. 2005, 118, 3371–3385. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix metalloproteinases in neuroinflammation. Glia 2002, 39, 279–291. [Google Scholar] [CrossRef]
- Wang, H.-H.; Hsieh, H.-L.; Wu, C.-Y.; Sun, C.-C.; Yang, C.-M. Oxidized low-density lipoprotein induces matrix metalloproteinase-9 expression via a p42/p44 and JNK-dependent AP-1 pathway in brain astrocytes. Glia 2009, 57, 24–38. [Google Scholar] [CrossRef]
- Speidl, W.S.; Kastl, S.P.; Hutter, R.; Katsaros, K.M.; Kaun, C.; Bauriedel, G.; Maurer, G.; Huber, K.; Badimon, J.J.; Wojta, J. The complement component C5a is present in human coronary lesions in vivo and induces the expression of MMP-1 and MMP-9 in human macrophages in vitro. FASEB J. 2011, 25, 35–44. [Google Scholar] [CrossRef]
- Hwang, Y.P.; Yun, H.J.; Choi, J.H.; Han, E.H.; Kim, H.G.; Song, G.Y.; Kwon, K.-I.; Jeong, T.C.; Jeong, H.G. Suppression of EGF-induced tumor cell migration and matrix metalloproteinase-9 expression by capsaicin via the inhibition of EGFR-mediated FAK/Akt, PKC/Raf/ERK, p38 MAPK, and AP-1 signaling. Mol. Nutr. Food Res. 2011, 55, 594–605. [Google Scholar] [CrossRef]
- Park, J.; Kwak, C.-H.; Ha, S.-H.; Kwon, K.-M.; Abekura, F.; Cho, S.-H.; Chang, Y.-C.; Lee, Y.-C.; Ha, K.-T.; Chung, T.-W.; et al. Ganglioside GM3 suppresses lipopolysaccharide-induced inflammatory responses in rAW 264.7 macrophage cells through NF-κB, AP-1, and MAPKs signaling. J. Cell Biochem. 2018, 119, 1173–1182. [Google Scholar] [CrossRef]
- Ryan, A.E.; Colleran, A.; O’Gorman, A.; O’Flynn, L.; Pindjacova, J.; Lohan, P.; O’Malley, G.; Nosov, M.; Mureau, C.; Egan, L.J. Targeting colon cancer cell NF-κB promotes an anti-tumour M1-like macrophage phenotype and inhibits peritoneal metastasis. Oncogene 2015, 34, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.G.; Bhatia, S.K.; Anderson, C.M.; Eichenberger-Gilmore, J.M.; Sibenaller, Z.A.; Mapuskar, K.A.; Schoenfeld, J.D.; Buatti, J.M.; Spitz, D.R.; Fath, M.A. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biol. 2014, 2, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, C.; Jin, L.; Zhang, R.; Wang, T.; Wang, Q.; Chen, J.; Yang, F.; Siebert, H.-C.; Zheng, X. Ketogenic Diet Elicits Antitumor Properties through Inducing Oxidative Stress, Inhibiting MMP-9 Expression, and Rebalancing M1/M2 Tumor-Associated Macrophage Phenotype in a Mouse Model of Colon Cancer. J. Agric. Food Chem. 2020, 68, 11182–11196. [Google Scholar] [CrossRef]
- Park, S.K.; Lin, H.L.; Murphy, S. Nitric oxide regulates nitric oxide synthase-2 gene expression by inhibiting NF-kappaB binding to DNA. Biochem. J. 1997, 322, 609–613. [Google Scholar] [CrossRef]
- Eberhardt, W.; Beeg, T.; Beck, K.F.; Walpen, S.; Gauer, S.; Böhles, H.; Pfeilschifter, J. Nitric oxide modulates expression of matrix metalloproteinase-9 in rat mesangial cells. Kidney Int. 2000, 57, 59–69. [Google Scholar] [CrossRef]
- Chen, H.-H.; Wang, D.L. Nitric oxide inhibits matrix metalloproteinase-2 expression via the induction of activating transcription factor 3 in endothelial cells. Mol. Pharmacol. 2004, 65, 1130–1140. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Windhausen, A.N.; Isenberg, J.S.; Yeung, N.; Thomas, D.D.; Vitek, M.P.; Roberts, D.D.; Wink, D.A. Nitric oxide regulates matrix metalloproteinase-9 activity by guanylyl-cyclase-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 16898–16903. [Google Scholar] [CrossRef]
- O’Sullivan, S.; Medina, C.; Ledwidge, M.; Radomski, M.W.; Gilmer, J.F. Nitric oxide-matrix metaloproteinase-9 interactions: Biological and pharmacological significance: NO and MMP-9 interactions. Biochim. Biophys. Acta 2014, 1843, 603–617. [Google Scholar] [CrossRef]
- Koodie, L.; Ramakrishnan, S.; Roy, S. Morphine Suppresses Tumor Angiogenesis through a HIF-1α/p38MAPK Pathway. Am. J. Pathol. 2010, 177, 984–997. [Google Scholar] [CrossRef]
- Gach, K.; Wyrębska, A.; Fichna, J.; Janecka, A. The role of morphine in regulation of cancer cell growth. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 384, 221–230. [Google Scholar] [CrossRef]
- Afsharimani, B.; Baran, J.; Watanabe, S.; Lindner, D.; Cabot, P.J.; Parat, M.-O. Morphine and breast tumor metastasis: The role of matrix-degrading enzymes. Clin. Exp. Metastasis 2014, 31, 149–158. [Google Scholar] [CrossRef]
- Koodie, L.; Yuan, H.; Pumper, J.A.; Yu, H.; Charboneau, R.; Ramkrishnan, S.; Roy, S. Morphine inhibits migration of tumor-infiltrating leukocytes and suppresses angiogenesis associated with tumor growth in mice. Am. J. Pathol. 2014, 184, 1073–1084. [Google Scholar] [CrossRef]
- Harimaya, Y.; Koizumi, K.; Andoh, T.; Nojima, H.; Kuraishi, Y.; Saiki, I. Potential ability of morphine to inhibit the adhesion, invasion and metastasis of metastatic colon 26-L5 carcinoma cells. Cancer Lett. 2002, 187, 121–127. [Google Scholar] [CrossRef]
- Gach, K.; Wyrebska, A.; Szemraj, J.; Janecka, A. The influence of opioid peptides on matrix metalloproteinase-9 and urokinase plasminogen activator expression in three cancer cell lines. Mol. Biol. 2012, 46, 894–899. [Google Scholar] [CrossRef]
- Bimonte, S.; Barbieri, A.; Palma, G.; Arra, C. The role of morphine in animal models of human cancer: Does morphine promote or inhibit the tumor growth? Biomed. Res. Int. 2013, 2013, 258141. [Google Scholar] [CrossRef]
- Roy, S.; Cain, K.J.; Chapin, R.B.; Charboneau, R.G.; Barke, R.A. Morphine Modulates NFκB Activation in Macrophages. Biochem. Biophys. Res. Commun. 1998, 245, 392–396. [Google Scholar] [CrossRef]
- Khabbazi, S.; Hassanshahi, M.; Hassanshahi, A.; Peymanfar, Y.; Su, Y.-W.; Xian, C.J. Opioids and matrix metalloproteinases: The influence of morphine on MMP-9 production and cancer progression. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 123–133. [Google Scholar] [CrossRef]
- Sawada, M.; Ichinose, M.; Stefano, G.B. Nitric oxide inhibits the dopamine-induced K+ current via guanylate cyclase in Aplysia neurons. J. Neurosci. Res. 1997, 50, 450–456. [Google Scholar] [CrossRef]
- Fimiani, C.; Arcuri, E.; Santoni, A.; Rialas, C.M.; Bilfinger, T.V.; Peter, D.; Salzet, B.; Stefano, G.B. Mu3 opiate receptor expression in lung and lung carcinoma: Ligand binding and coupling to nitric oxide release. Cancer Lett. 1999, 146, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Welters, I.D.; Menzebach, A.; Goumon, Y.; Cadet, P.; Menges, T.; Hughes, T.K.; Hempelmann, G.; Stefano, G.B. Morphine inhibits NF-kappaB nuclear binding in human neutrophils and monocytes by a nitric oxide-dependent mechanism. Anesthesiology 2000, 92, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Kream, R.M.; Mantione, K.J.; Sheehan, M.; Cadet, P.; Zhu, W.; Bilfinger, T.V.; Esch, T. Endogenous morphine/nitric oxide-coupled regulation of cellular physiology and gene expression: Implications for cancer biology. Semin. Cancer Biol. 2008, 18, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Gach, K.; Szemraj, J.; Wyrębska, A.; Janecka, A. The influence of opioids on matrix metalloproteinase-2 and-9 secretion and mRNA levels in MCF-7 breast cancer cell line. Mol. Biol. Rep. 2011, 38, 1231–1236. [Google Scholar] [CrossRef]
- Lee, Y.J.; Suh, S.-Y.; Song, J.; Lee, S.S.; Seo, A.-R.; Ahn, H.-Y.; Lee, M.A.; Kim, C.-M.; Klepstad, P. Serum and urine concentrations of morphine and morphine metabolites in patients with advanced cancer receiving continuous intravenous morphine: An observational study. BMC Palliat. Care 2015, 14, 53. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Nielsen, H.M.; Hansen, L.L. Epigenetics and cancer treatment. Eur. J. Pharmacol. 2009, 625, 131–142. [Google Scholar] [CrossRef]
- Sung, B.; Pandey, M.K.; Ahn, K.S.; Yi, T.; Chaturvedi, M.M.; Liu, M.; Aggarwal, B.B. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood 2008, 111, 4880–4891. [Google Scholar] [CrossRef]
- Tan, J.; Chen, B.; He, L.; Tang, Y.; Jiang, Z.; Yin, G.; Wang, J.; Jiang, X. Anacardic acid (6-pentadecylsalicylic acid) induces apoptosis of prostate cancer cells through inhibition of androgen receptor and activation of p53 signaling. Chin. J. Cancer Res. 2012, 24, 275–283. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Coolen, M.; Katz, S.; Bally-Cuif, L. miR-9: A versatile regulator of neurogenesis. Front. Cell. Neurosci. 2013, 7, 220. [Google Scholar] [CrossRef]
- Fuziwara, C.S.; Kimura, E.T. Insights into Regulation of the miR-17-92 Cluster of miRNAs in Cancer. Front. Med. 2015, 2, 64. [Google Scholar] [CrossRef]
- Pfeffer, S.R.; Yang, C.H.; Pfeffer, L.M. The Role of miR-21 in Cancer. Drug Dev. Res. 2015, 76, 270–277. [Google Scholar] [CrossRef]
- Sheedy, P.; Medarova, Z. The fundamental role of miR-10b in metastatic cancer. Am. J. Cancer Res. 2018, 8, 1674–1688. [Google Scholar]
- Li, X.; Gao, L.; Cui, Q.; Gary, B.D.; Dyess, D.L.; Taylor, W.; Shevde, L.A.; Samant, R.S.; Dean-Colomb, W.; Piazza, G.A.; et al. Sulindac inhibits tumor cell invasion by suppressing NF-κB-mediated transcription of microRNAs. Oncogene 2012, 31, 4979–4986. [Google Scholar] [CrossRef]
- van der Donk, W.A.; Tsai, A.-L.; Kulmacz, R.J. The cyclooxygenase reaction mechanism. Biochemistry 2002, 41, 15451–15458. [Google Scholar] [CrossRef]
- Hata, A.N.; Breyer, R.M. Pharmacology and signaling of prostaglandin receptors: Multiple roles in inflammation and immune modulation. Pharmacol. Ther. 2004, 103, 147–166. [Google Scholar] [CrossRef]
- Rolland, P.H.; Martin, P.M.; Jacquemier, J.; Rolland, A.M.; Toga, M. Prostaglandin in human breast cancer: Evidence suggesting that an elevated prostaglandin production is a marker of high metastatic potential for neoplastic cells. J. Natl. Cancer Inst. 1980, 64, 1061–1070. [Google Scholar]
- Kargman, S.L.; O’Neill, G.P.; Vickers, P.J.; Evans, J.F.; Mancini, J.A.; Jothy, S. Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res. 1995, 55, 2556–2559. [Google Scholar]
- Surh, Y.-J.; Chun, K.-S.; Cha, H.-H.; Han, S.S.; Keum, Y.-S.; Park, K.-K.; Lee, S.S. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: Down-regulation of COX-2 and iNOS through suppression of NF-κB activation. Mutat. Res. /Fundam. Mol. Mech. Mutagen. 2001, 480, 243–268. [Google Scholar] [CrossRef]
- Kolev, Y.; Uetake, H.; Iida, S.; Ishikawa, T.; Kawano, T.; Sugihara, K. Prognostic significance of VEGF expression in correlation with COX-2, microvessel density, and clinicopathological characteristics in human gastric carcinoma. Ann. Surg. Oncol. 2007, 14, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.; Zhao, C.; Dai, X. Involvement of COX-2/PGE(2) Pathway in the Upregulation of MMP-9 Expression in Pancreatic Cancer. Gastroenterol. Res. Pract. 2011, 2011, 214269. [Google Scholar] [CrossRef] [PubMed]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Sawaoka, H.; Hori, M.; DuBois, R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998, 93, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Larkins, T.L.; Nowell, M.; Singh, S.; Sanford, G.L. Inhibition of cyclooxygenase-2 decreases breast cancer cell motility, invasion and matrix metalloproteinase expression. BMC Cancer 2006, 6, 181. [Google Scholar] [CrossRef] [PubMed]
- Koontongkaew, S. The Tumor Microenvironment Contribution to Development, Growth, Invasion and Metastasis of Head and Neck Squamous Cell Carcinomas. J. Cancer 2013, 4, 66–83. [Google Scholar] [CrossRef]
- Alfonso, L.; Ai, G.; Spitale, R.C.; Bhat, G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer 2014, 111, 61–67. [Google Scholar] [CrossRef]
- Liao, X.; Lochhead, P.; Nishihara, R.; Morikawa, T.; Kuchiba, A.; Yamauchi, M.; Imamura, Y.; Qian, Z.R.; Baba, Y.; Shima, K. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N. Engl. J. Med. 2012, 367, 1596–1606. [Google Scholar] [CrossRef]
- Cheng, C.-Y.; Hsieh, H.-L.; Hsiao, L.-D.; Yang, C.-M. PI3-K/Akt/JNK/NF-κB is essential for MMP-9 expression and outgrowth in human limbal epithelial cells on intact amniotic membrane. Stem Cell Res. 2012, 9, 9–23. [Google Scholar] [CrossRef]
- Xue, J.; Hua, Y.N.; Xie, M.L.; Gu, Z.L. Aspirin inhibits MMP-9 mRNA expression and release via the PPARα/γ and COX-2/mPGES-1-mediated pathways in macrophages derived from THP-1 cells. Biomed. Pharmacother. 2010, 64, 118–123. [Google Scholar] [CrossRef]
- Wan Mohd Tajuddin, W.N.B.; Lajis, N.H.; Abas, F.; Othman, I.; Naidu, R. Mechanistic Understanding of Curcumin’s Therapeutic Effects in Lung Cancer. Nutrients 2019, 11, 2989. [Google Scholar] [CrossRef]
- Lev-Ari, S.; Starr, A.; Vexler, A.; Karaush, V.; Loew, V.; Greif, J.; Fenig, E.; Aderka, D.; Ben-Yosef, R. Inhibition of pancreatic and lung adenocarcinoma cell survival by curcumin is associated with increased apoptosis, down-regulation of COX-2 and EGFR and inhibition of Erk1/2 activity. Anticancer Res. 2006, 26, 4423–4430. [Google Scholar]
- Jobin, C.; Bradham, C.A.; Russo, M.P.; Juma, B.; Narula, A.S.; Brenner, D.A.; Sartor, R.B. Curcumin blocks cytokine-mediated NF-kappa B activation and proinflammatory gene expression by inhibiting inhibitory factor I-kappa B kinase activity. J. Immunol. 1999, 163, 3474–3483. [Google Scholar] [CrossRef]
- Lin, S.-S.; Lai, K.-C.; Hsu, S.-C.; Yang, J.-S.; Kuo, C.-L.; Lin, J.-P.; Ma, Y.-S.; Wu, C.-C.; Chung, J.-G. Curcumin inhibits the migration and invasion of human A549 lung cancer cells through the inhibition of matrix metalloproteinase-2 and -9 and Vascular Endothelial Growth Factor (VEGF). Cancer Lett. 2009, 285, 127–133. [Google Scholar] [CrossRef]
- Lee, J.; Pilch, P.F. The insulin receptor: Structure, function, and signaling. Am. J. Physiol. 1994, 266, C319–C334. [Google Scholar] [CrossRef]
- Pilch, P.F.; Czech, M.P. The subunit structure of the high affinity insulin receptor. Evidence for a disulfide-linked receptor complex in fat cell and liver plasma membranes. J. Biol. Chem. 1980, 255, 1722–1731. [Google Scholar] [CrossRef]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.C.; Tsubokawa, M. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.H.; Masiarz, F.; Kan, Y.W.; Goldfine, I.D. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell 1985, 40, 747–758. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef]
- Cefalu, W.T. Insulin resistance: Cellular and clinical concepts. Exp. Biol. Med. 2001, 226, 13–26. [Google Scholar] [CrossRef]
- Reaven, G. The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol. Metab. Clin. N. Am. 2004, 33, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Wheatcroft, S.B.; Williams, I.L.; Shah, A.M.; Kearney, M.T. Pathophysiological implications of insulin resistance on vascular endothelial function. Diabet. Med. 2003, 20, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Blaise, S.; Romier, B.; Kawecki, C.; Ghirardi, M.; Rabenoelina, F.; Baud, S.; Duca, L.; Maurice, P.; Heinz, A.; Schmelzer, C.E.H.; et al. Elastin-derived peptides are new regulators of insulin resistance development in mice. Diabetes 2013, 62, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Dridi, L.; Seyrantepe, V.; Fougerat, A.; Pan, X.; Bonneil, E.; Thibault, P.; Moreau, A.; Mitchell, G.A.; Heveker, N.; Cairo, C.W.; et al. Positive regulation of insulin signaling by neuraminidase 1. Diabetes 2013, 62, 2338–2346. [Google Scholar] [CrossRef]
- Schlein, M.; Havelund, S.; Kristensen, C.; Dunn, M.F.; Kaarsholm, N.C. Ligand-induced conformational change in the minimized insulin receptor. J. Mol. Biol. 2000, 303, 161–169. [Google Scholar] [CrossRef]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell Physiol. 2007, 213, 589–602. [Google Scholar] [CrossRef]
- Isami, S.; Kishikawa, H.; Araki, E.; Uehara, M.; Kaneko, K.; Shirotani, T.; Todaka, M.; Ura, S.; Motoyoshi, S.; Matsumoto, K.; et al. Bradykinin enhances GLUT4 translocation through the increase of insulin receptor tyrosine kinase in primary adipocytes: Evidence that bradykinin stimulates the insulin signalling pathway. Diabetologia 1996, 39, 412–420. [Google Scholar] [CrossRef]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Kasuya, J.; Paz, I.B.; Maddux, B.A.; Goldfine, I.D.; Hefta, S.A.; Fujita-Yamaguchi, Y. Characterization of human placental insulin-like growth factor-I/insulin hybrid receptors by protein microsequencing and purification. Biochemistry 1993, 32, 13531–13536. [Google Scholar] [CrossRef]
- Belfiore, A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr. Pharm. Des. 2007, 13, 671–686. [Google Scholar] [CrossRef]
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of insulin/IGF hybrid receptors: Contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 2007, 403, 603–613. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef]
- Bailyes, E.M.; Navé, B.T.; Soos, M.A.; Orr, S.R.; Hayward, A.C.; Siddle, K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: Quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem. J. 1997, 327, 209–215. [Google Scholar] [CrossRef]
- White, M.F. The IRS-signalling system: A network of docking proteins that mediate insulin action. Mol. Cell. Biochem. 1998, 182, 3–11. [Google Scholar] [CrossRef]
- Milazzo, G.; Giorgino, F.; Damante, G.; Sung, C.; Stampfer, M.R.; Vigneri, R.; Goldfine, I.D.; Belfiore, A. Insulin receptor expression and function in human breast cancer cell lines. Cancer Res. 1992, 52, 3924–3930. [Google Scholar]
- Rozengurt, E.; Sinnett-Smith, J.; Kisfalvi, K. Crosstalk between Insulin/IGF-1 and GPCR Signaling Systems: A Novel Target for the Anti-diabetic Drug Metformin in Pancreatic Cancer. Clin. Cancer Res. 2010, 16, 2505–2511. [Google Scholar] [CrossRef]
- Kisfalvi, K.; Rey, O.; Young, S.H.; Sinnett-Smith, J.; Rozengurt, E. Insulin potentiates Ca2+ signaling and phosphatidylinositol 4,5-bisphosphate hydrolysis induced by Gq protein-coupled receptor agonists through an mTOR-dependent pathway. Endocrinology 2007, 148, 3246–3257. [Google Scholar] [CrossRef]
- Kisfalvi, K.; Eibl, G.; Sinnett-Smith, J.; Rozengurt, E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009, 69, 6539–6545. [Google Scholar] [CrossRef]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Zhang, C.; Ruan, W.; Schwarz, M.A.; Schwarz, R.E. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol. Cancer Ther. 2012, 11, 2644–2653. [Google Scholar] [CrossRef] [PubMed]
- Stratikopoulos, E.E.; Dendy, M.; Szabolcs, M.; Khaykin, A.J.; Lefebvre, C.; Zhou, M.-M.; Parsons, R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015, 27, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Kawano, Y.; Sakakibara, T.; Hazeki, O.; Ui, M. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J. Biol. Chem. 1994, 269, 3568–3573. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef]
- White, M.F. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E413–E422. [Google Scholar] [CrossRef]
- Dong, X.; Park, S.; Lin, X.; Copps, K.; Yi, X.; White, M.F. Irs1 and Irs2 signaling is essential for hepatic glucose homeostasis and systemic growth. J. Clin. Investig. 2006, 116, 101–114. [Google Scholar] [CrossRef]
- Shaw, L.M. The insulin receptor substrate (IRS) proteins: At the intersection of metabolism and cancer. Cell Cycle 2011, 10, 1750–1756. [Google Scholar] [CrossRef]
- Zhao, J.; Li, Z.; Chen, Y.; Zhang, S.; Guo, L.; Gao, B.; Jiang, Y.; Tian, W.; Hao, S.; Zhang, X. MicroRNA-766 inhibits papillary thyroid cancer progression by directly targeting insulin receptor substrate 2 and regulating the PI3K/Akt pathway. Int. J. Oncol. 2019, 54, 315–325. [Google Scholar] [CrossRef]
- Xu, L.; Tan, Y.; Xu, F.; Zhang, Y. Long noncoding RNA ADIRF antisense RNA 1 upregulates insulin receptor substrate 1 to decrease the aggressiveness of osteosarcoma by sponging microRNA-761. Bioengineered 2022, 13, 2028–2043. [Google Scholar] [CrossRef]
- Amar, L.; Benoit, C.; Beaumont, G.; Vacher, C.M.; Crepin, D.; Taouis, M.; Baroin-Tourancheau, A. MicroRNA expression profiling of hypothalamic arcuate and paraventricular nuclei from single rats using Illumina sequencing technology. J. Neurosci. Methods 2012, 209, 134–143. [Google Scholar] [CrossRef]
- Eguchi, T.; Watanabe, K.; Hara, E.S.; Ono, M.; Kuboki, T.; Calderwood, S.K. OstemiR: A Novel Panel of MicroRNA Biomarkers in Osteoblastic and Osteocytic Differentiation from Mesencymal Stem Cells. PLoS ONE 2013, 8, e58796. [Google Scholar] [CrossRef]
- Liu, M.-M.; Li, Z.; Han, X.-D.; Shi, J.-H.; Tu, D.-Y.; Song, W.; Zhang, J.; Qiu, X.-L.; Ren, Y.; Zhen, L.-L. MiR-30e inhibits tumor growth and chemoresistance via targeting IRS1 in Breast Cancer. Sci. Rep. 2017, 7, 15929. [Google Scholar] [CrossRef]
- Garofalo, C.; Capristo, M.; Mancarella, C.; Reunevi, H.; Picci, P.; Scotlandi, K. Preclinical Effectiveness of Selective Inhibitor of IRS-1/2 NT157 in Osteosarcoma Cell Lines. Front. Endocrinol. 2015, 6, 74. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bunsick, D.A.; Matsukubo, J.; Szewczuk, M.R. Cannabinoids Transmogrify Cancer Metabolic Phenotype via Epigenetic Reprogramming and a Novel CBD Biased G Protein-Coupled Receptor Signaling Platform. Cancers 2023, 15, 1030. https://doi.org/10.3390/cancers15041030
Bunsick DA, Matsukubo J, Szewczuk MR. Cannabinoids Transmogrify Cancer Metabolic Phenotype via Epigenetic Reprogramming and a Novel CBD Biased G Protein-Coupled Receptor Signaling Platform. Cancers. 2023; 15(4):1030. https://doi.org/10.3390/cancers15041030
Chicago/Turabian StyleBunsick, David A., Jenna Matsukubo, and Myron R. Szewczuk. 2023. "Cannabinoids Transmogrify Cancer Metabolic Phenotype via Epigenetic Reprogramming and a Novel CBD Biased G Protein-Coupled Receptor Signaling Platform" Cancers 15, no. 4: 1030. https://doi.org/10.3390/cancers15041030
APA StyleBunsick, D. A., Matsukubo, J., & Szewczuk, M. R. (2023). Cannabinoids Transmogrify Cancer Metabolic Phenotype via Epigenetic Reprogramming and a Novel CBD Biased G Protein-Coupled Receptor Signaling Platform. Cancers, 15(4), 1030. https://doi.org/10.3390/cancers15041030