
Correlations between Molecular Alterations, Histopathological Characteristics, and Poor Prognosis in Esophageal Adenocarcinoma
 , , , , , , ,
, , , , , , ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Recruitment
2.2. Custom EAC Panel: Library Preparation, Hybridization, Sequencing, and Bioinformatic Analysis
2.3. RNA Analysis
2.4. Immunohistochemistry Analysis
2.5. Statistical Analysis
3. Results
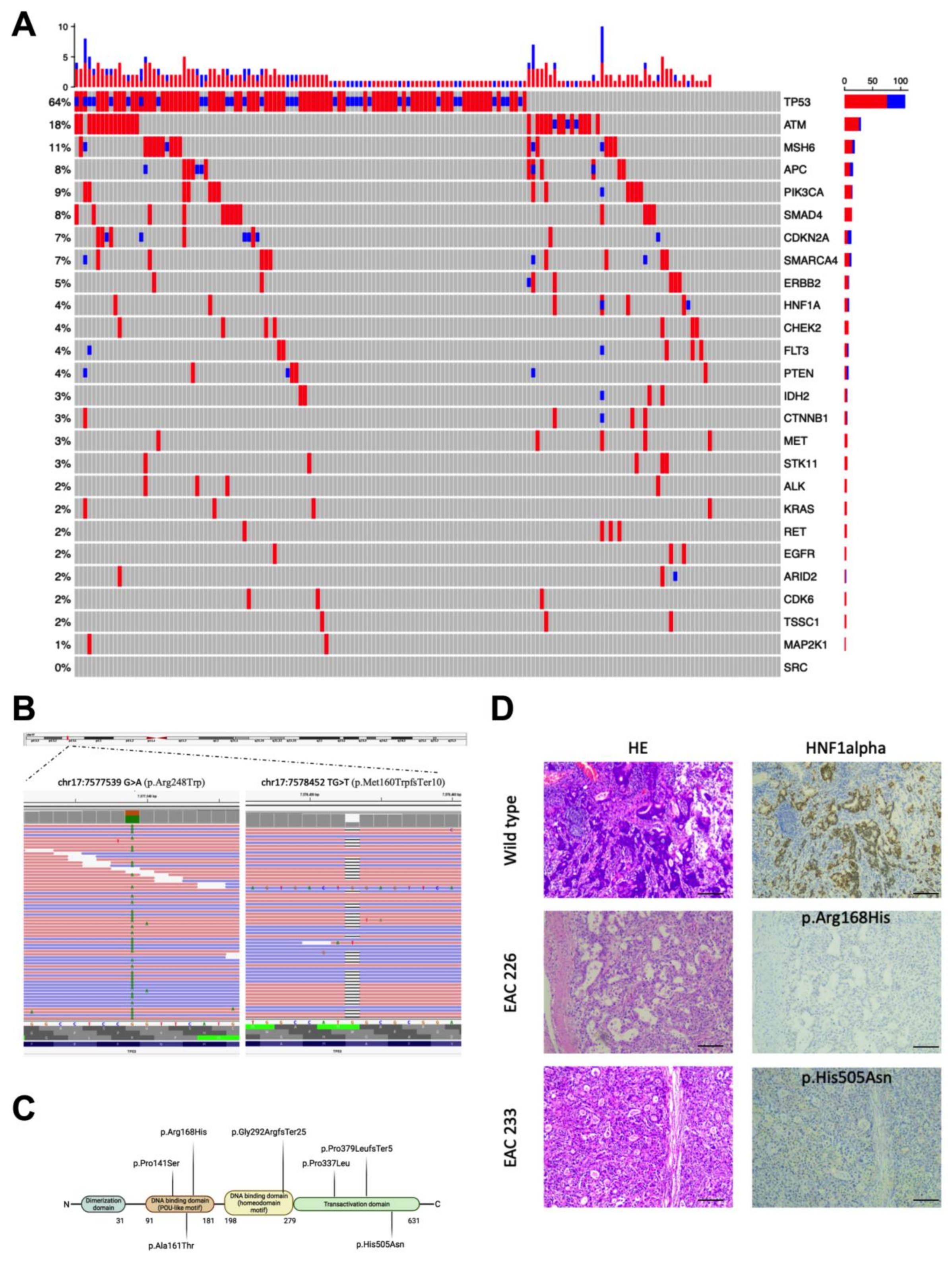
3.1. Genetic Alterations Identified in the EAC Samples
3.2. HNF1alpha Mutations in EAC
3.3. Correlation of Variants in Different Genes
3.4. Evaluating Associations between Genetic Variants and Histopathological and Clinical Phenotypes
3.5. SMAD4 Expression Loss and EAC Survival
3.6. Univariate and Multivariate Cox Regression Analysis
3.7. Gene Fusion Analysis from RNA Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coleman, H.G.; Xie, S.H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Curtius, K.; Rubenstein, J.H.; Chak, A.; Inadomi, J.M. Computational Modelling Suggests That Barrett’s Oesophagus May Be the Precursor of All Oesophageal Adenocarcinomas. Gut 2020, 70, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Yadlapati, R.; Gyawali, C.P.; Pandolfino, J.E.; Chang, K.; Kahrilas, P.J.; Katz, P.O.; Katzka, D.; Komaduri, S.; Lipham, J.; Menard-Katcher, P.; et al. AGA Clinical Practice Update on the Personalized Approach to the Evaluation and Management of GERD: Expert Review. Clin. Gastroenterol. Hepatol. 2022, 20, 984–994.e1. [Google Scholar] [CrossRef] [PubMed]
- Ronkainen, J.; Aro, P.; Storskrubb, T.; Johansson, S.E.; Lind, T.; Bolling-Sternevald, E.; Vieth, M.; Stolte, M.; Talley, N.J.; Agréus, L. Prevalence of Barrett’s Esophagus in the General Population: An Endoscopic Study. Gastroenterology 2005, 129, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Coleman, H.G.; Yousef, F.; Johnston, B.T.; McManus, D.T.; Gavin, A.T.; Murray, L.J. Risk of Malignant Progression in Barrett’s Esophagus Patients: Results from a Large Population-Based Study. J. Natl. Cancer Inst. 2011, 103, 1049–1057. [Google Scholar] [CrossRef]
- Katz-Summercorn, A.C.; Jammula, S.; Frangou, A.; Peneva, I.; O’Donovan, M.; Tripathi, M.; Malhotra, S.; di Pietro, M.; Abbas, S.; Devonshire, G.; et al. Multi-Omic Cross-Sectional Cohort Study of Pre-Malignant Barrett’s Esophagus Reveals Early Structural Variation and Retrotransposon Activity. Nat. Commun. 2022, 13, 1407. [Google Scholar] [CrossRef]
- Fiocca, R.; Mastracci, L.; Lugaresi, M.; Grillo, F.; D’errico, A.; Malvi, D.; Spaggiari, P.; Tomezzoli, A.; Albarello, L.; Ristimäki, A.; et al. The Prognostic Impact of Histology in Esophageal and Esophago-Gastric Junction Adenocarcinoma. Cancers 2021, 13, 5211. [Google Scholar] [CrossRef]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated Genomic Characterization of Oesophageal Carcinoma. Nature 2017, 541, 169–174. [Google Scholar] [CrossRef]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; Macrae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational Signatures in Esophageal Adenocarcinoma Define Etiologically Distinct Subgroups with Therapeutic Relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef]
- Isidori, F.; Malvi, D.; Fittipaldi, S.; Forcato, C.; Bozzarelli, I.; Sala, C.; Raulli, G.; D’Errico, A.; Fiorentino, M.; Seri, M.; et al. Genomic Profiles of Primary and Metastatic Esophageal Adenocarcinoma Identified via Digital Sorting of Pure Cell Populations: Results from a Case Report. BMC Cancer 2018, 18, 889. [Google Scholar] [CrossRef]
- Frankell, A.M.; Jammula, S.G.; Li, X.; Contino, G.; Killcoyne, S.; Abbas, S.; Perner, J.; Bower, L.; Devonshire, G.; Ococks, E.; et al. The Landscape of Selection in 551 Esophageal Adenocarcinomas Defines Genomic Biomarkers for the Clinic. Nat. Genet. 2019, 51, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Isidori, F.; Bozzarelli, I.; Mastracci, L.; Malvi, D.; Lugaresi, M.; Molinari, C.; Söderström, H.; Räsänen, J.; D’Errico, A.; Fiocca, R.; et al. Targeted Sequencing of Sorted Esophageal Adenocarcinoma Cells Unveils Known and Novel Mutations in the Separated Subpopulations. Clin. Transl. Gastroenterol. 2020, 11, e00202. [Google Scholar] [CrossRef] [PubMed]
- Bragoni, A.; Gambella, A.; Pigozzi, S.; Grigolini, M.; Fiocca, R.; Mastracci, L.; Grillo, F. Quality control in diagnostic immunohistochemistry: Integrated on-slide positive controls. Histochem. Cell. Biol. 2017, 148, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A Flexible Statistical Power Analysis Program for the Social, Behavioral, and Biomedical Sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP Eliminates a Majority of Variants of Uncertain Significance in Clinical Exomes at High Sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Cohen, J. A Power Primer. Psychol. Bull. 1992, 112, 155–159. [Google Scholar] [CrossRef]
- Chittenden, T.W.; Pak, J.; Rubio, R.; Cheng, H.; Holton, K.; Prendergast, N.; Glinskii, V.; Cai, Y.; Culhane, A.; Bentink, S.; et al. Therapeutic implications of GIPC1 silencing in cancer. PLoS ONE 2010, 5, e15581. [Google Scholar] [CrossRef]
- Katoh, M. Functional proteomics, human genetics and cancer biology of GIPC family members. Exp. Mol. Med. 2013, 45, e26. [Google Scholar] [CrossRef]
- Abdelhamed, W.; El-Kassas, M. Fibrolamellar Hepatocellular Carcinoma: A Rare but Unpleasant Event. World J. Gastrointest. Oncol. 2022, 14, 1103–1114. [Google Scholar] [CrossRef]
- Jiang, X.; Huang, X.; Zheng, G.; Jia, G.; Li, Z.; Ding, X.; Lei, L.; Yuan, L.; Xu, S.; Gao, N. Targeting PI4KA Sensitizes Refractory Leukemia to Chemotherapy by Modulating the ERK/AMPK/OXPHOS Axis. Theranostics 2022, 12, 6972–6988. [Google Scholar] [CrossRef]
- Umair, M.; Shah, K.; Alhaddad, B.; Haack, T.B.; Graf, E.; Strom, T.M.; Meitinger, T.; Ahmad, W. Exome Sequencing Revealed a Splice Site Variant in the IQCE Gene Underlying Post-Axial Polydactyly Type A Restricted to Lower Limb. Eur. J. Hum. Genet. 2017, 25, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, Y.; Lu, J.; Wang, X.; Wang, L.; Liang, J.; Sun, Z.S. Identification and Characterization of Novel Fusion Genes in Prostate Cancer by Targeted RNA Capture and Next-Generation Sequencing. Acta Biochim. Biophys. Sin. 2018, 50, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic Catastrophes Frequently Arise in Esophageal Adenocarcinoma and Drive Tumorigenesis. Nat. Commun. 2014, 5, 5224. [Google Scholar] [CrossRef]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and Whole-Genome Sequencing of Esophageal Adenocarcinoma Identifies Recurrent Driver Events and Mutational Complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.; Gu, J.; Hawk, E.T.; Wang, K.K.; Lai, M.; Huang, M.; Ajani, J.; Wu, X. Genome-Wide Methylation Analysis Shows Similar Patterns in Barrett’s Esophagus and Esophageal Adenocarcinoma. Carcinogenesis 2013, 34, 2750–2756. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, H.; Opalinska, J.; Zhou, L.; Sohal, D.; Fazzari, M.J.; Yu, Y.; Montagna, C.; Montgomery, E.A.; Canto, M.; Dunbar, K.B.; et al. Widespread Hypomethylation Occurs Early and Synergizes with Gene Amplification during Esophageal Carcinogenesis. PLoS Genet. 2011, 7, e1001356. [Google Scholar] [CrossRef]
- Hoefnagel, S.J.M.; Boonstra, J.J.; Russchen, M.J.A.M.; Krishnadath, K.K. Towards Personalized Treatment Strategies for Esophageal Adenocarcinoma; A Review on the Molecular Characterization of Esophageal Adenocarcinoma and Current Research Efforts on Individualized Curative Treatment Regimens. Cancers 2021, 13, 4811. [Google Scholar] [CrossRef]
- Luo, Z.; Li, Y.; Wang, H.; Fleming, J.; Li, M.; Kang, Y.; Zhang, R.; Li, D. Hepatocyte Nuclear Factor 1A (HNF1A) as a Possible Tumor Suppressor in Pancreatic Cancer. PLoS ONE 2015, 10, e0121082. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Lee, J.J.K.; Melloni, G.E.M.; Cortés-Ciriano, I.; Park, P.J. Detecting the Mutational Signature of Homologous Recombination Deficiency in Clinical Samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef]
- Sason, I.; Chen, Y.; Leiserson, M.D.M.; Sharan, R. A Mixture Model for Signature Discovery from Sparse Mutation Data. Genome Med. 2021, 13, 173. [Google Scholar] [CrossRef]
- Contino, G.; Vaughan, T.L.; Whiteman, D.; Fitzgerald, R.C. The Evolving Genomic Landscape of Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastroenterology 2017, 153, 657–673.e1. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. Mutant P53 Reactivation by PRIMA-1MET Induces Multiple Signaling Pathways Converging on Apoptosis. Oncogene 2009, 29, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Milano, F.; van Baal, J.W.P.M.; Buttar, N.S.; Rygiel, A.M.; de Kort, F.; DeMars, C.J.; Rosmolen, W.D.; Bergman, J.J.G.H.M.; van Marle, J.; Wang, K.K.; et al. Bone Morphogenetic Protein 4 Expressed in Esophagitis Induces a Columnar Phenotype in Esophageal Squamous Cells. Gastroenterology 2007, 132, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Foxwell, T.J.; Nason, K.; Cressman, K.L.; McGrath, K.M.; Sun, W.; Bahary, N.; Zeh, H.J.; Levy, R.M.; Luketich, J.D.; et al. Smad4 Loss in Esophageal Adenocarcinoma Is Associated with an Increased Propensity for Disease Recurrence and Poor Survival. Am. J. Surg. Pathol. 2015, 39, 487. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.H.; Burrows, A.D.; Anderson, R.L. Bone Morphogenetic Protein Signaling in Breast Cancer Progression. Growth Factors 2019, 37, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Gotovac, J.R.; Kader, T.; Milne, J.v.; Fujihara, K.M.; Lara-Gonzalez, L.E.; Gorringe, K.L.; Kalimuthu, S.N.; Jayawardana, M.W.; Duong, C.P.; Phillips, W.A.; et al. Loss of SMAD4 Is Sufficient to Promote Tumorigenesis in a Model of Dysplastic Barrett’s Esophagus. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 689–713. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hoefnagel, S.J.M.; Read, M.; Meijer, S.; van Berge Henegouwen, M.I.; Gisbertz, S.S.; Bonora, E.; Liu, D.S.H.; Phillips, W.A.; Calpe, S.; et al. Selective Targeting BMP2 and 4 in SMAD4 Negative Esophageal Adenocarcinoma Inhibits Tumor Growth and Aggressiveness in Preclinical Models. Cell Oncol. 2022, 45, 639–658. [Google Scholar] [CrossRef]
- Sorokin, M.; Rabushko, E.; Rozenberg, J.M.; Mohammad, T.; Seryakov, A.; Sekacheva, M.; Buzdin, A. Clinically relevant fusion oncogenes: Detection and practical implications. Adv. Med. Oncol. 2022, 14, 17588359221144108. [Google Scholar] [CrossRef]
- Ng, A.W.T.; Contino, G.; Killcoyne, S.; Devonshire, G.; Hsu, R.; Abbas, S.; Su, J.; Redmond, A.M.; Weaver, J.M.J.; Eldridge, M.D.; et al. Rearrangement Processes and Structural Variations Show Evidence of Selection in Oesophageal Adenocarcinomas. Commun. Biol. 2022, 5, 335. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orsini, A.; Mastracci, L.; Bozzarelli, I.; Ferrari, A.; Isidori, F.; Fiocca, R.; Lugaresi, M.; D’Errico, A.; Malvi, D.; Cataldi-Stagetti, E.; et al. Correlations between Molecular Alterations, Histopathological Characteristics, and Poor Prognosis in Esophageal Adenocarcinoma. Cancers 2023, 15, 1408. https://doi.org/10.3390/cancers15051408
Orsini A, Mastracci L, Bozzarelli I, Ferrari A, Isidori F, Fiocca R, Lugaresi M, D’Errico A, Malvi D, Cataldi-Stagetti E, et al. Correlations between Molecular Alterations, Histopathological Characteristics, and Poor Prognosis in Esophageal Adenocarcinoma. Cancers. 2023; 15(5):1408. https://doi.org/10.3390/cancers15051408
Chicago/Turabian StyleOrsini, Arianna, Luca Mastracci, Isotta Bozzarelli, Anna Ferrari, Federica Isidori, Roberto Fiocca, Marialuisa Lugaresi, Antonietta D’Errico, Deborah Malvi, Erica Cataldi-Stagetti, and et al. 2023. "Correlations between Molecular Alterations, Histopathological Characteristics, and Poor Prognosis in Esophageal Adenocarcinoma" Cancers 15, no. 5: 1408. https://doi.org/10.3390/cancers15051408
APA StyleOrsini, A., Mastracci, L., Bozzarelli, I., Ferrari, A., Isidori, F., Fiocca, R., Lugaresi, M., D’Errico, A., Malvi, D., Cataldi-Stagetti, E., Spaggiari, P., Tomezzoli, A., Albarello, L., Ristimäki, A., Bottiglieri, L., Krishnadath, K. K., Rosati, R., Fumagalli Romario, U., De Manzoni, G., ... on behalf of the EACSGE Consortium. (2023). Correlations between Molecular Alterations, Histopathological Characteristics, and Poor Prognosis in Esophageal Adenocarcinoma. Cancers, 15(5), 1408. https://doi.org/10.3390/cancers15051408