

Candidate Genes and Pathways in Cervical Cancer: A Systematic Review and Integrated Bioinformatic Analysis



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Inclusion Criteria
2.3. Exclusion Criteria
2.4. Screening of Articles for Eligibility
2.5. Data Extraction
2.6. Study Quality
2.7. Venn Diagram Analysis
2.8. Protein–Protein Interaction (PPI) Network, Clustering, and Visualization
2.9. Gene Ontology (GO) and Pathway Enrichment Analysis
3. Results
3.1. Patient Recruitment and Sample Collection Details
3.2. Study Quality
3.3. Identification of DEGs in Cervical Cancer
3.4. Common DEGs among the Studies Identified via Venn Diagram Analysis
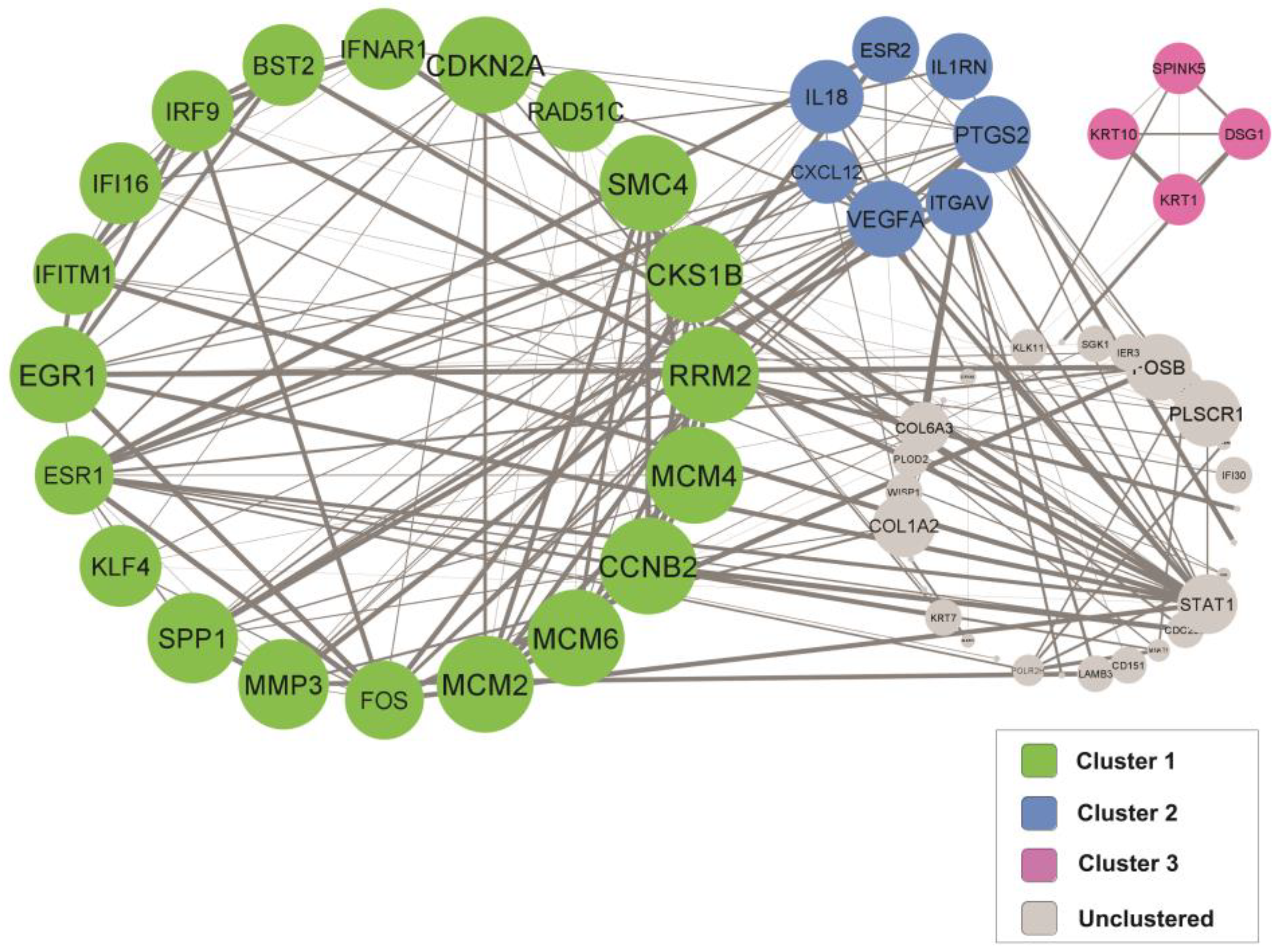
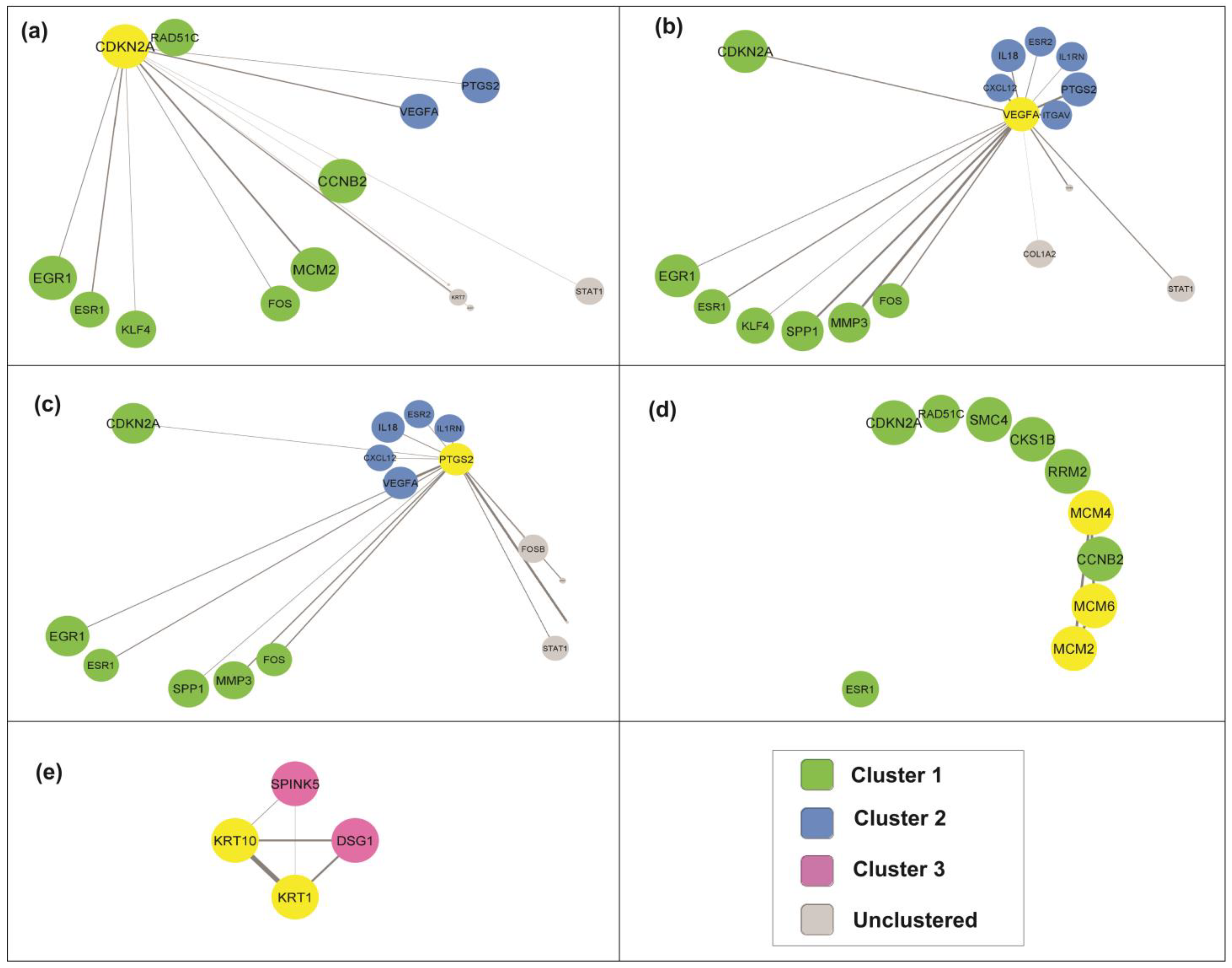
3.5. Identification of Key Candidate Genes and Pathways via Protein–Protein Interaction (PPI) Network and Modular Analysis
3.6. Gene Ontology (GO) and Pathway Enrichment Analysis of the Identified Clusters
3.7. Selection of Candidate Genes in Cervical Cancer Pathogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arbyn, M.; Weiderpass, E.; Bruni, L. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- Fontham, E.T.H.; Wolf, A.M.D.; Church, T.R.; Etzioni, R.; Flowers, C.R.; Herzig, A.; Guerra, C.E.; Oeffinger, K.C.; Shih, Y.-C.T.; Walter, L.C.; et al. Cervical cancer screening for individuals at average risk: 2020 guideline update from the American Cancer Society. CA Cancer J. Clin. 2020, 70, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rustum, N.R.; Yashar, C.M.; Bean, S.; Bradley, K.; Campos, S.M.; Chon, H.S.; Chu, C.; Cohn, D.; Crispens, M.A.; Damast, S.; et al. NCCN Guidelines Insights: Cervical Cancer, Version 1.2020: Featured Updates to the NCCN Guidelines. J. Natl. Compr. Cancer Netw. 2020, 18, 660–666. [Google Scholar] [CrossRef] [PubMed]
- US Preventive Services Task Force. Screening for Cervical Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 320, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Qian, Z.; Gong, Y.; Wang, Y.; Guan, Y.; Han, Y.; Yi, X.; Huang, W.; Ji, L.; Xu, J.; et al. Comprehensive genomic variation profiling of cervical intraepithelial neoplasia and cervical cancer identifies potential targets for cervical cancer early warning. J. Med. Genet. 2019, 56, 186. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, C.K.; Tye, G.J.; Balasubramaniam, S.D.; Kaur, G. CD74 and HLA-DRA in Cervical Carcinogenesis: Potential Targets for Antitumour Therapy. Medicina 2022, 58, 190. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhai, J.; Wang, W.; Liu, T.; Liu, C.; Zhu, X.; Wang, Q.; Tian, W.; Zhang, F. Identification of Tumor Microenvironment and DNA Methylation-Related Prognostic Signature for Predicting Clinical Outcomes and Therapeutic Responses in Cervical Cancer. Front. Mol. Biosci. 2022, 9, 872932. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, J.; Sun, X.; Long, W. Transcriptome sequencing reveals pathways and genes dysregulated in HPV infected cervical cancer. Eur. J. Gynaecol. Oncol. 2020, 41, 996–1003. [Google Scholar] [CrossRef]
- Balasubramaniam, S.D.; Balakrishnan, V.; Oon, C.E.; Kaur, G. Gene expression profiling of HPV-associated cervical carcinogenesis in formalin-fixed paraffin-embedded (FFPE) tissues using the NanoString nCounterTM platform. Gene 2022, 825, 146385. [Google Scholar] [CrossRef]
- Hao, Y.; Ye, M.; Chen, X.; Zhao, H.; Hasim, A.; Guo, X. Discovery and validation of FBLN1 and ANT3 as potential biomarkers for early detection of cervical cancer. Cancer Cell Int. 2021, 21, 125. [Google Scholar] [CrossRef]
- Teng, F.; Ruan, H.-J.; Xu, J.; Ni, J.; Qian, B.; Shen, R.; Gao, L.-J. RBBP6 promotes human cervical carcinoma malignancy via JNK signaling pathway. Biomed. Pharmacother. 2018, 101, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Manawapat-Klopfer, A.; Thomsen, L.T.; Martus, P.; Munk, C.; Russ, R.; Gmuender, H.; Frederiksen, K.; Haedicke-Jarboui, J.; Stubenrauch, F.; Kjaer, S.K.; et al. TMEM45A, SERPINB5 and p16INK4A transcript levels are predictive for development of high-grade cervical lesions. Am. J. Cancer Res. 2016, 6, 1524–1536. [Google Scholar] [PubMed]
- Munn, Z.; Barker, T.H.; Moola, S.; Tufanaru, C.; Stern, C.; McArthur, A.; Stephenson, M.; Aromataris, E. Methodological Quality of Case Series Studies: An Introduction to the Jbi Critical Appraisal Tool. JBI Evid Synth. 2020, 18, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Genomics, B.E. Calculate and Draw Custom Venn Diagrams; Bioinformatics & Evolutionary Genomics: Ghent, Belgium, 2022. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Annapurna, S.D.; Pasumarthi, D.; Pasha, A.; Doneti, R.; Sheela, B.; Botlagunta, M.; Vijaya, L.B.; Pawar, S.C. Identification of Differentially Expressed Genes in Cervical Cancer Patients by Comparative Transcriptome Analysis. BioMed Res. Int. 2021, 2021, 8810074. [Google Scholar] [CrossRef]
- Kim, Y.W.; Bae, S.M.; Kim, Y.W.; Park, D.C.; Lee, K.H.; Liu, H.B.; Kim, I.W.; Jang, C.K.; Ahn, W.S. Target-based molecular signature characteristics of cervical adenocarcinoma and squamous cell carcinoma. Int. J. Oncol. 2013, 43, 539–547. [Google Scholar] [CrossRef]
- Rajkumar, T.; Sabitha, K.; Vijayalakshmi, N.; Shirley, S.; Bose, M.V.; Gopal, G.; Selvaluxmy, G. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer 2011, 11, 80. [Google Scholar] [CrossRef]
- Miyatake, T.; Ueda, Y.; Nakashima, R.; Yoshino, K.; Kimura, T.; Murata, T.; Nomura, T.; Fujita, M.; Buzard, G.S.; Enomoto, T. Down-regulation of insulin-like growth factor binding protein-5 (IGFBP-5): Novel marker for cervical carcinogenesis. Int. J. Cancer 2007, 120, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Gius, D.; Funk, M.C.; Chuang, E.Y.; Feng, S.; Huettner, P.C.; Nguyen, L.; Bradbury, C.M.; Mishra, M.; Gao, S.; Buttin, B.M.; et al. Profiling microdissected epithelium and stroma to model genomic signatures for cervical carcinogenesis accommodating for covariates. Cancer Res. 2007, 67, 7113–7123. [Google Scholar] [CrossRef]
- Wong, Y.F.; Cheung, T.H.; Tsao, G.S.W.; Lo, K.W.K.; Yim, S.F.; Wang, V.W.; Heung, M.M.S.; Chan, S.C.S.; Chan, L.K.Y.; Ho, T.W.F.; et al. Genome-wide gene expression profiling of cervical cancer in Hong Kong women by oligonucleotide microarray. Int. J. Cancer 2006, 118, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yang, S.; Xu, J.; Lu, W.; Xie, X. Transcriptome Sequencing Profiles of Cervical Cancer Tissues and Siha Cells. Funct. Integr. Genom. 2020, 20, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Øvestad, I.T.; Engesæter, B.; Halle, M.K.; Akbari, S.; Bicskei, B.; Lapin, M.; Austdal, M.; Janssen, E.A.M.; Krakstad, C.; Lillesand, M.; et al. High-Grade Cervical Intraepithelial Neoplasia (Cin) Associates with Increased Proliferation and Attenuated Immune Signaling. Int. J. Mol. Sci. 2022, 23, 373. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Luan, Y.; Zhang, W.; Xie, J.; Mao, J. CDKN2A inhibits cell proliferation and invasion in cervical cancer through LDHA-mediated AKT/mTOR pathway. Clin. Transl. Oncol. 2021, 23, 222–228. [Google Scholar] [CrossRef]
- Truong, P.K.; Lao, T.D.; Le, T.A.H. Cdkn2a Methylation—An Epigenetic Biomarker for Cervical Cancer Risk: A Meta-Analysis. Pharmacophore 2020, 11, 21–29. [Google Scholar]
- Sudha, B.; Poornima, A.; Suganya, K.; Swathi, K.; Kumar, N.S.; Sumathi, S.; Chellapandi, P. Identification of hub genes and role of CDKN2A as a biomarker in cervical cancer: An in-silico approach. Hum. Gene 2022, 33, 201048. [Google Scholar] [CrossRef]
- Patel, K.A.; Patel, B.M.; Thobias, A.R.; Gokani, R.A.; Chhikara, A.B.; Desai, A.D.; Patel, P.S. Overexpression of VEGF165 is associated with poor prognosis of cervical cancer. J. Obstet. Gynaecol. Res. 2020, 46, 2397–2406. [Google Scholar] [CrossRef]
- Guo, H.; Li, J.; Fan, F.; Zhou, P. LINC00707 Regulates miR-382-5p/VEGFA Pathway to Enhance Cervical Cancer Progression. J. Immunol. Res. 2021, 2021, 5524632. [Google Scholar] [CrossRef] [PubMed]
- Ying, T.-H.; Lin, C.-L.; Chen, P.-N.; Wu, P.-J.; Liu, C.-J.; Hsieh, Y.-H. Angelol-A exerts anti-metastatic and anti-angiogenic effects on human cervical carcinoma cells by modulating the phosphorylated-ERK/miR-29a-3p that targets the MMP2/VEGFA axis. Life Sci. 2022, 296, 120317. [Google Scholar] [CrossRef] [PubMed]
- Hugo de Almeida, V.; Guimarães, I.D.S.; Almendra, L.R.; Rondon, A.M.R.; Tilli, T.M.; de Melo, A.C.; Sternberg, C.; Monteiro, R.Q. Positive crosstalk between EGFR and the TF-PAR2 pathway mediates resistance to cisplatin and poor survival in cervical cancer. Oncotarget 2018, 9, 30594–30609. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, Q.; Liu, Z.; Shen, G.; Sun, X.; Di, X. Identification of Immune and Hypoxia Risk Classifier to Estimate Immune Microenvironment and Prognosis in Cervical Cancer. J. Oncol. 2022, 2022, 6906380. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Balasubramaniam, S.D.; Lee, Y.J.; Balakrishnan, V.; Oon, C.E. Minichromosome Maintenance Complex (MCM) Genes Profiling and MCM2 Protein Expression in Cervical Cancer Development. Asian Pac. J. Cancer Prev. 2019, 20, 3043–3049. [Google Scholar] [CrossRef]
- Wu, B.; Xi, S. Bioinformatics analysis of the transcriptional expression of minichromosome maintenance proteins as potential indicators of survival in patients with cervical cancer. BMC Cancer 2021, 21, 928. [Google Scholar] [CrossRef]
- Han, W.; Hu, C.; Fan, Z.-J.; Shen, G.-L. Transcript levels of keratin 1/5/6/14/15/16/17 as potential prognostic indicators in melanoma patients. Sci. Rep. 2021, 11, 1023. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, L.; He, J.; He, Z.; Yue, C.; Jin, X.; Gao, M.; Xiao, S.; Zhou, Y. Fra-1 Inhibits Cell Growth and the Warburg Effect in Cervical Cancer Cells via STAT1 Regulation of the p53 Signaling Pathway. Front. Cell Dev. Biol. 2020, 8, 579629. [Google Scholar] [CrossRef]
- Niu, G.; Wang, D.; Pei, Y.; Sun, L. Systematic identification of key genes and pathways in the development of invasive cervical cancer. Gene 2017, 618, 28–41. [Google Scholar] [CrossRef]
- Pirim, D. Integrative analyses of molecular pathways and key candidate biomarkers associated with colorectal cancer. Cancer Biomark 2020, 27, 555–568. [Google Scholar] [CrossRef]
- Mollica, V.; Marchetti, A.; Rosellini, M.; Nuvola, G.; Rizzo, A.; Santoni, M.; Cimadamore, A.; Montironi, R.; Massari, F. An Insight on Novel Molecular Pathways in Metastatic Prostate Cancer: A Focus on DDR, MSI and AKT. Int. J. Mol. Sci. 2021, 22, 13519. [Google Scholar] [CrossRef] [PubMed]
- Miricescu, D.; Totan, A.; Stanescu, S., II; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Khan, Z.; Alloghbi, A.; Said Ahmed, T.S.; Ashraf, M.; Hammouda, D.M. Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies. Medicina 2019, 55, 526. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year (References) | Country | Study Design | Sampling | Sample Size (n) | Gene-Expression Analysis | Upregulated Genes | Downregulated Genes |
|---|---|---|---|---|---|---|---|
| Annapurna et al., 2021 [19] | India | Case–control | Cervical tissue | 7 patients, 3 normal | Microarray | 78 | 26 |
| Kim et al., 2013 [20] | Korea | Case–control | Cervical tissue | 28 patients, 17 control | Microarray | 208 | 201 |
| Rajkumar et al., 2011 [21] | India | Case–control | Cervical tissue | 28 patients, 5 normal | Microarray | 47 | 19 |
| Miyatake et al., 2007 [22] | Japan | Cross-sectional | Cervical tissue | 2 patients | Microarray | 8 | 14 |
| Gius et al., 2007 [23] | USA | Cross-sectional | Cervical tissue | 85 patients | Microarray | 536 | |
| Wong et al., 2006 [24] | Hong Kong | Case–control | Cervical tissue | 29 patients, 18 controls | Microarray | 19 | 83 |
| Cluster | Term | Description | Genes | p-Value |
|---|---|---|---|---|
| 1 | CC_ GO:0005654 | Nucleoplasm | EGR1, CDKN2A, FOS, KLF4, SMC4, ESR1, CKS1B, RAD51C, IFI16, MCM4, MCM6, IRF9, MCM2 | 6.06 ×10−6 |
| CC_GO:0005829 | Cytosol | BST2, CCNB2, RAD51C, IFI16, CDKN2A, FOS, KLF4, SMC4, ESR1, IRF9 | 1.96 × 10−2 | |
| CC_GO:0000785 | Chromatin | EGR1, FOS, KLF4, ESR1, IRF9, MCM2 | 1.32 × 10−3 | |
| BP_GO:0045944 | Positive regulation of transcription from RNA polymerase II promoter | EGR1, IFI16, CDKN2A, FOS, KLF4, ESR1, IRF9 | 3.96 × 10−4 | |
| BP_GO:0007049 | Cell cycle | CDKN2A, MCM4, MCM6, SMC4, MCM2, CKS1B | 1.11 × 10−5 | |
| BP_GO:0045893 | Positive regulation of transcription, DNA-templated | EGR1, CDKN2A, SPP1, FOS, KLF4, ESR1 | 3.13 × 10−4 | |
| MF_ GO:0005524 | ATP binding | RAD51C, MCM4, MCM6, SMC4, MCM2 | 4.50 × 10−2 | |
| MF_ GO:0003697 | Single-stranded DNA binding | MCM4, MCM6, SMC4, MCM2 | 1.41 × 10−4 | |
| MF_GO:0008134 | Transcription factor binding | IFI16, CDKN2A, FOS, ESR1 | 8.12 × 10−4 | |
| hsa04110 | Cell cycle | CCNB2, CDKN2A, MCM4, MCM6, MCM2 | 8.53 × 10−5 | |
| 2 | CC_GO:0005737 | Cytoplasm | IL1RN, CXCL12, IL18, PTGS2, VEGFA | 0.049 |
| BP_ GO:0045785 | Positive regulation of cell adhesion | CXCL12, ITGAV, VEGFA | 1.37 × 10−4 | |
| BP_GO:0030335 | Positive regulation of cell migration | CXCL12, ITGAV, VEGFA | 0.002 | |
| BP_ GO:0006954 | Inflammatory response | IL1RN, IL18, PTGS2 | 0.006 | |
| MF_ GO:0005125 | Cytokine activity | IL1RN, IL18, VEGFA | 0.002 | |
| MF_ GO:0050840 | Extracellular matrix binding | ITGAV, VEGFA | 0.009 | |
| hsa05165 | Human papillomavirus infection | ITGAV, PTGS2, VEGFA | 0.022 | |
| 3 | CC_GO:0005829 | Cytosol | KRT1, SPINK5, DSG1, KRT10 | 0.019 |
| CC_ GO:0045095 | Keratin filament | KRT1, KRT10 | 0.015 | |
| BP_ GO:0051290 | Protein heterotetramerization | KRT1, KRT10 | 0.002 | |
| BP_ GO:0018149 | Peptide cross-linking | KRT1, KRT10 | 0.005 | |
| MF_ GO:0030280 | Structural constituent of epidermis | KRT1, KRT10 | 0.006 |
| Candidate Gene | Functional Annotation | ||
|---|---|---|---|
| Upregulated | Downregulated | Term | Description |
| CDKN2A | GO:0000079 | Regulation of cyclin-dependent protein serine/threonine kinase activity | |
| hsa01522 | Endocrine resistance | ||
| hsa05166 | Human T-cell leukemia virus 1 infection | ||
| GO:0008134 | Transcription factor binding | ||
| hsa05200 | Pathways in cancer | ||
| hsa04110 | Cell cycle | ||
| VEGFA | PTGS2 | hsa04370 | VEGF signaling pathway |
| GO:0071456 | Cellular response to hypoxia | ||
| hsa05165 | Human papillomavirus infection | ||
| GO:0043154 | Negative regulation of cysteine-type endopeptidase activity involved in apoptotic process | ||
| MCM2, MCM6 | MCM4 | GO:0006270 | DNA replication initiation |
| hsa03030 | DNA replication | ||
| GO:0003678 | DNA helicase activity | ||
| GO:0000727 | Double-strand break repair via break-induced replication | ||
| KRT1 | KRT10 | GO:0030280 | Structural constituent of epidermis |
| GO:0018149 | Peptide cross-linking | ||
| 113800 | Epidermolytic hyperkeratosis | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elias, M.H.; Das, S.; Abdul Hamid, N. Candidate Genes and Pathways in Cervical Cancer: A Systematic Review and Integrated Bioinformatic Analysis. Cancers 2023, 15, 853. https://doi.org/10.3390/cancers15030853
Elias MH, Das S, Abdul Hamid N. Candidate Genes and Pathways in Cervical Cancer: A Systematic Review and Integrated Bioinformatic Analysis. Cancers. 2023; 15(3):853. https://doi.org/10.3390/cancers15030853
Chicago/Turabian StyleElias, Marjanu Hikmah, Srijit Das, and Nazefah Abdul Hamid. 2023. "Candidate Genes and Pathways in Cervical Cancer: A Systematic Review and Integrated Bioinformatic Analysis" Cancers 15, no. 3: 853. https://doi.org/10.3390/cancers15030853
APA StyleElias, M. H., Das, S., & Abdul Hamid, N. (2023). Candidate Genes and Pathways in Cervical Cancer: A Systematic Review and Integrated Bioinformatic Analysis. Cancers, 15(3), 853. https://doi.org/10.3390/cancers15030853