
Pediatric Acute Myeloid Leukemia Post Cytotoxic Therapy—Retrospective Analysis of the Patients Treated in Poland from 2005 to 2022

 , , , , , , , , , , , add
Show full author list
, , , , , , , , , , , add
Show full author list
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
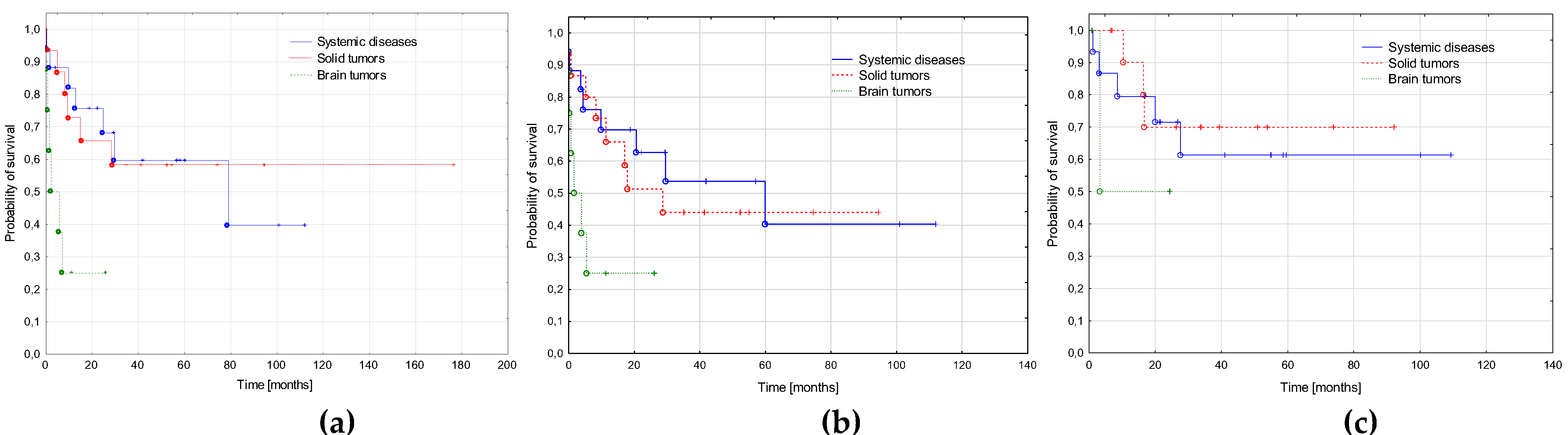
3.1. Primary Cancer
3.2. Latency Period
3.3. AML-pCT Characteristics
3.4. AML-pCT Treatement
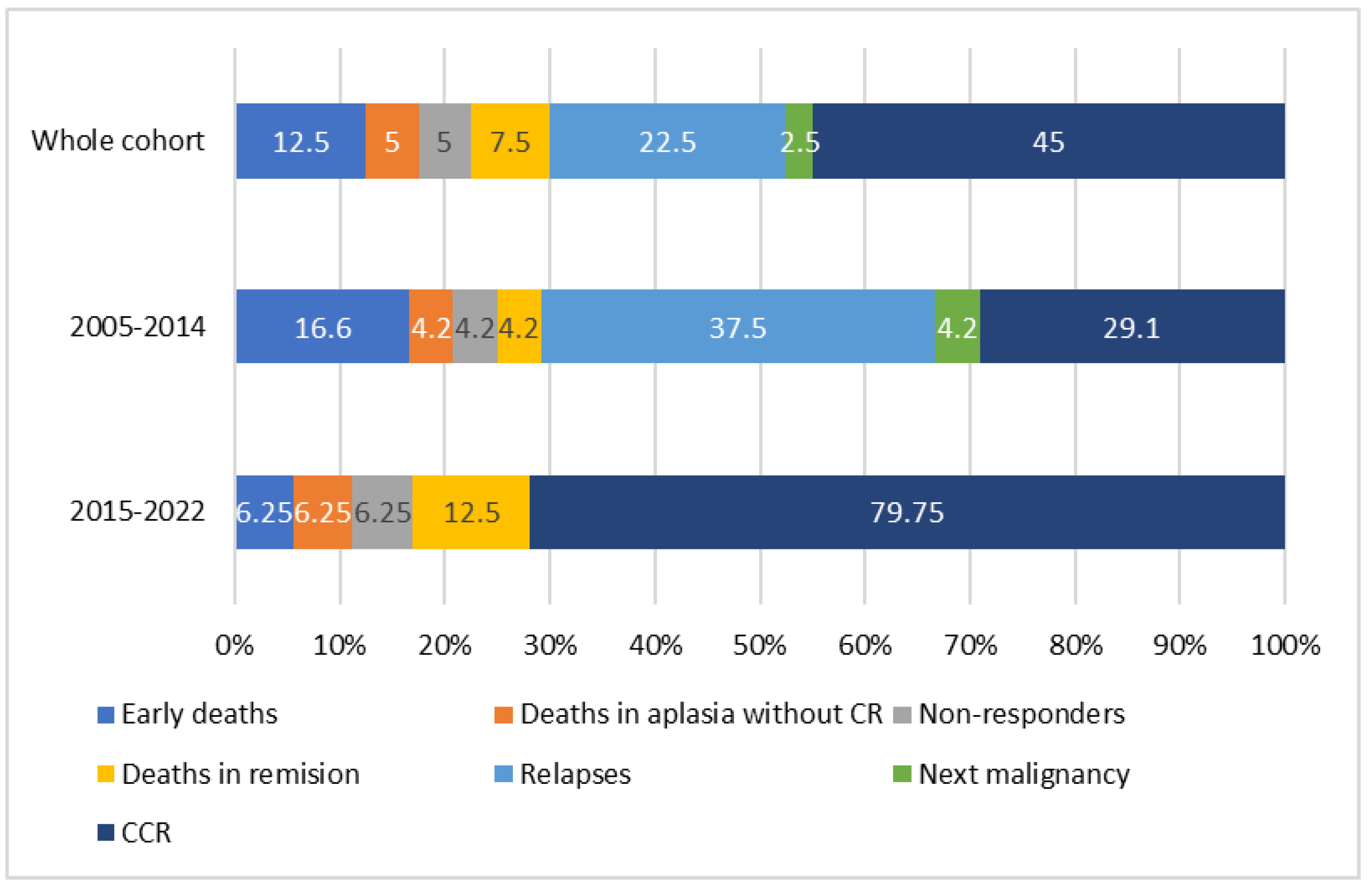
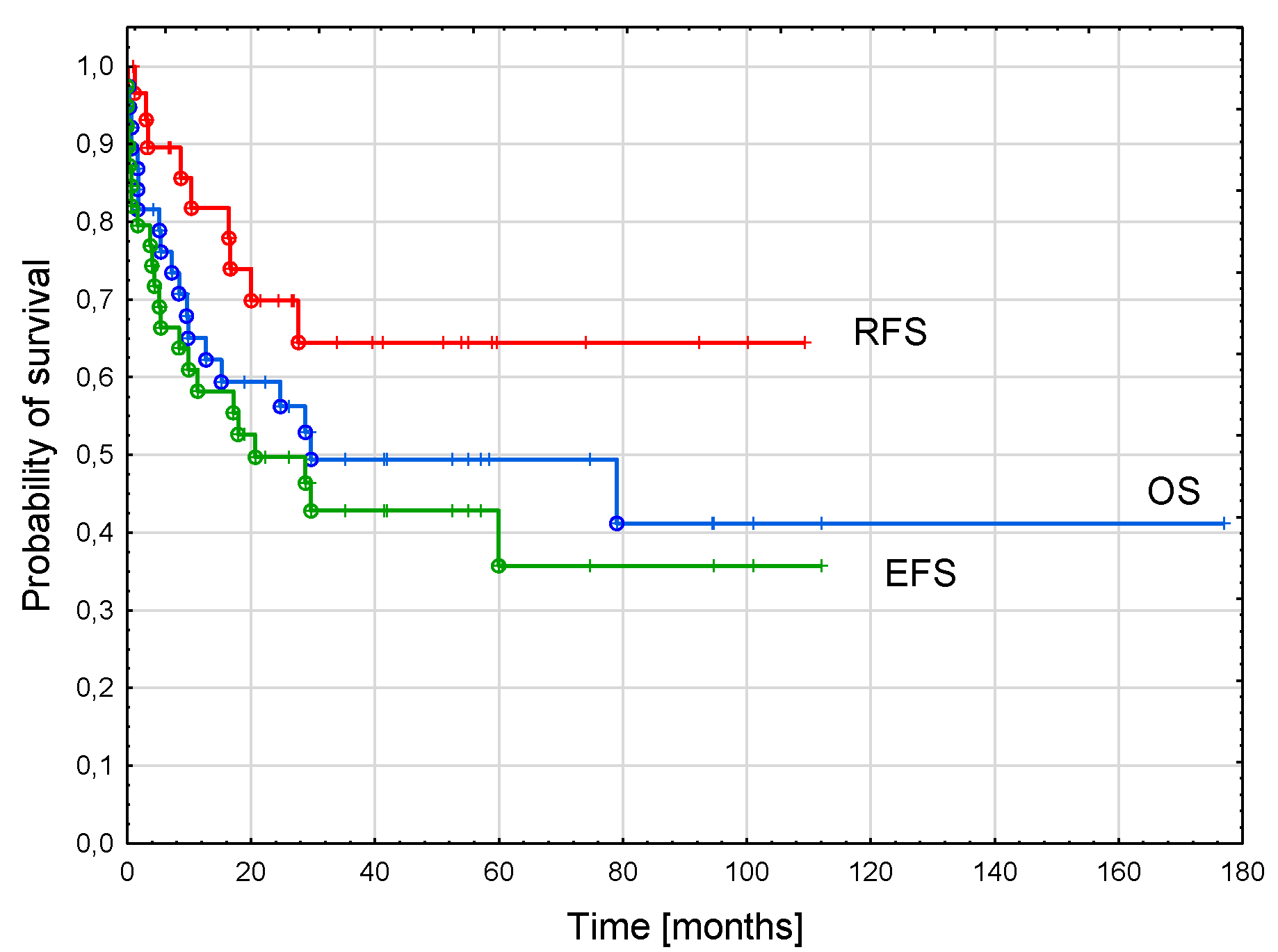
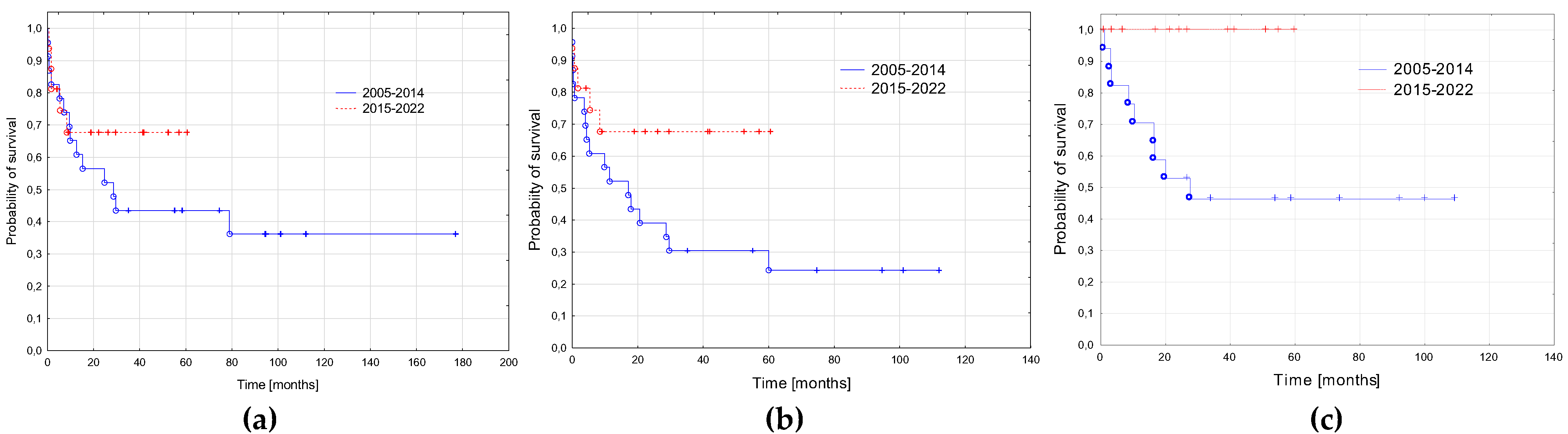
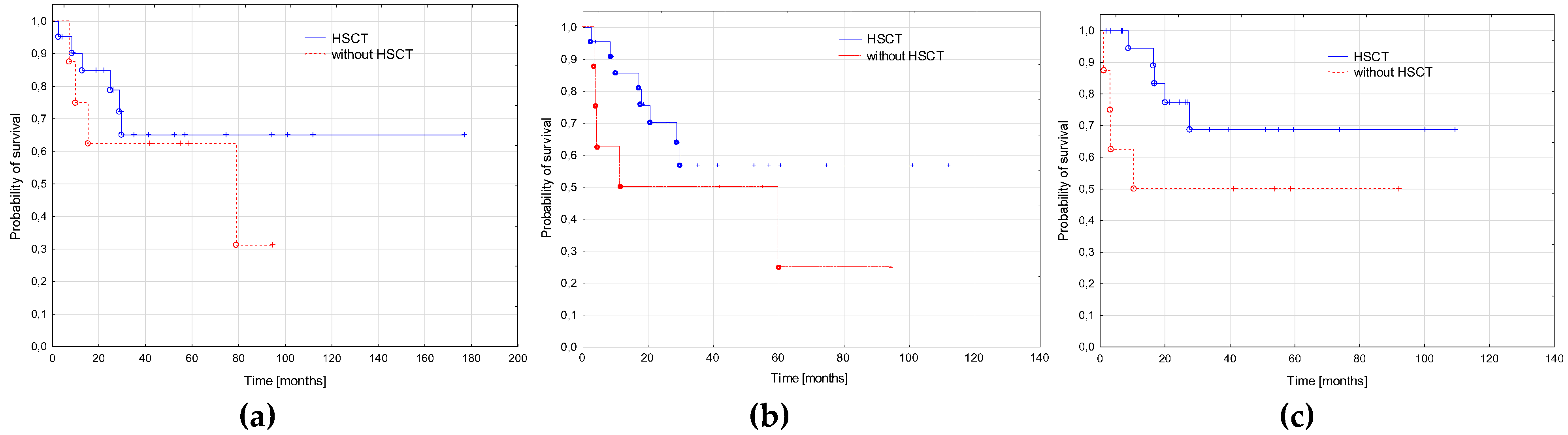
3.5. AML-pCT Outcome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pedersen-Bjergaard, J.; Andersen, M.T.; Andersen, M.K. Genetic pathways in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Hematology Am. Soc. Hematol. Educ. Program 2007, 2007, 392–397. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Brown, C.A.; Youlden, D.R.; Aitken, J.F.; Moore, A.S. Therapy-related acute myeloid leukemia following treatment for cancer in childhood: A population-based registry study. Pediatr. Blood Cancer 2018, 65, e27410. [Google Scholar] [CrossRef]
- Sandoval, C.; Pui, C.H.; Bowman, L.C.; Heaton, D.; Hurwitz, C.A.; Raimondi, S.C.; Behm, F.G.; Head, D.R. Secondary acute myeloid leukemia in children previously treated with alkylating agents, intercalating topoisomerase II inhibitors, and irradiation. J. Clin. Oncol. 1993, 11, 1039–1045. [Google Scholar] [CrossRef]
- Barnard, D.R.; Woods, W.G. Treatment-related myelodysplastic syndrome/acute myeloid leukemia in survivors of childhood cancer—An update. Leuk. Lymphoma 2005, 46, 651–663. [Google Scholar] [CrossRef]
- Schwartz, J.R.; Ma, J.; Kamens, J.; Westover, T.; Walsh, M.P.; Brady, S.W.; Michael, J.R.; Chen, X.; Montefiori, L.; Song, G.; et al. The acquisition of molecular drivers in pediatric therapy-related myeloid neoplasms. Nat. Commun. 2021, 12, 985. [Google Scholar] [CrossRef]
- Aguilera, D.G.; Vaklavas, C.; Tsimberidou, A.M.; Wen, S.; Medeiros, L.J.; Corey, S.J. Pediatric therapy-related myelodysplastic syndrome/acute myeloid leukemia: The MD Anderson Cancer Center experience. J. Pediatr. Hematol. Oncol. 2009, 31, 803–811. [Google Scholar] [CrossRef]
- Waack, K.; Röllecke, K.; Rasche, M.; Walter, C.; Creutzig, U.; Reinhardt, D. Treatment-Related Acute Myeloid Leukemia in Children. Blood 2019, 134 (Suppl. S1), 1322. [Google Scholar] [CrossRef]
- Imamura, T.; Taga, T.; Takagi, M.; Kawasaki, H.; Koh, K.; Taki, T.; Adachi, S.; Manabe, A.; Ishida, Y.; Leukemia/Lymphoma Committee; et al. Nationwide survey of therapy-related leukemia in childhood in Japan. Int. J. Hematol. 2018, 108, 91–97. [Google Scholar] [CrossRef]
- Kowalczyk, J. Epidemiologia nowotworów złośliwych u dzieci. In Onkologia I Hematologia Dziecięca; Chybicka, A., Sawicz-Birkowska, K., Kazanowska, B., Eds.; PZWL Wydawnictwo Lekarskie: Warszawa, Poland, 2021; Volume 1, pp. 19–26. [Google Scholar]
- Tragiannidis, A.; Gombakis, N.; Papageorgiou, M.; Hatzipantelis, E.; Papageorgiou, T.; Hatzistilianou, M. Treatment-related myelodysplastic syndrome (t-MDS)/acute myeloid leukemia (AML) in children with cancer: A single-center experience. Int. J. Immunopathol. Pharmacol. 2016, 29, 729–730. [Google Scholar] [CrossRef]
- Cho, H.W.; Choi, J.B.; Yi, E.S.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Therapy-related myeloid neoplasms in children and adolescents. Blood Res. 2016, 51, 242–248. [Google Scholar] [CrossRef]
- Tsurusawa, M.; Manabe, A.; Hayashi, Y.; Akiyama, Y.; Kigasawa, H.; Inada, H.; Noguchi, Y.; Sawai, N.; Kobayashi, R.; Nagatoshi, Y.; et al. Therapy-related myelodysplastic syndrome in childhood: A retrospective study of 36 patients in Japan. Leuk. Res. 2005, 29, 625–632. [Google Scholar] [CrossRef]
- Strickland, S.A.; Vey, N. Diagnosis and treatment of therapy-related acute myeloid leukemia. Crit. Rev. Oncol. /Hematol. 2022, 171, 103607. [Google Scholar] [CrossRef]
- Cowell, I.G.; Austin, C.A. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int. J. Environ. Res. Public Health 2012, 9, 2075–2091. [Google Scholar] [CrossRef]
- Olney, H.J.; Mitelman, F.; Johansson, B.; Mrozek, K.; Berger, R.; Rowley, J.D. Unique balanced chromosome abnormalities in treatment-related myelodysplastic syndromes and acute myeloid leukemia: Report from an international workshop. Genes Chromosomes Cancer 2002, 33, 413–423. [Google Scholar] [CrossRef]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef]
- Hale, G.A.; Heslop, H.E.; Bowman, L.C.; Rochester, R.A.; Pui, C.H.; Brenner, M.K.; Krance, R.A. Bone marrow transplantation for therapy-induced acute myeloid leukemia in children with previous lymphoid malignancies. Bone Marrow Transplant. 1999, 24, 735–739. [Google Scholar] [CrossRef]
- Woodard, P.; Barfield, R.; Hale, G.; Horwitz, E.; Leung, W.; Ribeiro, R.; Rubnitz, J.; Srivistava, D.K.; Tong, X.; Yusuf, U.; et al. Outcome of hematopoietic stem cell transplantation for pediatric patients with therapy-related acute myeloid leukemia or myelodysplastic syndrome. Pediatr. Blood Cancer. 2006, 47, 931–935. [Google Scholar] [CrossRef]
- Czogała, M.; Balwierz, W.; Pawińska-Wąsikowska, K.; Książek, T.; Bukowska-Strakova, K.; Czogała, W.; Sikorska-Fic, B.; Matysiak, M.; Skalska-Sadowska, J.; Wachowiak, J.; et al. Advances in the First Line Treatment of Pediatric Acute Myeloid Leukemia in the Polish Pediatric Leukemia and Lymphoma Study Group from 1983 to 2019. Cancers 2021, 13, 4536. [Google Scholar] [CrossRef]
- Schmiegelow, K.; Levinsen, M.F.; Attarbaschi, A.; Baruchel, A.; Devidas, M.; Escherich, G.; Gibson, B.; Heydrich, C.; Horibe, K.; Ishida, Y.; et al. Second malignant neoplasms after treatment of childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2013, 31, 2469–2476. [Google Scholar] [CrossRef]
- Hu, Y.; Caldwell, K.J.; Onciu, M.; Federico, S.M.; Salek, M.; Lewis, S.; Lei, S.; Zhang, J.; Nichols, K.E.; Takemoto, C.M.; et al. CPX-351 induces remission in newly diagnosed pediatric secondary myeloid malignancies. Blood Adv. 2022, 6, 521–527. [Google Scholar] [CrossRef]
- Oliai, C.; Schiller, G. How to address second and therapy-related acute myelogenous leukaemia. Br. J. Haematol. 2020, 188, 116–128. [Google Scholar] [CrossRef]
- Fianchi, L.; Criscuolo, M.; Lunghi, M.; Gaidano, G.; Breccia, M.; Levis, A.; Finelli, C.; Santini, V.; Musto, P.; Oliva, E.N.; et al. Outcome of therapy-related myeloid neoplasms treated with azacitidine. J. Hematol. Oncol. 2012, 5, 44. [Google Scholar] [CrossRef]
- Reinhardt, D.; Hasle, H.; Nysom, K.; Baruchel, A.; Locatelli, F.; Benettaib, B.; Biserna, N.; Patturajan, M.; Simcoc, M.; Gaud, A.; et al. Efficacy, Safety, and Pharmacokinetics (PK) of Azacitidine (AZA) in Children and Young Adults with AcuteMyeloid Leukemia(AML) inthePhase2AZA-AML-004Trial. Blood 2020, 136 (Suppl. S1), 10–11. [Google Scholar] [CrossRef]
- Sun, W.; Triche, T., Jr.; Malvar, J.; Gaynon, P.; Sposto, R.; Yang, X.; Bittencourt, H.; Place, A.E.; Messinger, Y.; Fraser, C.; et al. A phase 1 study of azacitidine combined with chemotherapy in child hood leukemia: A report from the TACL consortium. Blood 2018, 131, 1145–1148. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Di Nardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Döhner, H.; Fenaux, P.; Recher, C.; Konopleva, M.; et al. A randomized, double-blind, placebo-controlled study of venetoclax with azacitidine vs azacitidine in treatment-naïve patients with acute myeloid leukemia ineligible for intensive therapy: VIALE-A. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Venditti, A.; Voso, M.T.; Mazzone, C.; Magro, D.; et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: Results of the randomized phase III EORTC-GIMEMA AML-19 trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Weber, D.; Benner, A.; Bullinger, L.; Heuser, M.; Gaidzik, V.; Thol, F.; Agrawal, M.; Teleanu, V.; et al. Adding dasatinib to intensive treatment in core-binding factor acute myeloid leukemia-results of the AMLSG 11-08 trial. Leukemia 2018, 32, 1621–1630. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Period | 2005–2014 | 2015–2022 | All Patients |
|---|---|---|---|
| Number of patients | 24 | 16 | 40 |
| Observation end-point | 31/12/2020 | 31/12/2021 | |
| Median observation time (range) [months] | 94.4 (35.2–176.9) | 29.5 (4.2–57.1) | 53.7 (7.0–176.9) |
| Gender: male/female | 10/14 | 9/7 | 19/21 |
| Age at primary cancer diagnosis—median (range) [years] | 9.4 (0.5–15.7) | 9.3 (0.7–15.9) | 9.4 (0.5–15.9) |
| Age at AML-pCT diagnosis—median (range) [years] | 13.0 (2.7–18.2) | 12.4 (2.8–18.5) | 12.8 (2.7–18.5) |
| Latency period—median (range) [years] | 3.1 (1.5–10.5) | 2.7 (0.7–12.9) | 2.9 (0.7–12.9) |
| Primary neoplasm n (%) | |||
| ALL | 8 (33.3) | 5 (31.2) | 13 (32.5)) |
| Brain tumors | 4 (16.7) | 4 (25) | 8 (20) |
| NBL | 4 (16.7) | 0 | 4 (10) |
| RMS | 3 (12.5) | 1 (6.25) | 4 (10) |
| Osteosarcoma | 2 (8.3) | 1 (6.25) | 3 (7.5)) |
| RBL | 1 (4.2) | 1 (6.25) | 2 (5) |
| HLH | 0 | 2 (12.5) | 2 (5) |
| JMML | 1 (4.2) | 0 | 1 (2.5) |
| Nephroblastoma | 0 | 1 (6.250 | 1 (2.5) |
| Immature teratoma | 1 (4.2) | 0 | 1 (2.5) |
| AML | 0 | 1 (6.25) | 1 (2.5) |
| Alkylating Agents | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cyclophosphamide | Ifosfamide | Melphalan | Busulfan | Lomustine | Temozolomide | Cisplatin | Carboplatin | |
| Number of patients (%) | 25 (62.5) | 14 (35) | 3 (7.5) | 3 (7.5) | 7 (17.5) | 1 (2.5) | 16 (4) | 18 (4.5) |
| Topoisomerase II inhibitors | ||||||||
| Daunorubicin | Doxorubicin | Idarubicin | Etoposide | |||||
| Number of patients (%) | 15 (37.5) | 19 (47.5) | 1 (2.5) | 24 (60) | ||||
| Primary Neoplasm—Number of Patients (%) | Latency Period—Median (Range) [Months] | Genetics —Number of Patients (% of Patients with Available Result) | |||
|---|---|---|---|---|---|
| Complex Karyotype | Aneuploidies | KMT2A Rearrangements | CBF Mutations | ||
| ALL—13 (32.5) | 24.8 (16.2–117.1) | 2 (25.0) | 3 (37.5) | 4 (40) | 1 (10) |
| Brain tumors—8 (20) | 30.4 (23.2–54.8) | 0 | 5 (71.4) | 2 (33.3) | 0 |
| NBL—4 (10) | 52.9 (50.6–125.9) | 1 (33.3) | 1 (33.3) | 0 | 2 (66.6) |
| RMS—4 (10) | 35.7 (22.4–45.7) | 1 (50) | 1 (50) | 0 | 1 (50) |
| Osteosarcoma—3 (7.5) | 45.6 (42.2–48.9) | 1 (50) | 1 (50) | 1 (33.3) | 0 |
| RBL—2 (5) | 95.0 (35.1–154.8) | 0 | 1 (50) | 0 | 1 (50) |
| HLH—2 (5) | 20.1 (7.3–32.8) | 0 | 1 (50) | 1 (50) | 0 |
| JMML—1 (2.5) | 52.1 | 0 | 0 | 0 | 0 |
| Nephroblastoma—1 (2.5) | 25.4 | 0 | 0 | 1 | 0 |
| Immature teratoma—1 (2.5) | 121.3 | 0 | 1 | 0 | 0 |
| AML—1 (2.5) | 77.9 | 0 | 0 | 0 | 0 |
| Period | 2005–2014 | 2015–2022 | All Patients |
|---|---|---|---|
| Number of patients | 24 | 16 | 40 |
| FAB types n (%) | |||
| M0 | 5 (20.9) | 0 | 5 (12.5) |
| M1 | 0 | 3 (18.7) | 3 (7.5) |
| M2 | 4 (16.7) | 1 (6.2) | 5 (12.5) |
| M3 | 0 | 0 | 0 |
| M4 | 4 (16.7) | 0 | 4 (10) |
| M5 | 7 (29.2) | 3 (18.7) | 10 (25) |
| M6 | 0 | 0 | 0 |
| M7 | 0 | 1 (6.2) | 1 (2.5) |
| Non defined | 4 (16.7) | 8 (50) | 12 (30) |
| WBC at diagnosis—median (range) [103/µL] | 5.2 (1.5–120.0) | 3.0 (1.1–309.8) | 4.5 (1.1–309.8) |
| Cytogenetics—number of results (%) | 16 (66.7) | 11 (68.7) | 27 (67.5) |
| Normal karyotype | 2 (12.5) | 2 (18.2) | 4 (14.8) |
| Complex karyotype | 3 (18.7) | 2 (18.2) | 5 (18.5)) |
| Monosomy 7 | 2 (12.5) | 2 (18.2) | 4 (14.8) |
| Monosomy Y | 0 | 3 (27.3) | 3 (11.1) |
| Trisomy 8 | 1 (6.2) | 1 (9.1) | 2 (7.4) |
| KMT2A rearrangements | 2 (12.5) | 4 (36.4) | 6 (22.2) |
| t(8;21)(q22;q22) | 2 (12.5) | 1 (9.1) | 3 (11.1) |
| inv16 | 1 (6.2) | 0 | 1 (3.7) |
| Molecular genetics—number of results (%) | 14 (87.5) | 16 (100) | 30 (75) |
| KMT2A rearrangements | 2 (14.3) | 7 (43.7) | 9 (30) |
| RUNX1::RUNX1T1 fusion | 2 (14.3) | 2 (12.5) | 4 (13.3) |
| CBFβ::MYH11 fusion | 1 (7.1) | 0 | 1 (3.3) |
| No fusion genes found | 9 (64.3) | 7 (43.7) | 16 (53.3) |
| Period | 2005–2014 | 2015–2022 |
|---|---|---|
| Number of patients | 24 | 16 |
| Standard AML therapy—number of treated patients (%) | AML-BFM 2004 Interim—23 (96) | AML-BFM 2012 Registry—7 (44) AML-BFM 2019—6 (38) AML-BFM 2019 + GO (6) |
| Other—number of treated patients (%) | IdaFLA+ FLA—(4) | IdaFLA + FLA, Venetoclax, Azacitidine—1 (6), Venetoclax, Azacitidine—1 (6) |
| Number of chemotherapy cycles before SCT—median (range) | 4 (2–4) | 2 (2–4) |
| SCT—number of patients (%) | 11 (46) | 11 (69) |
| Primary Diagnosis (N) | Non-Responders | Early Deaths | Deaths in Aplasia | Complete Remission | CCR | Deaths in Remission | Relapses | Deaths after Relapse | II CCR |
|---|---|---|---|---|---|---|---|---|---|
| N (%) | |||||||||
| ALL (13) | 1 (7.7) | 1 (7.7) | 0 | 11 (84.6) | 6 (46.1) | 0 | 5 (38.5) | 4 (30.8) | 1 (7.7) |
| Brain tumors (8) | 0 | 3 (37.5) | 1 (12.5) | 4 (50) | 2 (25) | 1 (12.5) | 1 (12.5) | 1 (12.5) | 0 |
| NBL (4) | 1 (25) | 0 | 0 | 3 (75) | 2 (50) | 0 | 1 (25) | 0 | 1 (25) |
| RMS (4) | 0 | 1 (25) | 1 (25) | 2 (50) | 1 (25) | 0 | 1 (25) | 1 (25) | 0 |
| Osteosarcoma (3) | 0 | 0 | 0 | 3 (100) | 2 (66.7) | 1 (33.3) | 0 | 0 | 0 |
| RBL (2) | 0 | 0 | 0 | 2 (100) | 1 (50) | 0 | 1 (50) | 0 | 1 (50) |
| HLH (2) | 0 | 0 | 0 | 2 (100) | 2 (100) | 0 | 0 | 0 | 0 |
| JMML (1) | 0 | 0 | 0 | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 |
| Nephroblastoma (1) | 0 | 0 | 0 | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 |
| Immature teratoma (1) | 0 | 0 | 0 | 1 (100) | 0 | 1 (100) | 0 | 0 | 0 |
| AML (1) | 0 | 0 | 0 | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czogała, M.; Czogała, W.; Pawińska-Wąsikowska, K.; Książek, T.; Bukowska-Strakova, K.; Sikorska-Fic, B.; Łaguna, P.; Skalska-Sadowska, J.; Wachowiak, J.; Rodziewicz-Konarska, A.; et al. Pediatric Acute Myeloid Leukemia Post Cytotoxic Therapy—Retrospective Analysis of the Patients Treated in Poland from 2005 to 2022. Cancers 2023, 15, 734. https://doi.org/10.3390/cancers15030734
Czogała M, Czogała W, Pawińska-Wąsikowska K, Książek T, Bukowska-Strakova K, Sikorska-Fic B, Łaguna P, Skalska-Sadowska J, Wachowiak J, Rodziewicz-Konarska A, et al. Pediatric Acute Myeloid Leukemia Post Cytotoxic Therapy—Retrospective Analysis of the Patients Treated in Poland from 2005 to 2022. Cancers. 2023; 15(3):734. https://doi.org/10.3390/cancers15030734
Chicago/Turabian StyleCzogała, Małgorzata, Wojciech Czogała, Katarzyna Pawińska-Wąsikowska, Teofila Książek, Karolina Bukowska-Strakova, Barbara Sikorska-Fic, Paweł Łaguna, Jolanta Skalska-Sadowska, Jacek Wachowiak, Anna Rodziewicz-Konarska, and et al. 2023. "Pediatric Acute Myeloid Leukemia Post Cytotoxic Therapy—Retrospective Analysis of the Patients Treated in Poland from 2005 to 2022" Cancers 15, no. 3: 734. https://doi.org/10.3390/cancers15030734
APA StyleCzogała, M., Czogała, W., Pawińska-Wąsikowska, K., Książek, T., Bukowska-Strakova, K., Sikorska-Fic, B., Łaguna, P., Skalska-Sadowska, J., Wachowiak, J., Rodziewicz-Konarska, A., Moj-Hackemer, M., Kałwak, K., Muszyńska-Rosłan, K., Krawczuk-Rybak, M., Fałkowska, A., Drabko, K., Kozłowska, M., Irga-Jaworska, N., Bobeff, K., ... Skoczeń, S. (2023). Pediatric Acute Myeloid Leukemia Post Cytotoxic Therapy—Retrospective Analysis of the Patients Treated in Poland from 2005 to 2022. Cancers, 15(3), 734. https://doi.org/10.3390/cancers15030734