
MAPK Pathway Inhibitors in Thyroid Cancer: Preclinical and Clinical Data
 ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
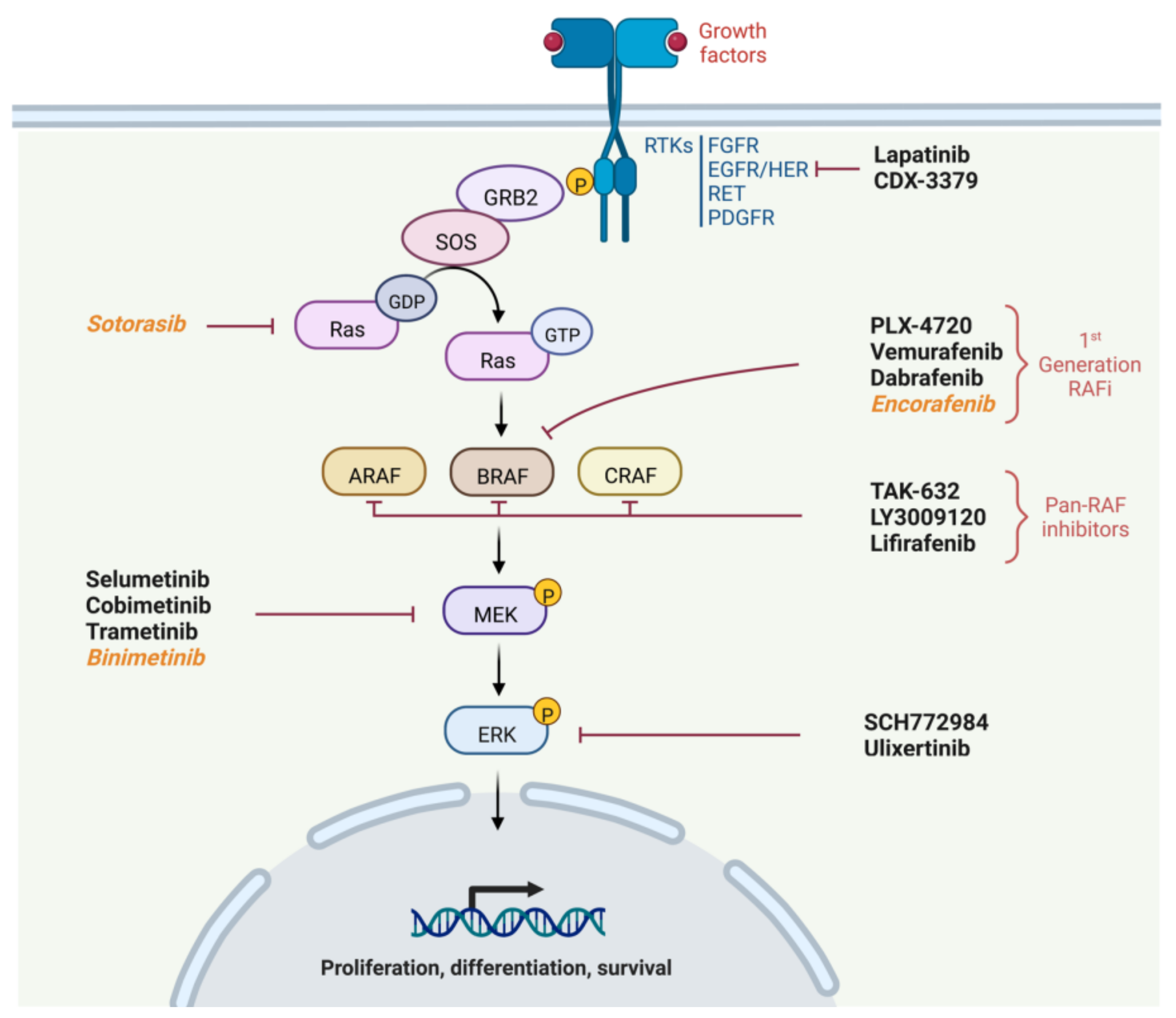
2. Physiology of the MAPK Pathway
- Recruitment of Growth factor Receptor-Bound protein 2 (GRB2) to the phosphorylated site of the receptor and then attachment of Son Of Sevenless (SOS) to GRB2.
- SOS, which is a GTP exchange factor, enables the activation of Ras-GDP to Ras-GTP.
- Ras is a GTPase including three isoforms coded by three genes (HRAS, NRAS or KRAS). It is anchored to the membrane and leads when in active form (Ras-GTP) to the fixation, dimerization and phosphorylation of RAF. The phosphorylation of RAF is not directly performed by RAS but by the SRC Kinase family (SKF) and Casein Kinase 2 (CK2) at the plasma membrane. RAS provides on the one hand the anchoring of RAF to the plasma membrane making it accessible to phosphorylation, and on the other hand, it allows CK2 activation.
- RAF is a protein kinase of which there are also three isoforms coded by three genes (ARAF, BRAF and CRAF). It activates MEK by phosphorylation on serines 218 and 222.
- Finally, MEK activates by phosphorylation ERK1 and ERK 2, the two isoforms of ERK. ERK1 is phosphorylated on threonine 202 and tyrosine 204, while ERK2 is phosphorylated on threonine 185 and tyrosine 187.
3. RAF and MEK Inhibitors in Clinical Studies of Thyroid Cancer without Redifferentiation Purpose
4. Mechanisms of Resistance to MAPKi
5. New Treatments Perspectives
6. Iodine Recaptation Approach in Thyroid Cancers Models
7. Clinical Redifferentiation Strategies in Radioactive Iodine Refractory Thyroid Cancers
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization 2020 Global Cancer Observatory. Available online: https://gco.iarc.fr/today/online-analysis-table?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=asr&sex=0&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&group_cancer=1&include_nmsc=1&include_nmsc_other=1 (accessed on 25 August 2022).
- Megwalu, U.C.; Moon, P.K. Thyroid Cancer Incidence and Mortality Trends in the United States: 2000–2018. Thyroid 2022, 32, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Pizzato, M.; Li, M.; Vignat, J.; Laversanne, M.; Singh, D.; La Vecchia, C.; Vaccarella, S. The Epidemiological Landscape of Thyroid Cancer Worldwide: GLOBOCAN Estimates for Incidence and Mortality Rates in 2020. Lancet Diabetes Endocrinol. 2022, 10, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in Thyroid Cancer Incidence and Mortality in the United States, 1974–2013. JAMA 2017, 317, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Baloch, Z.W.; Asa, S.L.; Barletta, J.A.; Ghossein, R.A.; Juhlin, C.C.; Jung, C.K.; LiVolsi, V.A.; Papotti, M.G.; Sobrinho-Simões, M.; Tallini, G.; et al. Overview of the 2022 WHO Classification of Thyroid Neoplasms. Endocr. Pathol. 2022, 33, 27–63. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.V.; Osamura, R.Y.; Klöppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs. In WHO Classification of Tumours; IARC: Lyon, France, 2017; Volume 10. [Google Scholar]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C.; Kebebew, E.; Brierley, J.; Brito, J.P.; Cabanillas, M.E.; Clark, T.J.; Di Cristofano, A.; Foote, R.; Giordano, T.; Kasperbauer, J.; et al. 2021 American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer: American Thyroid Association Anaplastic Thyroid Cancer Guidelines Task Force. Thyroid 2021, 31, 337–386. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK Signalling: A Master Regulator of Cell Behaviour, Life and Fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and Transcriptomic Hallmarks of Poorly Differentiated and Anaplastic Thyroid Cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Wang, L.Y.; Palmer, F.L.; Nixon, I.J.; Thomas, D.; Patel, S.G.; Shaha, A.R.; Shah, J.P.; Tuttle, R.M.; Ganly, I. Multi-Organ Distant Metastases Confer Worse Disease-Specific Survival in Differentiated Thyroid Cancer. Thyroid 2014, 24, 1594–1599. [Google Scholar] [CrossRef]
- Haq, M.; Harmer, C. Differentiated Thyroid Carcinoma with Distant Metastases at Presentation: Prognostic Factors and Outcome. Clin. Endocrinol. 2005, 63, 87–93. [Google Scholar] [CrossRef]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-Term Outcome of 444 Patients with Distant Metastases from Papillary and Follicular Thyroid Carcinoma: Benefits and Limits of Radioiodine Therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Fugazzola, L.; Elisei, R.; Fuhrer, D.; Jarzab, B.; Leboulleux, S.; Newbold, K.; Smit, J. 2019 European Thyroid Association Guidelines for the Treatment and Follow-Up of Advanced Radioiodine-Refractory Thyroid Cancer. Eur. Thyroid J. 2019, 8, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Gild, M.L.; Tsang, V.H.M.; Clifton-Bligh, R.J.; Robinson, B.G. Multikinase Inhibitors in Thyroid Cancer: Timing of Targeted Therapy. Nat. Rev. Endocrinol. 2021, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in Radioactive Iodine-Refractory, Locally Advanced or Metastatic Differentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 3 Trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Robinson, B.; Sherman, S.I.; Krajewska, J.; Lin, C.-C.; Vaisman, F.; Hoff, A.O.; Hitre, E.; Bowles, D.W.; Hernando, J.; et al. Cabozantinib for Radioiodine-Refractory Differentiated Thyroid Cancer (COSMIC-311): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2021, 22, 1126–1138. [Google Scholar] [CrossRef]
- Smallridge, R.C.; Copland, J.A. Anaplastic Thyroid Carcinoma: Pathogenesis and Emerging Therapies. Clin. Oncol. 2010, 22, 486–497. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Cook, F.A.; Cook, S.J. Inhibition of RAF Dimers: It Takes Two to Tango. Biochem. Soc. Trans. 2021, 49, 237–251. [Google Scholar] [CrossRef]
- Lavoie, H.; Therrien, M. Regulation of RAF Protein Kinases in ERK Signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.C.; Cabanillas, M.E.; Boran, A.; et al. Dabrafenib plus Trametinib in Patients with BRAF V600E-Mutant Anaplastic Thyroid Cancer: Updated Analysis from the Phase II ROAR Basket Study. Ann. Oncol. 2022, 33, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Filetti, S.; Durante, C.; Hartl, D.; Leboulleux, S.; Locati, L.D.; Newbold, K.; Papotti, M.G.; Berruti, A. Thyroid Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019, 30, 1856–1883. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Cabanillas, M.E.; Lazar, A.J.; Williams, M.D.; Sanders, D.L.; Ilagan, J.L.; Nolop, K.; Lee, R.J.; Sherman, S.I. Clinical Responses to Vemurafenib in Patients with Metastatic Papillary Thyroid Cancer Harboring BRAF V600E Mutation. Thyroid 2013, 23, 1277–1283. [Google Scholar] [CrossRef]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.W.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in Patients with BRAFV600E-Positive Metastatic or Unresectable Papillary Thyroid Cancer Refractory to Radioactive Iodine: A Non-Randomised, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef]
- Falchook, G.S.; Millward, M.; Hong, D.; Naing, A.; Piha-Paul, S.; Waguespack, S.G.; Cabanillas, M.E.; Sherman, S.I.; Ma, B.; Curtis, M.; et al. BRAF Inhibitor Dabrafenib in Patients with Metastatic BRAF -Mutant Thyroid Cancer. Thyroid 2015, 25, 71–77. [Google Scholar] [CrossRef]
- Busaidy, N.; Konda, B.; Wei, L.; Wirth, L.J.; Devine, C.; Daniels, G.A.; DeSouza, J.A.; Poi, M.; Seligson, N.D.; Cabanillas, M.; et al. Dabrafenib vs Dabrafenib + Trametinib in BRAF-Mutated Radioactive Iodine Refractory Differentiated Thyroid Cancer—Results of a Randomized, Phase 2, Open-Label, Multicenter Trial. Thyroid 2022, 32, 1184–1192. [Google Scholar] [CrossRef]
- Hayes, D.N.; Lucas, A.S.; Tanvetyanon, T.; Krzyzanowska, M.K.; Chung, C.H.; Murphy, B.A.; Gilbert, J.; Mehra, R.; Moore, D.T.; Sheikh, A.; et al. Phase II Efficacy and Pharmacogenomic Study of Selumetinib (AZD6244; ARRY-142886) in Iodine-131 Refractory Papillary Thyroid Carcinoma with or without Follicular Elements. Clin. Cancer Res. 2012, 18, 2056–2065. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus Binimetinib versus Vemurafenib or Encorafenib in Patients with BRAF -Mutant Melanoma (COLUMBUS): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Roelli, M.A.; Ruffieux-Daidié, D.; Stooss, A.; Phillips, W.A.; Dettmer, M.S.; Charles, R.-P. PIK3CAH1047R-Induced Paradoxical ERK Activation Results in Resistance to BRAFV600E Specific Inhibitors in BRAFV600E PIK3CAH1047R Double Mutant Thyroid Tumors. Oncotarget 2017, 8, 103207. [Google Scholar] [CrossRef] [PubMed]
- Duquette, M.; Sadow, P.M.; Husain, A.; Sims, J.N.; Antonello, Z.A.; Fischer, A.H.; Song, C.; Castellanos-Rizaldos, E.; Makrigiorgos, G.M.; Kurebayashi, J.; et al. Metastasis-Associated MCL1 and P16 Copy Number Alterations Dictate Resistance to Vemurafenib in a BRAFV600E Patient-Derived Papillary Thyroid Carcinoma Preclinical Model. Oncotarget 2015, 6, 42445–42467. [Google Scholar] [CrossRef]
- Danysh, B.P.; Rieger, E.Y.; Sinha, D.K.; Evers, C.V.; Cote, G.J.; Cabanillas, M.E.; Hofmann, M.-C. Long-Term Vemurafenib Treatment Drives Inhibitor Resistance through a Spontaneous KRAS G12D Mutation in a BRAF V600E Papillary Thyroid Carcinoma Model. Oncotarget 2016, 7, 30907–30923. [Google Scholar] [CrossRef] [PubMed]
- Antonello, Z.A.; Hsu, N.; Bhasin, M.; Roti, G.; Joshi, M.; Van Hummelen, P.; Ye, E.; Lo, A.S.; Karumanchi, S.A.; Bryke, C.R.; et al. Vemurafenib-Resistance via de Novo RBM Genes Mutations and Chromosome 5 Aberrations Is Overcome by Combined Therapy with Palbociclib in Thyroid Carcinoma with BRAFV600E. Oncotarget 2017, 8, 84743–84760. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Dadu, R.; Iyer, P.; Wanland, K.B.; Busaidy, N.L.; Ying, A.; Gule-Monroe, M.; Wang, J.R.; Zafereo, M.; Hofmann, M.-C. Acquired Secondary RAS Mutation in BRAF V600E -Mutated Thyroid Cancer Patients Treated with BRAF Inhibitors. Thyroid 2020, 30, 1288–1296. [Google Scholar] [CrossRef]
- Bagheri-Yarmand, R.; Busaidy, N.L.; McBeath, E.; Danysh, B.P.; Evans, K.W.; Moss, T.J.; Akcakanat, A.; Ng, P.K.S.; Knippler, C.M.; Golden, J.A.; et al. RAC1 Alterations Induce Acquired Dabrafenib Resistance in Association with Anaplastic Transformation in a Papillary Thyroid Cancer Patient. Cancers 2021, 13, 4950. [Google Scholar] [CrossRef]
- Knauf, J.A.; Luckett, K.A.; Chen, K.-Y.; Voza, F.; Socci, N.D.; Ghossein, R.; Fagin, J.A. Hgf/Met Activation Mediates Resistance to BRAF Inhibition in Murine Anaplastic Thyroid Cancers. J. Clin. Investig. 2018, 128, 4086–4097. [Google Scholar] [CrossRef]
- Byeon, H.K.; Na, H.J.; Yang, Y.J.; Kwon, H.J.; Chang, J.W.; Ban, M.J.; Kim, W.S.; Shin, D.Y.; Lee, E.J.; Koh, Y.W.; et al. C-Met-Mediated Reactivation of PI3K/AKT Signaling Contributes to Insensitivity of BRAF(V600E) Mutant Thyroid Cancer to BRAF Inhibition. Mol. Carcinog. 2016, 55, 1678–1687. [Google Scholar] [CrossRef] [PubMed]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of Feedback Inhibition of HER3 Transcription by RAF and MEK Inhibitors Attenuates Their Antitumor Effects in BRAF-Mutant Thyroid Carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Levin, R.S.; Gordan, J.D.; Oses-Prieto, J.A.; Webber, J.T.; Salt, M.; Hann, B.; Burlingame, A.L.; McCormick, F.; Bandyopadhyay, S.; et al. Oncogene Mimicry as a Mechanism of Primary Resistance to BRAF Inhibitors. Cell Rep. 2014, 8, 1037–1048. [Google Scholar] [CrossRef]
- Notarangelo, T.; Sisinni, L.; Trino, S.; Calice, G.; Simeon, V.; Landriscina, M. IL6/STAT3 Axis Mediates Resistance to BRAF Inhibitors in Thyroid Carcinoma Cells. Cancer Lett. 2018, 433, 147–155. [Google Scholar] [CrossRef]
- Wang, N.; Wen, J.; Ren, W.; Wu, Y.; Deng, C. Upregulation of TRIB2 by Wnt/β-Catenin Activation in BRAFV600E Papillary Thyroid Carcinoma Cells Confers Resistance to BRAF Inhibitor Vemurafenib. Cancer Chemother. Pharmacol. 2021, 88, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Notarangelo, T.; Sisinni, L.; Condelli, V.; Landriscina, M. Dual EGFR and BRAF Blockade Overcomes Resistance to Vemurafenib in BRAF Mutated Thyroid Carcinoma Cells. Cancer Cell Int. 2017, 17, 86. [Google Scholar] [CrossRef]
- Zhi, J.; Yi, J.; Hou, X.; Wang, W.; Yang, W.; Hu, L.; Huang, J.; Ruan, X.; Gao, M.; Zheng, X. Targeting SHP2 Sensitizes Differentiated Thyroid Carcinoma to the MEK Inhibitor. Am. J. Cancer Res. 2022, 12, 247. [Google Scholar] [PubMed]
- Gianì, F.; Russo, G.; Pennisi, M.; Sciacca, L.; Frasca, F.; Pappalardo, F. Computational Modeling Reveals MAP3K8 as Mediator of Resistance to Vemurafenib in Thyroid Cancer Stem Cells. Bioinformatics 2019, 35, 2267–2275. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, J.; Tian, M.; Kang, N.; Xu, G.; Zhi, J.; Ruan, X.; Hou, X.; Zhang, W.; Yi, J.; et al. Pharmacological Inhibition of Ref-1 Enhances the Therapeutic Sensitivity of Papillary Thyroid Carcinoma to Vemurafenib. Cell Death Dis. 2022, 13, 124. [Google Scholar] [CrossRef]
- Run, L.; Wang, L.; Nong, X.; Li, N.; Huang, X.; Xiao, Y. Involvement of HMGB1 in Vemurafenib Resistance in Thyroid Cancer Cells Harboring BRAF (V600E) Mutation by Regulating Excessive Autophagy. Endocrine 2021, 71, 418–426. [Google Scholar] [CrossRef]
- Giuffrida, R.; Adamo, L.; Iannolo, G.; Vicari, L.; Giuffrida, D.; Eramo, A.; Gulisano, M.; Memeo, L.; Conticello, C. Resistance of Papillary Thyroid Cancer Stem Cells to Chemotherapy. Oncol. Lett. 2016, 12, 687–691. [Google Scholar] [CrossRef]
- Holderfield, M.; Merritt, H.; Chan, J.; Wallroth, M.; Tandeske, L.; Zhai, H.; Tellew, J.; Hardy, S.; Hekmat-Nejad, M.; Stuart, D.D.; et al. RAF Inhibitors Activate the MAPK Pathway by Relieving Inhibitory Autophosphorylation. Cancer Cell 2013, 23, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF Inhibitors Transactivate RAF Dimers and ERK Signalling in Cells with Wild-Type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Karoulia, Z.; Wu, Y.; Ahmed, T.A.; Xin, Q.; Bollard, J.; Krepler, C.; Wu, X.; Zhang, C.; Bollag, G.; Herlyn, M.; et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 2016, 30, 485–498. [Google Scholar] [CrossRef]
- Noeparast, A.; Giron, P.; De Brakeleer, S.; Eggermont, C.; De Ridder, U.; Teugels, E.; De Grève, J. Type II RAF Inhibitor Causes Superior ERK Pathway Suppression Compared to Type I RAF Inhibitor in Cells Expressing Different BRAF Mutant Types Recurrently Found in Lung Cancer. Oncotarget 2018, 9, 16110–16123. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.-J.; Sun, Z.-K.; Shen, C.-T.; Song, H.-J.; Zhang, X.-Y.; Qiu, Z.-L.; Luo, Q.-Y. Obatoclax and LY3009120 Efficiently Overcome Vemurafenib Resistance in Differentiated Thyroid Cancer. Theranostics 2017, 7, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Hicks, H.M.; McKenna, L.R.; Espinoza, V.L.; Pozdeyev, N.; Pike, L.A.; Sams, S.B.; LaBarbera, D.; Reigan, P.; Raeburn, C.D.; Schweppe, R. Inhibition of BRAF and ERK1/2 Has Synergistic Effects on Thyroid Cancer Growth in Vitro and in Vivo. Mol. Carcinog. 2021, 60, 201–212. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hollebecque, A.; Flaherty, K.T.; Shapiro, G.I.; Rodon Ahnert, J.; Millward, M.J.; Zhang, W.; Gao, L.; Sykes, A.; Willard, M.D.; et al. A Phase I Study of LY3009120, a Pan-RAF Inhibitor, in Patients with Advanced or Metastatic Cancer. Mol. Cancer Ther. 2020, 19, 460–467. [Google Scholar] [CrossRef]
- Desai, J.; Gan, H.; Barrow, C.; Jameson, M.; Atkinson, V.; Haydon, A.; Millward, M.; Begbie, S.; Brown, M.; Markman, B.; et al. Phase I, Open-Label, Dose-Escalation/Dose-Expansion Study of Lifirafenib (BGB-283), an RAF Family Kinase Inhibitor, in Patients with Solid Tumors. J. Clin. Oncol. 2020, 38, 2140–2150. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic Non-Small Cell Lung Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Dohán, O.; Baloch, Z.; Bánrévi, Z.; Livolsi, V.; Carrasco, N. RAPID COMMUNICATION: Predominant Intracellular Overexpression of the Na+/I− Symporter (NIS) in a Large Sampling of Thyroid Cancer Cases. J. Clin. Endocrinol. Metab. 2001, 86, 2697–2700. [Google Scholar] [CrossRef] [PubMed]
- Bonaldi, E.; Gargiuli, C.; De Cecco, L.; Micali, A.; Rizzetti, M.G.; Greco, A.; Borrello, M.G.; Minna, E. BRAF Inhibitors Induce Feedback Activation of RAS Pathway in Thyroid Cancer Cells. Int. J. Mol. Sci. 2021, 22, 5744. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hu, S.; Hou, P.; Jiang, D.; Condouris, S.; Xing, M. Suppression of BRAF/MEK/MAP Kinase Pathway Restores Expression of Iodide-Metabolizing Genes in Thyroid Cells Expressing the V600E BRAF Mutant. Clin. Cancer Res. 2007, 13, 1341–1349. [Google Scholar] [CrossRef]
- Nagarajah, J.; Le, M.; Knauf, J.A.; Ferrandino, G.; Montero-Conde, C.; Pillarsetty, N.; Bolaender, A.; Irwin, C.; Krishnamoorthy, G.P.; Saqcena, M.; et al. Sustained ERK Inhibition Maximizes Responses of BrafV600E Thyroid Cancers to Radioiodine. J. Clin. Investig. 2016, 126, 4119–4124. [Google Scholar] [CrossRef]
- Ullmann, T.M.; Liang, H.; Moore, M.D.; Al-Jamed, I.; Gray, K.D.; Limberg, J.; Stefanova, D.; Buicko, J.L.; Finnerty, B.; Beninato, T.; et al. Dual Inhibition of BRAF and MEK Increases Expression of Sodium Iodide Symporter in Patient-Derived Papillary Thyroid Cancer Cells in Vitro. Surgery 2020, 167, 56–63. [Google Scholar] [CrossRef]
- Fu, H.; Cheng, L.; Jin, Y.; Cheng, L.; Liu, M.; Chen, L. MAPK Inhibitors Enhance HDAC Inhibitor-Induced Redifferentiation in Papillary Thyroid Cancer Cells Harboring BRAFV600E: An In Vitro Study. Mol. Ther. Oncolytics 2019, 12, 235–245. [Google Scholar] [CrossRef]
- Fu, H.; Cheng, L.; Sa, R.; Jin, Y.; Chen, L. Combined Tazemetostat and MAPKi Enhances Differentiation of Papillary Thyroid Cancer Cells Harbouring BRAFV600E by Synergistically Decreasing Global Trimethylation of H3K27. J. Cell. Mol. Med. 2020, 24, 3336–3345. [Google Scholar] [CrossRef]
- Cheng, L.; Jin, Y.; Liu, M.; Ruan, M.; Chen, L. HER Inhibitor Promotes BRAF/MEK Inhibitor-Induced Redifferentiation in Papillary Thyroid Cancer Harboring BRAFV600E. Oncotarget 2017, 8, 19843–19854. [Google Scholar] [CrossRef]
- Luckett, K.A.; Cracchiolo, J.R.; Krishnamoorthy, G.P.; Leandro-Garcia, L.J.; Nagarajah, J.; Saqcena, M.; Lester, R.; Im, S.Y.; Zhao, Z.; Lowe, S.W.; et al. Co-Inhibition of SMAD and MAPK Signaling Enhances 124I Uptake in BRAF-Mutant Thyroid Cancers. Endocr. Relat. Cancer 2021, 28, 391–402. [Google Scholar] [CrossRef]
- Riesco-Eizaguirre, G.; Rodríguez, I.; De la Vieja, A.; Costamagna, E.; Carrasco, N.; Nistal, M.; Santisteban, P. The BRAFV600E Oncogene Induces Transforming Growth Factor β Secretion Leading to Sodium Iodide Symporter Repression and Increased Malignancy in Thyroid Cancer. Cancer Res. 2009, 69, 8317–8325. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, S.M.; McFadden, D.G.; Palmer, E.L.; Daniels, G.H.; Wirth, L.J. Redifferentiation of Iodine-Refractory BRAF V600E-Mutant Metastatic Papillary Thyroid Cancer with Dabrafenib. Clin. Cancer Res. 2015, 21, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.A.; Sherman, E.J.; Baxi, S.S.; Tchekmedyian, V.; Grewal, R.K.; Larson, S.M.; Pentlow, K.S.; Haque, S.; Tuttle, R.M.; Sabra, M.M.; et al. Vemurafenib Redifferentiation of BRAF Mutant, RAI-Refractory Thyroid Cancers. J. Clin. Endocrinol. Metab. 2019, 104, 1417–1428. [Google Scholar] [CrossRef]
- Jaber, T.; Waguespack, S.G.; Cabanillas, M.E.; Elbanan, M.; Vu, T.; Dadu, R.; Sherman, S.I.; Amit, M.; Santos, E.B.; Zafereo, M.; et al. Targeted Therapy in Advanced Thyroid Cancer to Resensitize Tumors to Radioactive Iodine. J. Clin. Endocrinol. Metab. 2018, 103, 3698–3705. [Google Scholar] [CrossRef] [PubMed]
- Iravani, A.; Solomon, B.; Pattison, D.A.; Jackson, P.; Ravi Kumar, A.; Kong, G.; Hofman, M.S.; Akhurst, T.; Hicks, R.J. Mitogen-Activated Protein Kinase Pathway Inhibition for Redifferentiation of Radioiodine Refractory Differentiated Thyroid Cancer: An Evolving Protocol. Thyroid 2019, 29, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-Enhanced Radioiodine Uptake in Advanced Thyroid Cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef]
- Leboulleux, S.; Cao, C.; Zerdoud, S.; Attard, M.; Bournaud, C.; Benisvy, D.; Taieb, D.; Bardet, S.; Terroir-Cassou-Mounat, M.; Betrian, S.; et al. MERAIODE: A Redifferentiation Phase II Trial with Trametinib and Dabrafenib Followed by Radioactive Iodine Administration for Metastatic Radioactive Iodine Refractory Differentiated Thyroid Cancer Patients with a BRAFV600E Mutation (NCT03244956). J. Endocr. Soc. 2021, 5, A876. [Google Scholar] [CrossRef]
- Weber, M.; Kersting, D.; Riemann, B.; Brandenburg, T.; Führer-Sakel, D.; Grünwald, F.; Kreissl, M.C.; Dralle, H.; Weber, F.; Schmid, K.W.; et al. Enhancing Radioiodine Incorporation into Radioiodine-Refractory Thyroid Cancer with MAPK Inhibition (ERRITI): A Single-Center Prospective Two-Arm Study. Clin. Cancer Res. 2022, 28, 4194–4202. [Google Scholar] [CrossRef]
- Tchekmedyian, V.; Dunn, L.; Sherman, E.; Baxi, S.S.; Grewal, R.K.; Larson, S.M.; Pentlow, K.S.; Haque, S.; Tuttle, R.M.; Sabra, M.M.; et al. Enhancing Radioiodine Incorporation in BRAF-Mutant, Radioiodine-Refractory Thyroid Cancers with Vemurafenib and the Anti-ErbB3 Monoclonal Antibody CDX-3379: Results of a Pilot Clinical Trial. Thyroid 2022, 32, 273–282. [Google Scholar] [CrossRef]
- Leboulleux, S.; Dupuy, C.; Lacroix, L.; Attard, M.; Grimaldi, S.; Corre, R.; Ricard, M.; Nasr, S.; Berdelou, A.; Hadoux, J.; et al. Redifferentiation of a BRAF K601E-Mutated Poorly Differentiated Thyroid Cancer Patient with Dabrafenib and Trametinib Treatment. Thyroid 2019, 29, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Groussin, L.; Bessiene, L.; Arrondeau, J.; Garinet, S.; Cochand-Priollet, B.; Lupo, A.; Zerbit, J.; Clerc, J.; Huillard, O. Selpercatinib-Enhanced Radioiodine Uptake in RET-Rearranged Thyroid Cancer. Thyroid 2021, 31, 1603–1604. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Dedecjus, M.; Wirth, L.J.; Tuttle, R.M.; Inabnet, W.B.; Tennvall, J.; Vaisman, F.; Bastholt, L.; Gianoukakis, A.G.; Rodien, P.; et al. Selumetinib Plus Adjuvant Radioactive Iodine in Patients with High-Risk Differentiated Thyroid Cancer: A Phase III, Randomized, Placebo-Controlled Trial (ASTRA). J. Clin. Oncol. 2022, 40, 1870–1878. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Thyroid Cancer Types | Drug Targets | Therapies | Patients Number | Study Design | ORR | Median Duration of Response (Months) | Median PFS (Months) | Median OS (Months) | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Locally advanced or metastatic BRAF mutated ATC | BRAF + MEK1/2 | Dabrafenib + trametinib | 36 | Open-label, phase II trial | 56% (3 CR, 17 PR) | 12-months DoR: 50% | 6.7 | 14.5 | [24] |
| Metastatic BRAF mutated PTC | BRAF | Vemurafenib | 3 | Phase I trial | 33.3% (1 PR) | NA | n1 = 11.4 n2 = 11.7 n3 = 13.2 | n1 = 15 n2 = 21 n3 = 31.7 | [26] |
| Metastatic or recurrent BRAF mutated PTC | BRAF | Vemurafenib | Total: 51 Naïve, cohort 1 (C1): 26 Previous TKI, cohort 2 (C2): 25 | Open-label, phase II trial | C1: 38.5% (10 PR) C2: 27.3% (6 PR) | C1: 16.5 C2: 7.4 | C1: 18.2 C2: 8.9 | C1: NR C2: 14.4 | [27] |
| Metastatic BRAF mutated PDTC or DTC | BRAF | Dabrafenib | 14 | Phase I trial | 29% (4 PR) | NA | 11.3 | NA | [28] |
| BRAF Mutated RAIR PTC | BRAF | Dabrafenib | 26 | Randomized phase II trial | 35% (9 PR) | 18.3 | 10.7 | 37.9 | [29] |
| BRAF Mutated RAIR PTC | BRAF + MEK1/2 | Dabrafenib + trametinib | 27 | Randomized phase II trial | 30% (8 PR) | 17.0 | 15.1 | 47.5 | |
| BRAF Mutated or WT RAIR PTC | MEK1/2 | Selumetinib | 32 | Open-label, phase II trial | 3% (1 PR) | NA | 8 | NA | [30] |
| Type of Resistance Mechanism | Drugs Used to Study Resistance (Target) | Thyroid Cancer Models | Mechanism of Resistance (Intrinsic or Acquired Resistance) | Drug Used to Overcome Resistance (target) | Resistance Overcome | Ref |
|---|---|---|---|---|---|---|
| Genomic instability | PLX-4720 (BRAF) | - BRAFV600E ATC cell line - BRAFV600E and double mutant BRAFV600E + PIK3CAH1047R TC mouse models | PIK3CAH1047R mutation (intrinsic resistance) leading to: - MAPK pathway paradoxical activation | GDC-0941 (PIK3CA) | Yes | [36] |
| Vemurafenib (BRAF) | - BRAFV600E PTC cell lines - Samples derived from BRAF-mutated PTC patient - Primary cell culture of BRAFV600E metastatic or recurrent PTC | Copy number gain of MCL1 and loss of CDKN2A (intrinsic resistance) leading to: - Impairment of the BCL2-regulated apoptotic pathway - CDK4/6 pathway activation | Obatoclax (BCL2/MCL1) | Yes | [37] | |
| BRAFV600E PTC cell line | KRASG12D mutation (acquired resistance) leading to: - PI3K/AKT pathway activation - MAPK pathway paradoxical activation | NA | NA | [38] | ||
| BRAFV600E PTC cell line | Amplification of chromosome 5 and de novo mutations in the RBM genes family (intrinsic and acquired resistance) leading to: - Chromosome instability and deregulation of cell cycle checkpoints in response to DNA-damage | Palbociclib (CDK4/6) | Yes | [39] | ||
| Vemurafenib (BRAF) Dabrafenib (BRAF) + Trametinib (MEK) | 2 PTC patients and 2 ATC patients with BRAF mutation | Acquired KRASG12V (n = 2), NRASQ61K (n = 1), and NRASG13D (n = 1) mutations on progressive metastatic lesions after treatment with MAPKi | NA | NA | [40] | |
| Dabrafenib (BRAF) | - BRAFV600E PTC cell lines - PTC BRAF-mutated patient - Patient derived cell line | RAC1 mutation and copy number gain (acquired resistance) leading to: - RAC1/PAK1 pathway activation | EHop-016 (RAC1) | Yes | [41] | |
| Autocrine loop | PLX-4720 (BRAF) | - Transgenic p53- and BRAFV600E ATC mouse model - Mouse derived cell lines | c-Met overexpression and HGF increased secretion (acquired resistance) leading to: - PI3K/AKT pathway activation - MAPK pathway paradoxical activation | PF-04217903 and crizotinib (c-Met) | Yes | [42] |
| Vemurafenib (BRAF) | - BRAFV600E PTC and ATC cell lines - ATC xenograft mouse model | c-Met overexpression and HGF increased secretion (acquired resistance) leading to: - PI3K/AKT pathway activation | PHA665752 (c-Met) | Yes | [43] | |
| BRAFV600E PTC and ATC cell lines | HER3 overexpression and activation by NRG1 secretion (acquired resistance) leading to: - PI3K/AKT pathway activation - MAPK pathway paradoxical activation | Lapatinib (HER) | Yes | [44] | ||
| Autocrine loop | Vemurafenib (BRAF) | BRAFV600E PTC and ATC cell lines | IL6 secretion (acquired resistance) leading to: - STAT3/JAK pathway activation | Tofacitinib (JAK) | Yes | [45] |
| Tocilizumab (IL6-R) | [46] | |||||
| Upregulation of proteins operating synergistically with the MAPK and PI3K/AKT pathways | Vemurafenib (BRAF) | BRAFV600E PTC cell lines | TRIB2 upregulation induced by activation of the Wnt/β-catenin pathway (acquired resistance) leading to: - PI3K/AKT pathway activation - MAPK pathway paradoxical activation | ICG-001 (β-catenin) | Yes | [47] |
| BRAFV600E PTC and ATC cell lines | EGFR overactivation (acquired resistance) leading to: - PI3K/AKT pathway activation - MAPK pathway paradoxical activation | Gefitinib (EGFR) | Yes | [48] | ||
| Selumetinib (MEK) | - BRAFV600E PTC cell lines - PTC xenograft mouse models - Transgenic BRAFV600E mouse models | SHP2 upregulation and activation (acquired resistance) induced by upregulation and activation of multiple RTKs (RET, FGFR, HER2…) leading to: - MAPK pathway paradoxical activation | SHP099 (SHP2) | Yes | [49] | |
| Cancer Stem Cells (CSCs) mediated resistance | Vemurafenib (BRAF) | CSCs selected from BRAFV600E ATC cell lines | TPL2 overexpression in CSCs (acquired resistance) leading to: - PI3K/AKT pathway activation - MAPK pathway paradoxical activation | (TPL2) | Yes | [50] |
| Oxidative stress mediated resistance | Vemurafenib (BRAF) | - BRAFV600E PTC cell lines - Samples derived from BRAF mutated PTC patient | Ref-1 upregulation (intrinsic resistance) leading to: - MAPK pathway paradoxical activation | E3330 (Ref-1) | Yes | [51] |
| Autophagy mediated resistance | Vemurafenib (BRAF) | BRAFV600E PTC Cell line | HMGB1 upregulation (acquired resistance) leading to: - HMGB1-induced autophagy | 3-MA (Autophagy inhibitor) | Yes | [52] |
| Preclinical Stage | |||||
|---|---|---|---|---|---|
| Drug Targets | Therapies | Thyroid Cancer Model | Experimentation Type | Effectiveness criteria | Ref |
| ARAF, BRAF, CRAF | TAK-632 vs. vemurafenib | 3 ATC BRAFV600E cell lines | - Quantification of MAPK pathway inhibition - Proliferation assay | TAK-632 > vemurafenib: On MAPK inhibition On GI50 and IC50 | [56] |
| ARAF, BRAF, CRAF | LY3009120 vs. vemurafenib | - 3 PTC BRAFV600E cell lines - Mouse xenograft model | - Viability assay - Apoptosis assay - Cytotoxic assay - In vivo tumor growth |
On tumor growth inhibition in vivo | [58] |
| RAF + ERK1/2 | Dabrafenib + SCH772984 | - 5 BRAFV600E cell lines (ATC + DTC) - Mouse xenograft model | - Quantification of MAPK pathway inhibition - Viability assay - Apoptosis assay - In vivo tumor growth | Dabrafenib + SCH772984 avoid MAPK reactivation observed with dabrafenib alone
On tumor growth inhibition in vivo | [59] |
| Drug Targets | Therapies | Thyroid Cancer Model | Experimentation Type | Effectiveness | Ref |
|---|---|---|---|---|---|
| BRAF | Vemurafenib (V) Dabrafenib (D) | 3 PTC + 1 ATC BRAFV600E cell lines | - NIS expression (RT-qPCR) - Iodide uptake assay - Gene expression scores related to TCGA derived gene signatures | Monotherapy (V) or (D): ↑ NIS mRNA ↑ Iodide uptake capacity ↑ Thyroid differentiation score (done in 1 PTC Cell line) | [66] |
| MEK | U0126 | 1 BRAFV600E inducible rat thyroid derived cell line | - NIS expression (RT-qPCR) | ↑ NIS mRNA | [67] |
| BRAF or MEK | Vemurafenib (V) Selumetinib (S) U0126 (U) CKI (C) | - 1 BRAFV600E inducible rat thyroid derived cell line - Mouse model of BRAFV600E PTC | Cell line: - NIS expression (WB) Mouse model experience: - NIS expression (RT-qPCR) - Iodide uptake assay - Tumoral response to RAI-therapy (tumor volume evaluated by US) | Cell line experience: ↑ NIS protein with (V), (S), (U), (C) Mouse model experience (C) vs. (S): ↑ NIS mRNA with (C) > (S) ↑ Iodide uptake capacity with (C) > (S) (knowing S > CTL) Tumoral response to RAI-therapy with (C) > (S) (knowing S > CTL) | [68] |
| BRAF + MEK | Dabrafenib (D) Trametinib (T) | - 1 PTC BRAFV600E cell line - PTC-patient derived primary cell cultures | - NIS expression (RT-qPCR) | Cell line: No NIS re-expression with monotherapy (T) ↑ NIS mRNA with (D+T) PTC-Patient derived primary cell cultures: ↑ NIS mRNA with (T) Bi-therapy (D+T) even more efficient | [69] |
| BRAF + HDAC | Dabrafenib (D) Selumetinib (S) Panobinostat (P) | 2 PTC BRAFV600E cell lines | - NIS expression (RT-qPCR) - NIS localization (immunofluorescent microscopy) - Iodide uptake assay | Monotherapy (D) or (S): ↑ NIS mRNA ↑ NIS fluorescence to the cell membrane ↑ Iodide uptake capacity Bi-therapy (D+P) and (S+P) even more efficient on all experimentations | [70] |
| BRAF + EZH2 | Dabrafenib (D) Selumetinib (S) Tazemetostat (T) (EZH2 inhibitor) | 2 PTC BRAFV600E cell lines | - NIS expression (RT-qPCR, WB) - NIS localization (immunofluorescent microscopy) - Iodide uptake assay | Monotherapy (D) or (S): ↑ NIS mRNA and protein ↑ NIS fluorescence to the cell membrane ↑ Iodide uptake capacity Bi-therapy (D+T) and (S+T) even more efficient on all experimentations | [71] |
| BRAF + HER | Dabrafenib (D) Selumetinib (S) Lapatinib (L) | 2 PTC BRAFV600E cell lines | - NIS expression (RT-qPCR, WB) - NIS localization (immunofluorescent microscopy) - Iodide uptake assay | (Monotherapy (D) or (S): ↑ NIS mRNA and protein ↑ NIS fluorescence to the cell membrane ↑ Iodide uptake capacity Bi-therapy (D+L) and (S+L) even more efficient on all experimentations | [72] |
| MEK + ACVR1B/TGFBR1 | CKI (C) Vactosertib (V) | Mouse model of BRAFV600E PTC | - NIS expression (RT-qPCR) - NIS localization (immunohistochemistry) - Iodide uptake assay | (C): ↑ NIS mRNA ↑ NIS fluorescence in tumors ↑ Iodide uptake capacity Bi-therapy (C+V) more efficient on Iodide uptake capacity but not on NIS mRNA and NIS fluorescence in tumors | [73] |
| Drug Targets | Therapy (Duration of Treatment) | Thyroid Cancer Types | Oncogenic Driver | Study Design | N Total | Rate of RAI Uptake Restoration | RECIST Response (N Treated) | Ref |
|---|---|---|---|---|---|---|---|---|
| BRAF | Dabrafenib (6 weeks) | PTC | BRAF | - Prospective evaluation of RAI avidity restoration by diagnostic 131I-WBS - If avidity restored, treatment with fixed activity of 5.5 GBq | 10 | 60% | At 3 months (n = 6): 2 PR, 4 SD | [75] |
| BRAF | Vemurafenib (4 weeks) | PTC | BRAF | - Prospective evaluation of RAI avidity restoration by diagnostic 124I PET-scan - If specific dosimetry criteria met, treatment with maximum tolerable activity (mean activity 9.4 GBq) | 10 | 60% | At 6 months (n = 4): 2 PR, 2 SD | [76] |
| BRAF and/or MEK | - Dabrafenib +/− trametinib - Vemurafenib - Trametinib - Investigational MEKi (median 14 months, range 1–76.4) | 77% PTC 15% PDTC 8% FTC | 70% BRAF 23% RAS 7% WT | - Retrospective study including patients treated with MAPKi for RAIR-TC - Proof of RAI avidity restoration by 131I-WBS - Median administered activity: 7.5 GBq | 13 | 62% | Median time of follow-up after RAI: 8,3 months (n = 8): 3 PR, 5 SD | [77] |
| BRAF and/or MEK | - Trametinib +/− dabrafenib - Vemurafenib + cobimetinib (4 weeks) | 50% PTC 33% FTC 17% PDTC | 50% BRAF 50% RAS | - Retrospective study including patients treated with MAPKi for RAIR-TC - Proof of RAI avidity restoration by 124I PET-scan - Mean administered activity: 7.9 GBq | 6 | 67% | At 3 months (n = 4): 3 PR, 1 SD | [78] |
| MEK | Selumetinib (4 weeks) | 65% PTC 35% PDTC | 45% BRAF 25% RAS 15% RET/PTC 15% WT | - Prospective evaluation of RAI avidity restoration by 124I PET-scan - If specific dosimetry criteria met, treatment with maximum tolerable activity (NA mean activity) | 20 | 60% | At 6 months (n = 8): 5 PR, 3 SD | [79] |
| BRAF + MEK | Dabrafenib + Trametinib (6 weeks) | PTC | BRAF | - Prospective evaluation of RAI avidity restoration by diagnostic 131I-WBS systematically followed by fixed 131I activity of 5.5GBq | 21 | Dc-WBS: 65% Pt-WBS: 95% | At 6 months (n = 21): 8 PR, 11 SD | [80] |
| BRAF + MEK | Trametinib +/− dabrafenib (3 weeks) | 50% PTC 35% FTC 15% PDTC | 70% WT 30% BRAF | - Prospective evaluation of RAI avidity restoration by diagnostic 123I-WBS - If avidity restored, treatment with mean 131I activity of 11 GBq | 20 | 35% | Between 3–12 months (n = 7): 1 PR, 5 SD | [81] |
| BRAF + HER3 | Vemurafenib + CDX-3379 (5 weeks) | 50% PTC 50% PDTC | BRAF | - Prospective evaluation of RAI avidity restoration by 124I PET-scan - If specific dosimetry criteria met, treatment with maximum tolerable activity (mean activity 9.1 GBq) | 6 | 83% | At 6 months (n = 4): 2 PR | [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schubert, L.; Mariko, M.L.; Clerc, J.; Huillard, O.; Groussin, L. MAPK Pathway Inhibitors in Thyroid Cancer: Preclinical and Clinical Data. Cancers 2023, 15, 710. https://doi.org/10.3390/cancers15030710
Schubert L, Mariko ML, Clerc J, Huillard O, Groussin L. MAPK Pathway Inhibitors in Thyroid Cancer: Preclinical and Clinical Data. Cancers. 2023; 15(3):710. https://doi.org/10.3390/cancers15030710
Chicago/Turabian StyleSchubert, Louis, Mohamed Lamine Mariko, Jérôme Clerc, Olivier Huillard, and Lionel Groussin. 2023. "MAPK Pathway Inhibitors in Thyroid Cancer: Preclinical and Clinical Data" Cancers 15, no. 3: 710. https://doi.org/10.3390/cancers15030710
APA StyleSchubert, L., Mariko, M. L., Clerc, J., Huillard, O., & Groussin, L. (2023). MAPK Pathway Inhibitors in Thyroid Cancer: Preclinical and Clinical Data. Cancers, 15(3), 710. https://doi.org/10.3390/cancers15030710