

Tumour Necrosis Factor-Alpha (TNF-α)-Induced Metastatic Phenotype in Colorectal Cancer Epithelial Cells: Mechanistic Support for the Role of MicroRNA-21

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Wound Healing Assay
2.3. Transwell Migration and Invasion
2.4. Cell Proliferation
2.5. RNA and MiRNA Extraction
2.6. Reverse Transcription and RT-qPCR
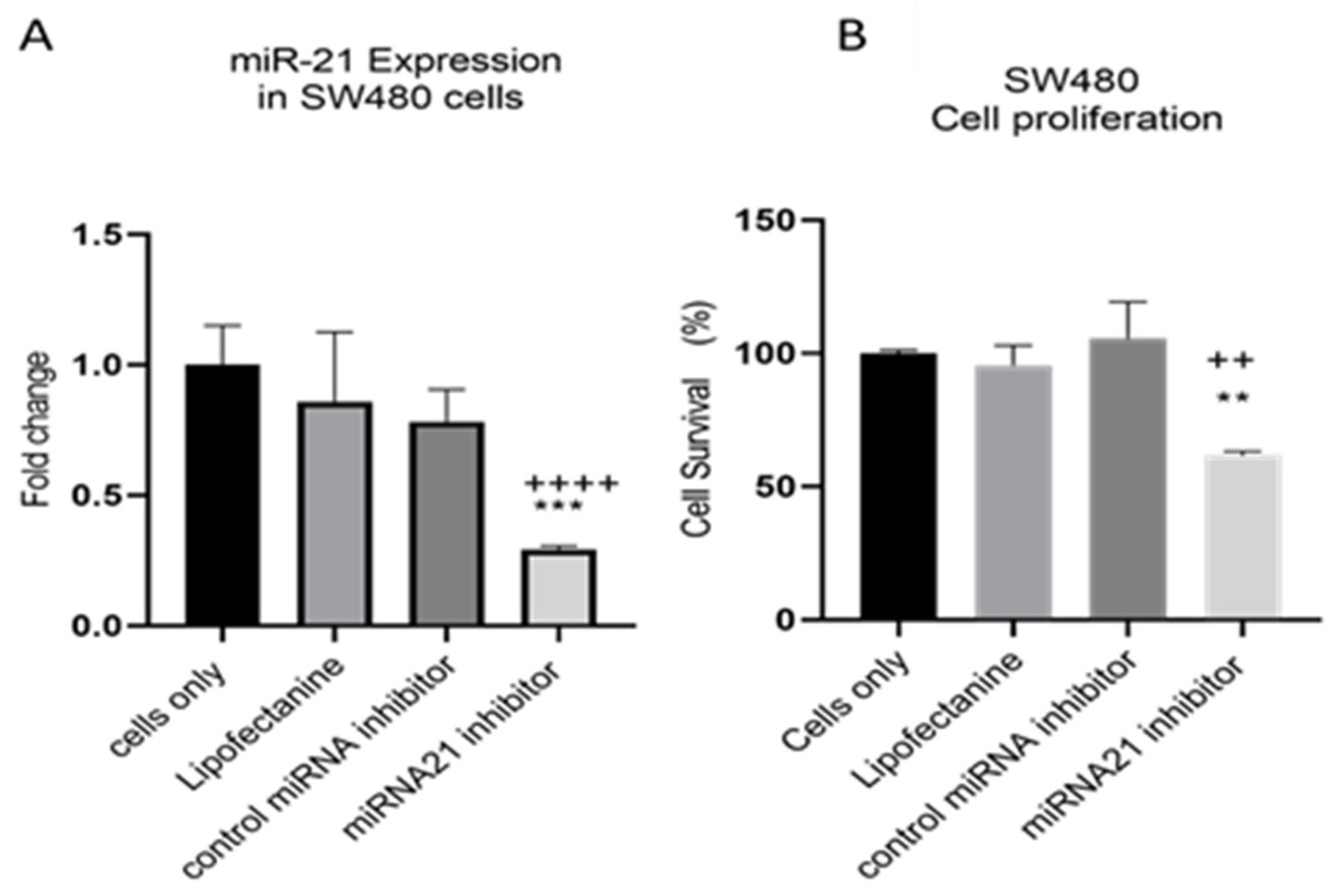
2.7. MiRNA Inhibitor Transfection
2.8. Statistical Analysis
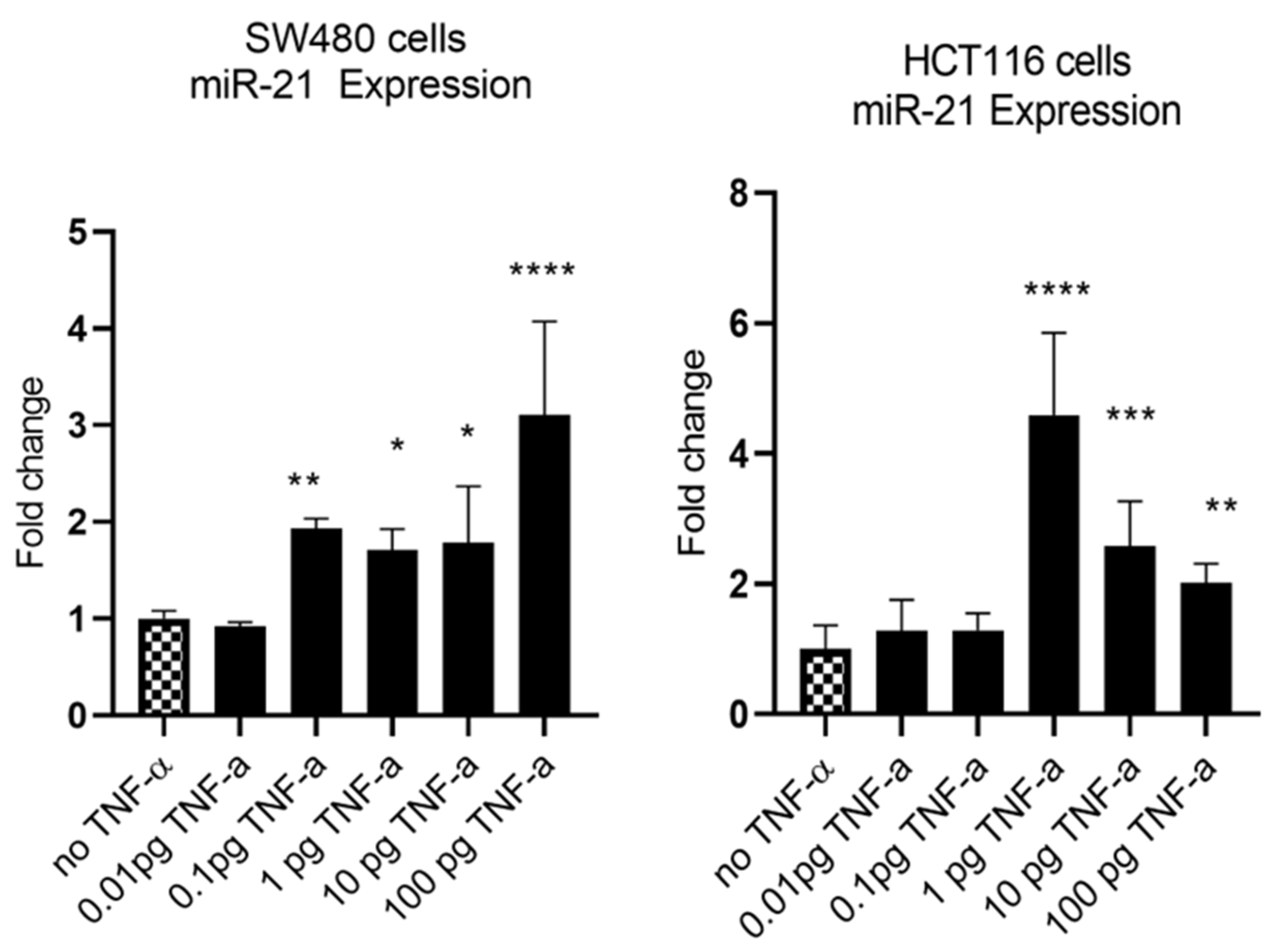
3. Results
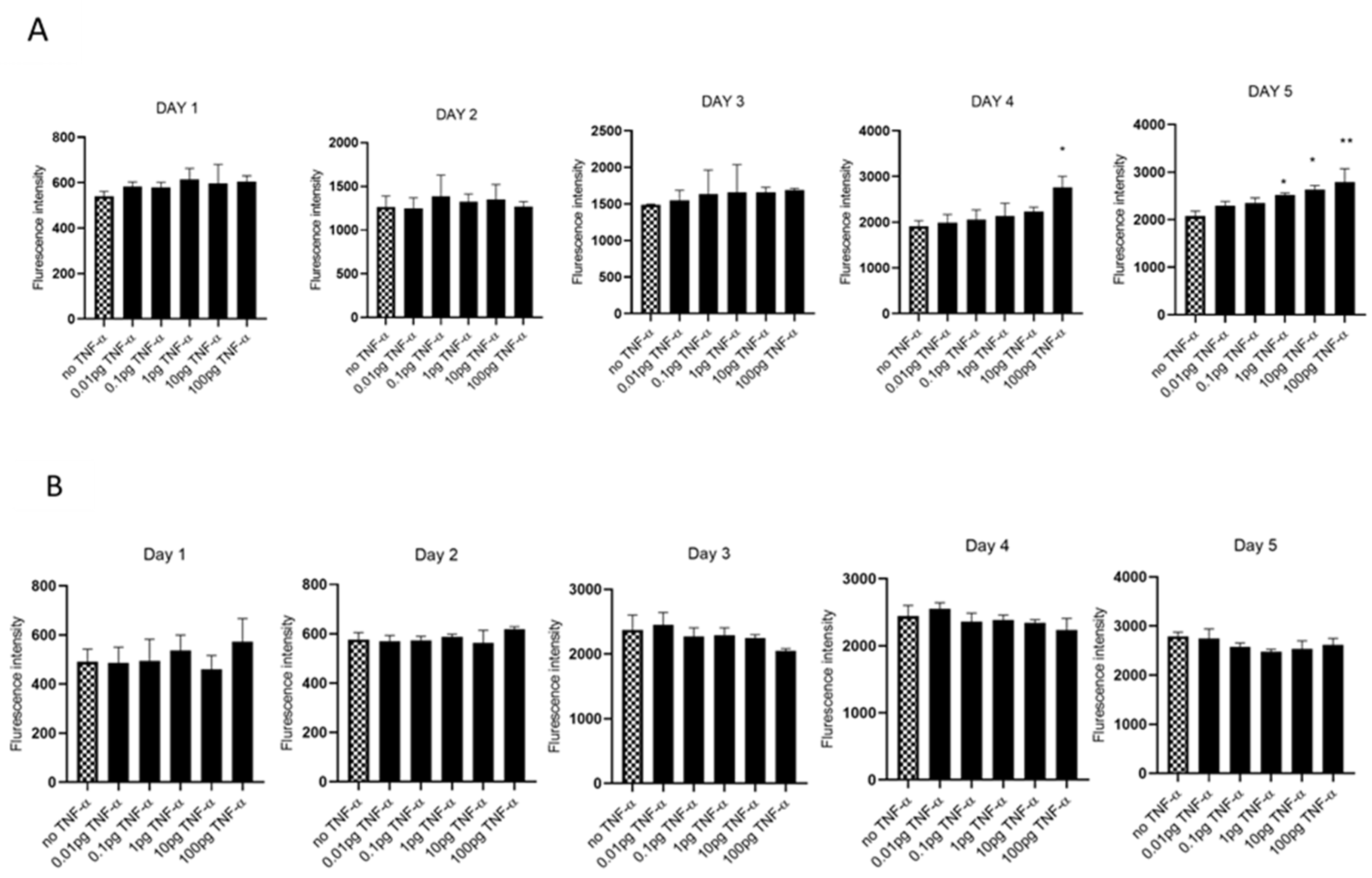
3.1. TNF-α Promotes Cell Proliferation
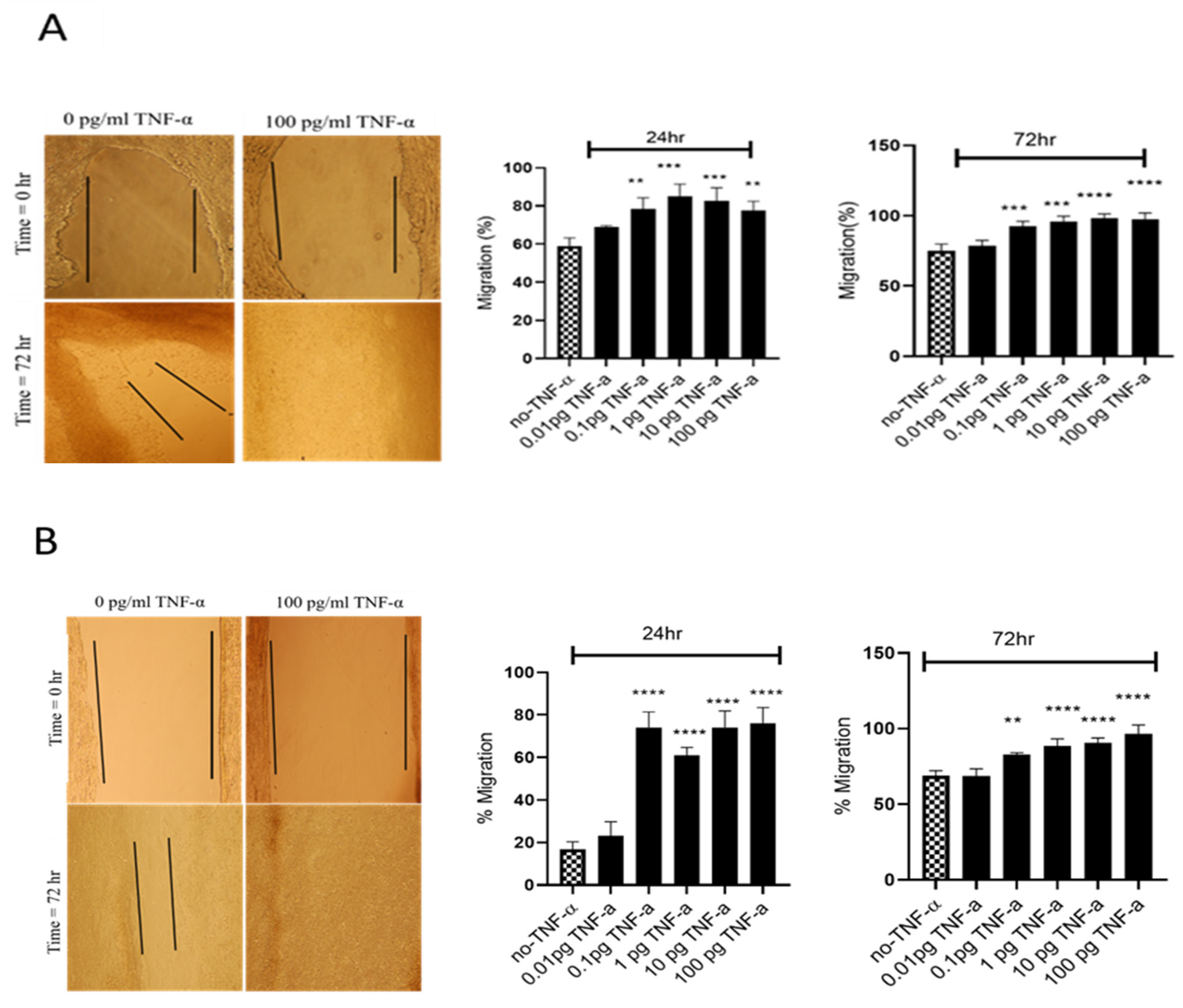
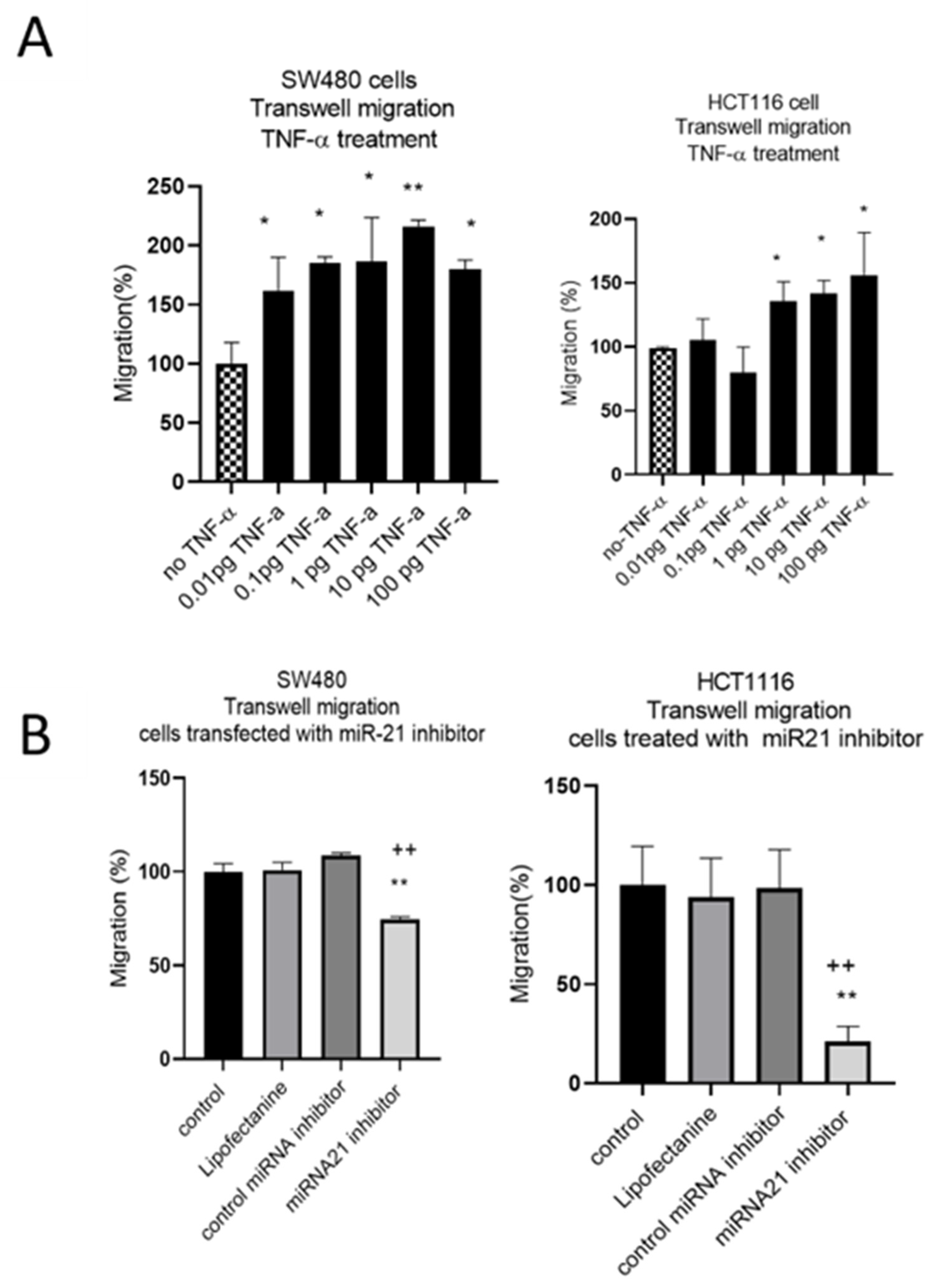
3.2. TNF-a Promotes Cell Migration
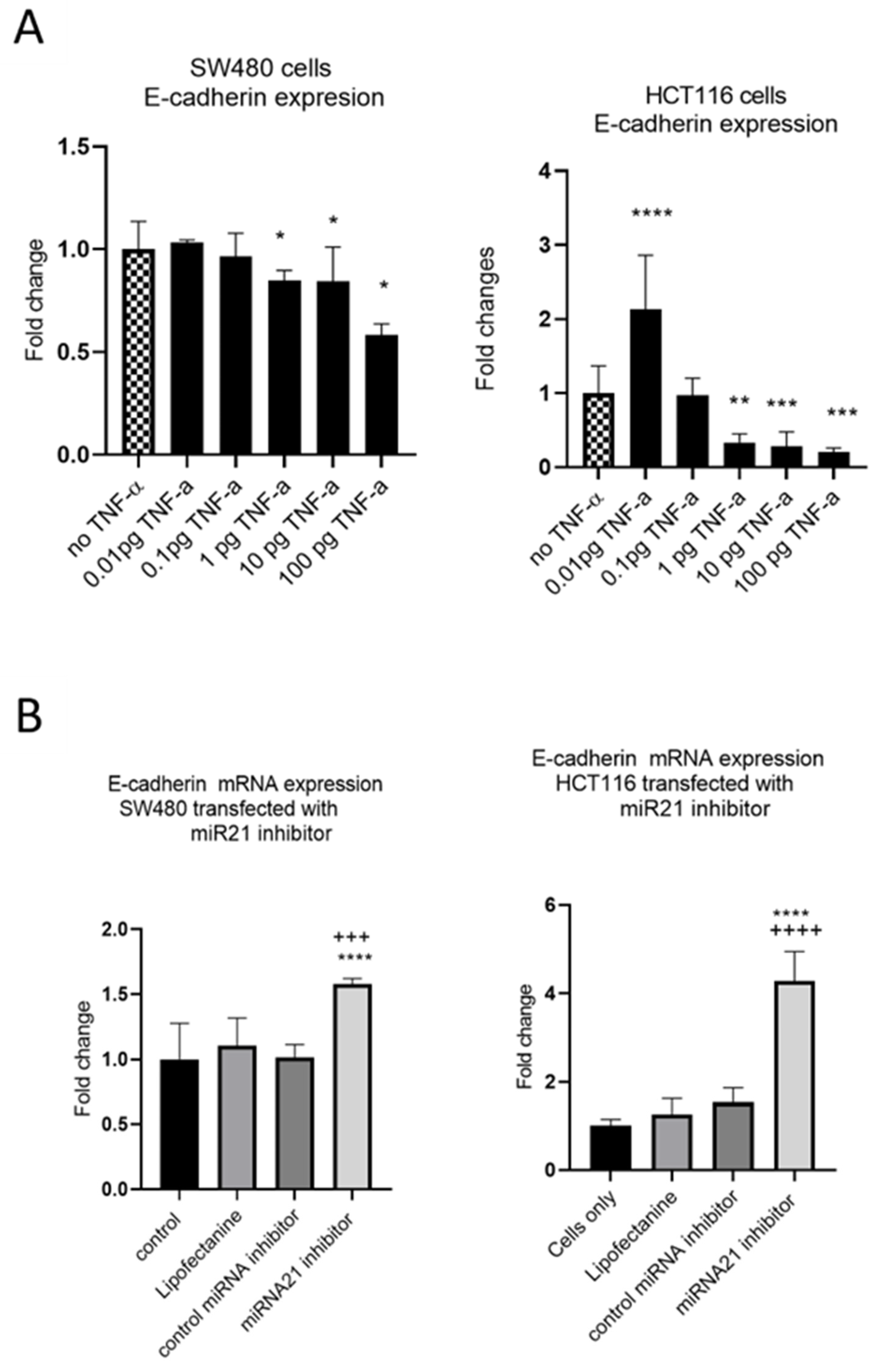
3.3. TNF-α Alters E-Cadherin Expression
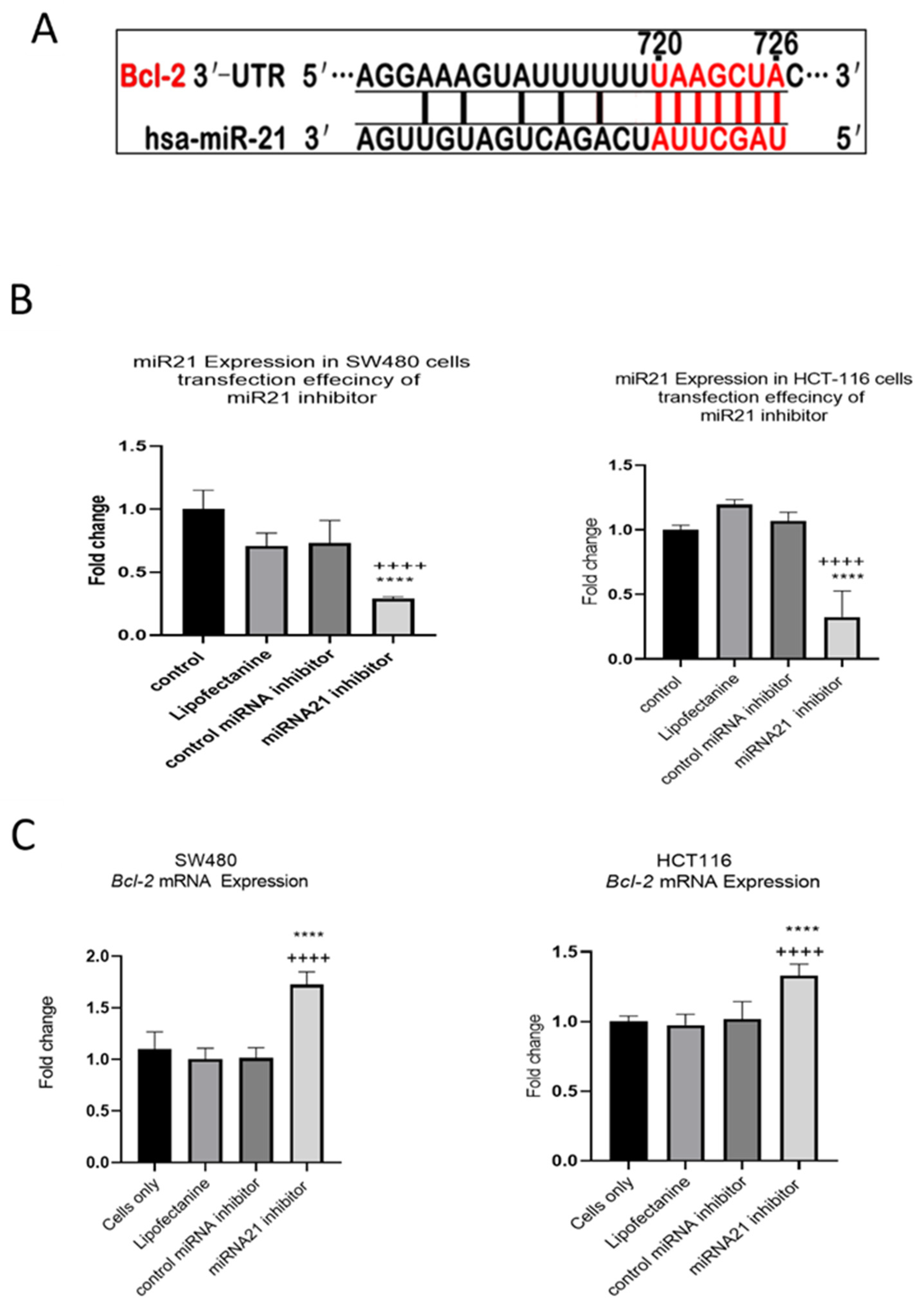
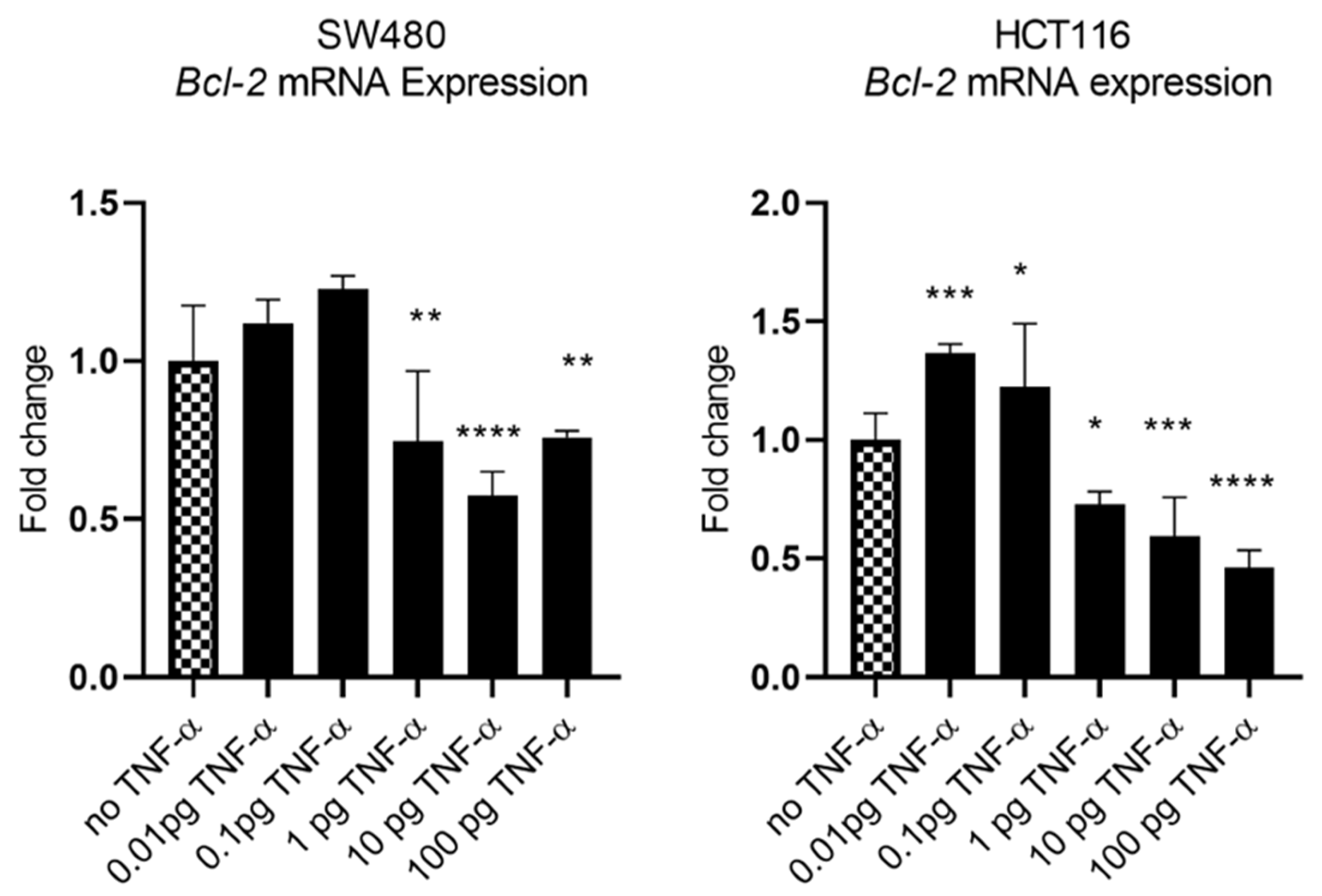
3.4. TNF-a Downregulates BCL-2 Expression
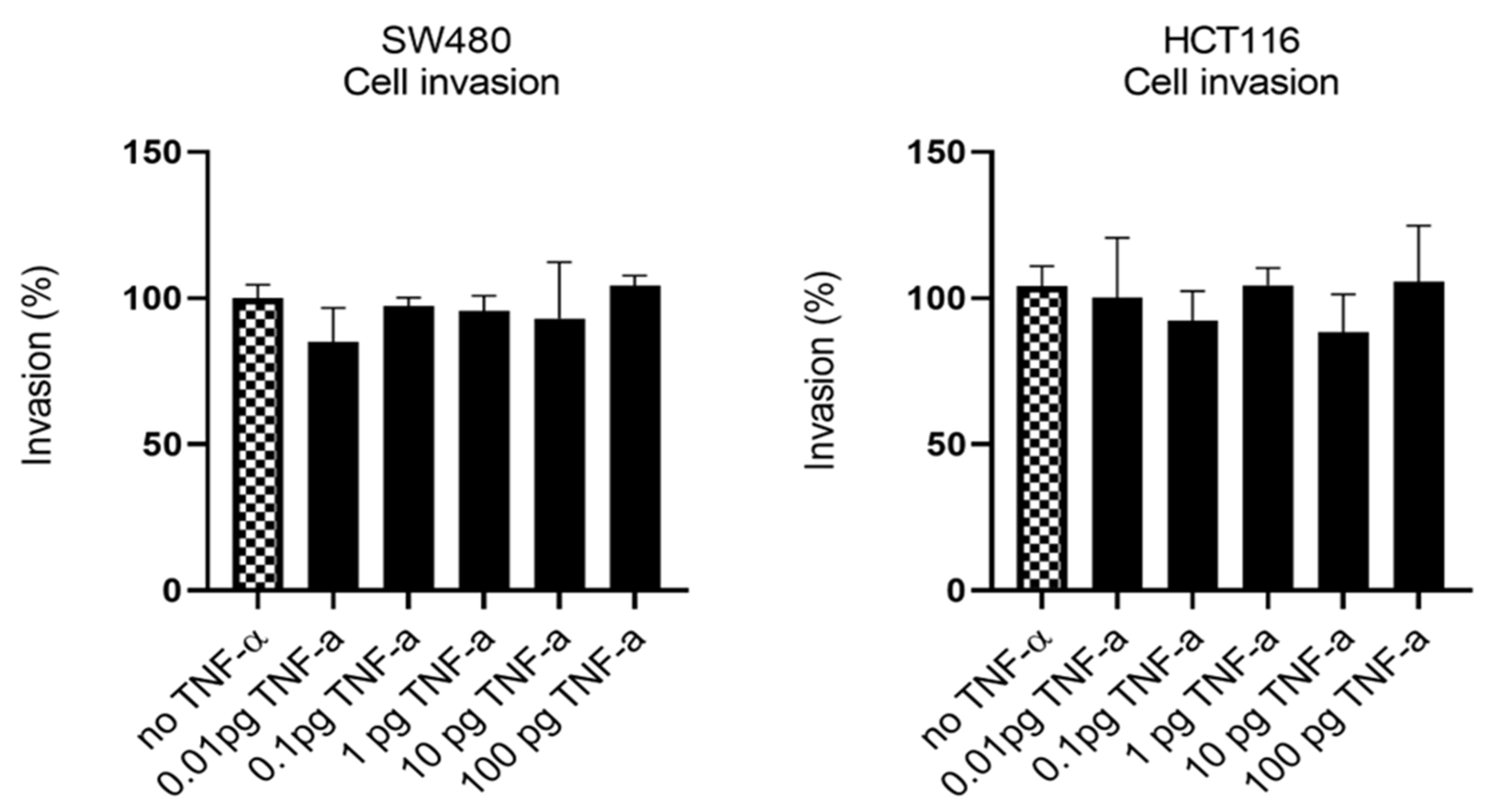
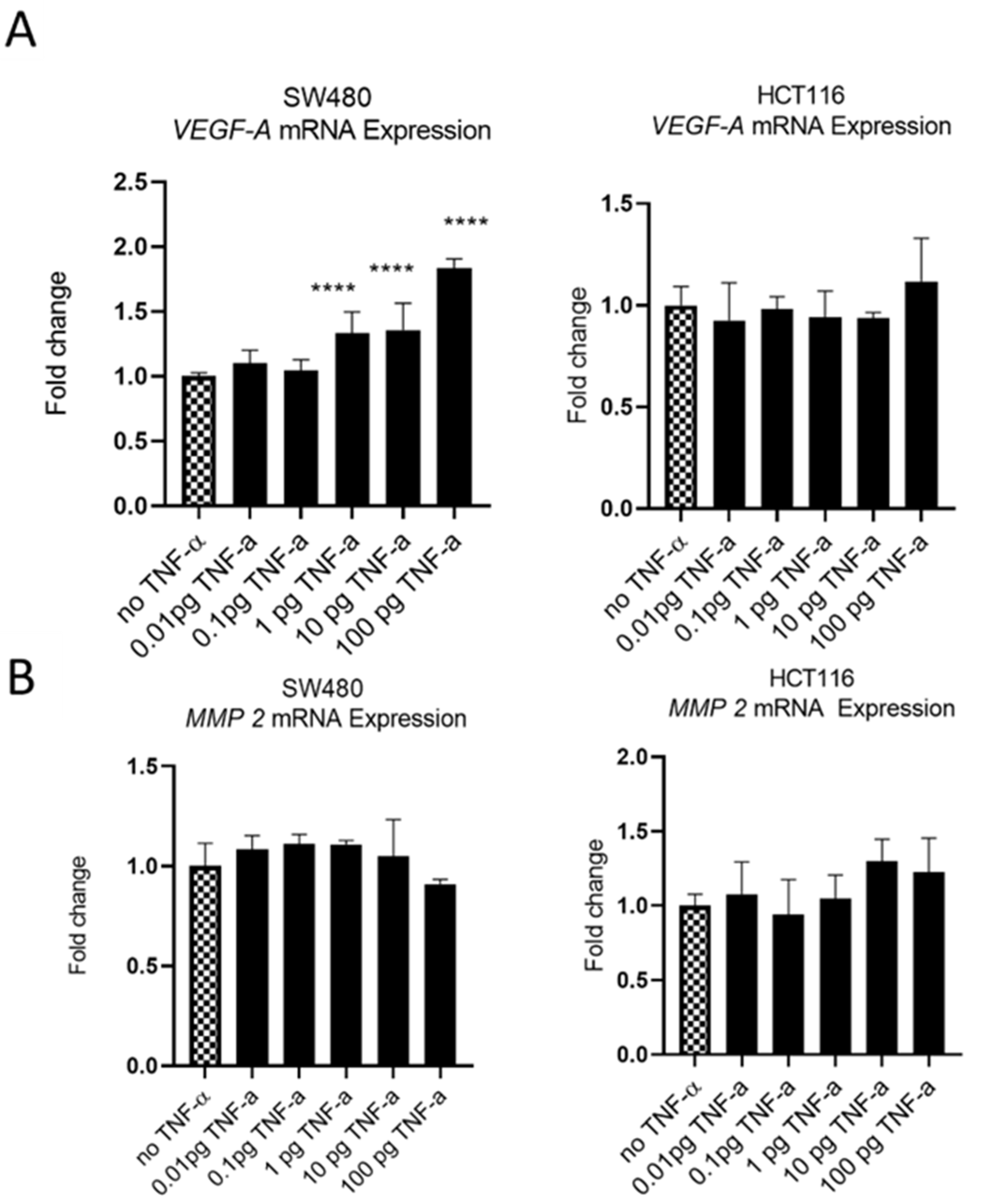
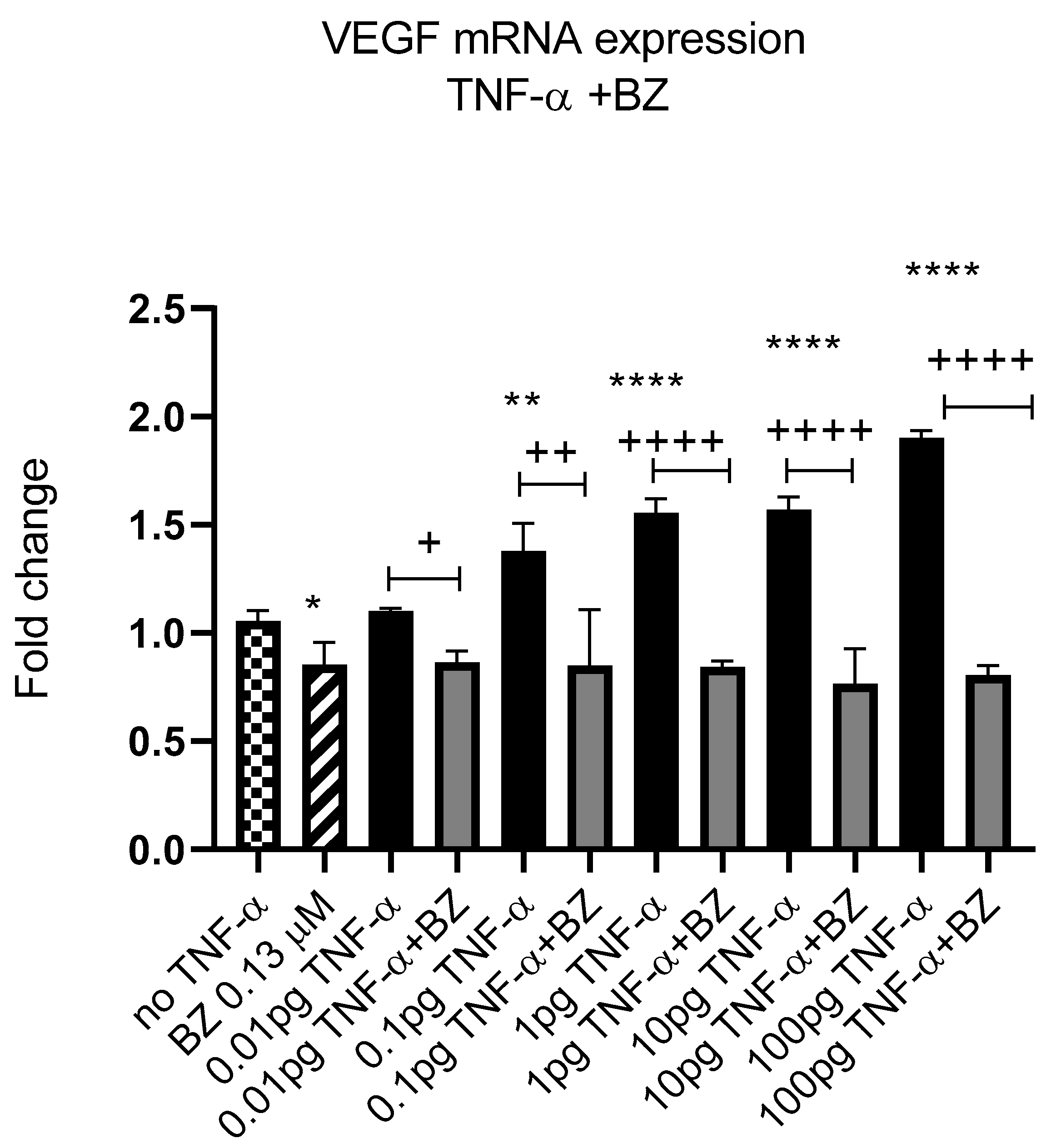
3.5. TNF-α Alters VEGF-A Expression, but Not MMP2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Wolf, A.M.D.; Fontham, E.T.H.; Church, T.R.; Flowers, C.R.; Guerra, C.E.; LaMonte, S.J.; Etzioni, R.; McKenna, M.T.; Oeffinger, K.C.; Shih, Y.T.; et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J. Clin. 2018, 68, 250–281. [Google Scholar] [CrossRef]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef]
- Schwager, S.C.; Taufalele, P.V.; Reinhart-King, C.A. Cell-Cell Mechanical Communication in Cancer. Cell. Mol. Bioeng. 2019, 12, 1–14. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Maihofner, C.; Charalambous, M.P.; Bhambra, U.; Lightfoot, T.; Geisslinger, G.; Gooderham, N.J.; Colorectal Cancer, G. Expression of cyclooxygenase-2 parallels expression of interleukin-1beta, interleukin-6 and NF-kappaB in human colorectal cancer. Carcinogenesis 2003, 24, 665–671. [Google Scholar] [CrossRef]
- Charalambous, M.P.; Lightfoot, T.; Speirs, V.; Horgan, K.; Gooderham, N.J. Expression of COX-2, NF-kappaB-p65, NF-kappaB-p50 and IKKalpha in malignant and adjacent normal human colorectal tissue. Br. J. Cancer 2009, 101, 106–115. [Google Scholar] [CrossRef]
- Charalambous, M.P.; Maihofner, C.; Bhambra, U.; Lightfoot, T.; Gooderham, N.J.; Colorectal Cancer Study, G. Upregulation of cyclooxygenase-2 is accompanied by increased expression of nuclear factor-kappa B and I kappa B kinase-alpha in human colorectal cancer epithelial cells. Br. J. Cancer 2003, 88, 1598–1604. [Google Scholar] [CrossRef]
- Al Obeed, O.A.; Alkhayal, K.A.; Al Sheikh, A.; Zubaidi, A.M.; Vaali-Mohammed, M.A.; Boushey, R.; McKerrow, J.H.; Abdulla, M.H. Increased expression of tumor necrosis factor-alpha is associated with advanced colorectal cancer stages. World J. Gastroenterol. 2014, 20, 18390–18396. [Google Scholar] [CrossRef]
- Stanilov, N.; Miteva, L.; Dobreva, Z.; Stanilova, S. Colorectal cancer severity and survival in correlation with tumour necrosis factor-alpha. Biotechnol. Biotechnol. Equip. 2014, 28, 911–917. [Google Scholar] [CrossRef]
- Li, W.; Xu, J.; Zhao, J.; Zhang, R. Oxaliplatin and Infliximab Combination Synergizes in Inducing Colon Cancer Regression. Med. Sci. Monit. 2017, 23, 780–789. [Google Scholar] [CrossRef]
- Patel, S.A. Can Interleukin-6 Promote Human Colorectal Cancer Progression through Changes in microRNA Expression? Imperial College London: London, UK, 2015. [Google Scholar]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2009, 1796, 75–90. [Google Scholar] [CrossRef]
- Zhu, Y.; Cheng, Y.; Guo, Y.; Chen, J.; Chen, F.; Luo, R.; Li, A. Protein kinase D2 contributes to TNF-alpha-induced epithelial mesenchymal transition and invasion via the PI3K/GSK-3beta/beta-catenin pathway in hepatocellular carcinoma. Oncotarget 2016, 7, 5327–5341. [Google Scholar] [CrossRef]
- Mikesh, L.M.; Kumar, M.; Erdag, G.; Hogan, K.T.; Molhoek, K.R.; Mayo, M.W.; Slingluff, C.L., Jr. Evaluation of molecular markers of mesenchymal phenotype in melanoma. Melanoma Res. 2010, 20, 485–495. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, Z. TNF-alpha promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol. Lett. 2018, 15, 3820–3827. [Google Scholar]
- Bates, R.C.; Mercurio, A.M. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell 2003, 14, 1790–1800. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Gao, J.; Li, L.; Wu, M.; Liu, M.; Xie, X.; Guo, J.; Tang, H.; Xie, X. MiR-26a inhibits proliferation and migration of breast cancer through repression of MCL-1. PLoS ONE 2013, 8, e65138. [Google Scholar] [CrossRef]
- Gao, W.; Xu, J.; Liu, L.; Shen, H.; Zeng, H.; Shu, Y. A systematic-analysis of predicted miR-21 targets identifies a signature for lung cancer. Biomed. Pharmacother. 2012, 66, 21–28. [Google Scholar] [CrossRef]
- Patel, S.A.; Bhambra, U.; Charalambous, M.P.; David, R.M.; Edwards, R.J.; Lightfoot, T.; Boobis, A.R.; Gooderham, N.J. Interleukin-6 mediated upregulation of CYP1B1 and CYP2E1 in colorectal cancer involves DNA methylation, miR27b and STAT3. Br. J. Cancer 2014, 111, 2287–2296. [Google Scholar] [CrossRef]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Gooderham, N.J.; Koufaris, C. Using microRNA profiles to predict and evaluate hepatic carcinogenic potential. Toxicol. Lett. 2014, 228, 127–132. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Patel, S.A.; Gooderham, N.J. IL6 Mediates Immune and Colorectal Cancer Cell Cross-talk via miR-21 and miR-29b. Mol. Cancer Res. 2015, 13, 1502–1508. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, W.; Wang, J.; Zhu, L.; Wang, Z.; Peng, Y. Exosomal noncoding RNAs in colorectal cancer. Cancer Lett. 2020, 493, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Schetter, A.J.; Leung, S.Y.; Sohn, J.J.; Zanetti, K.A.; Bowman, E.D.; Yanaihara, N.; Yuen, S.T.; Chan, T.L.; Kwong, D.L.; Au, G.K.; et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA 2008, 299, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Kjaer-Frifeldt, S.; Hansen, T.F.; Nielsen, B.S.; Joergensen, S.; Lindebjerg, J.; Soerensen, F.B.; dePont Christensen, R.; Jakobsen, A. The prognostic importance of miR-21 in stage II colon cancer: A population-based study. Br. J. Cancer 2012, 107, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Ribas, J.; Ni, X.; Haffner, M.; Wentzel, E.A.; Salmasi, A.H.; Chowdhury, W.H.; Kudrolli, T.A.; Yegnasubramanian, S.; Luo, J.; Rodriguez, R.; et al. miR-21: An androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res. 2009, 69, 7165–7169. [Google Scholar] [CrossRef]
- Markou, A.; Tsaroucha, E.G.; Kaklamanis, L.; Fotinou, M.; Georgoulias, V.; Lianidou, E.S. Prognostic value of mature microRNA-21 and microRNA-205 overexpression in non-small cell lung cancer by quantitative real-time RT-PCR. Clin. Chem. 2008, 54, 1696–1704. [Google Scholar] [CrossRef]
- Arisan, E.D.; Rencuzogullari, O.; Cieza-Borrella, C.; Miralles Arenas, F.; Dwek, M.; Lange, S.; Uysal-Onganer, P. MiR-21 Is Required for the Epithelial-Mesenchymal Transition in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 1557. [Google Scholar] [CrossRef]
- Xu, L.F.; Wu, Z.P.; Chen, Y.; Zhu, Q.S.; Hamidi, S.; Navab, R. MicroRNA-21 (miR-21) regulates cellular proliferation, invasion, migration, and apoptosis by targeting PTEN, RECK and Bcl-2 in lung squamous carcinoma, Gejiu City, China. PLoS ONE 2014, 9, e103698. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Alotaibi, A.G.; Li, J.V.; Gooderham, N.J. Tumour necrosis factor-alpha (TNF-alpha) enhances dietary carcinogen-induced DNA damage in colorectal cancer epithelial cells through activation of JNK signaling pathway. Toxicology 2021, 457, 152806. [Google Scholar] [CrossRef]
- Moller, T.; James, J.P.; Holmstrom, K.; Sorensen, F.B.; Lindebjerg, J.; Nielsen, B.S. Co-Detection of miR-21 and TNF-alpha mRNA in Budding Cancer Cells in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 1907. [Google Scholar] [CrossRef] [PubMed]
- Wyble, C.W.; Desai, T.R.; Clark, E.T.; Hynes, K.L.; Gewertz, B.L. Physiologic concentrations of TNFalpha and IL-1beta released from reperfused human intestine upregulate E-selectin and ICAM-1. J. Surg. Res. 1996, 63, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Riffo-Campos, A.L.; Riquelme, I.; Brebi-Mieville, P. Tools for Sequence-Based miRNA Target Prediction: What to Choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.Y.; Yeh, K.Y.; Liu, B.F.; Chang, T.M.; Chang, C.H.; Liao, Y.F.; Liu, Y.W.; Her, G.M. MicroRNA-21 Plays Multiple Oncometabolic Roles in Colitis-Associated Carcinoma and Colorectal Cancer via the PI3K/AKT, STAT3, and PDCD4/TNF-alpha Signaling Pathways in Zebrafish. Cancers 2021, 13, 5565. [Google Scholar] [CrossRef] [PubMed]
- Buscaglia, L.E.; Li, Y. Apoptosis and the target genes of microRNA-21. Chin. J. Cancer 2011, 30, 371–380. [Google Scholar] [CrossRef]
- Herzog, B.; Pellet-Many, C.; Britton, G.; Hartzoulakis, B.; Zachary, I.C. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol. Biol. Cell 2011, 22, 2766–2776. [Google Scholar] [CrossRef]
- Bodey, B.; Bodey, B., Jr.; Siegel, S.E.; Kaiser, H.E. Matrix metalloproteinase expression in malignant melanomas: Tumor-extracellular matrix interactions in invasion and metastasis. Vivo 2001, 15, 57–64. [Google Scholar]
- Laha, D.; Grant, R.; Mishra, P.; Nilubol, N. The Role of Tumor Necrosis Factor in Manipulating the Immunological Response of Tumor Microenvironment. Front. Immunol. 2021, 12, 656908. [Google Scholar] [CrossRef]
- Pan, Z.; Tian, Y.; Niu, G.; Cao, C. Role of microRNAs in remodeling the tumor microenvironment (Review). Int. J. Oncol. 2020, 56, 407–416. [Google Scholar] [CrossRef]
- Malik, D.E.; David, R.M.; Gooderham, N.J. Interleukin-6 selectively induces drug metabolism to potentiate the genotoxicity of dietary carcinogens in mammary cells. Arch. Toxicol. 2019, 93, 3005–3020. [Google Scholar] [CrossRef]
- Koufaris, C.; Wright, J.; Osborne, M.; Currie, R.A.; Gooderham, N.J. Time and dose-dependent effects of phenobarbital on the rat liver miRNAome. Toxicology 2013, 314, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, M.D.; Koufaris, C.; Gooderham, N.J. The cooked meat-derived mammary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) elicits estrogenic-like microRNA responses in breast cancer cells. Toxicol. Lett. 2014, 229, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Segal, C.V.; Koufaris, C.; Powell, C.; Gooderham, N.J. Effects of treatment with androgen receptor ligands on microRNA expression of prostate cancer cells. Toxicology 2015, 333, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Malik, D.E.; David, R.M.; Gooderham, N.J. Mechanistic evidence that benzo[a]pyrene promotes an inflammatory microenvironment that drives the metastatic potential of human mammary cells. Arch. Toxicol. 2018, 92, 3223–3239. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef]
- Yoshino, T.; Van Cutsem, E.; Li, J.; Shen, L.; Kim, T.W.; Sriuranpong, V.; Xuereb, L.; Aubel, P.; Fougeray, R.; Cattan, V.; et al. Effect of KRAS codon 12 or 13 mutations on survival with trifluridine/tipiracil in pretreated metastatic colorectal cancer: A meta-analysis. ESMO Open 2022, 7, 100511. [Google Scholar] [CrossRef]
- Zhong, Z.; Dong, Z.; Yang, L.; Gong, Z. miR-21 induces cell cycle at S phase and modulates cell proliferation by down-regulating hMSH2 in lung cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 1781–1788. [Google Scholar] [CrossRef]
- Liu, F.; Zheng, S.; Liu, T.; Liu, Q.; Liang, M.; Li, X.; Sheyhidin, I.; Lu, X.; Liu, W. MicroRNA-21 promotes the proliferation and inhibits apoptosis in Eca109 via activating ERK1/2/MAPK pathway. Mol. Cell. Biochem. 2013, 381, 115–125. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Grimshaw, M.J.; Kulbe, H.; Wilson, J.L.; Wilbanks, G.D.; Burke, F.; Balkwill, F.R. Expression and regulation of tumor necrosis factor alpha in normal and malignant ovarian epithelium. Mol. Cancer Ther. 2006, 5, 382–390. [Google Scholar] [CrossRef]
- Michalaki, V.; Syrigos, K.; Charles, P.; Waxman, J. Serum levels of IL-6 and TNF-alpha correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer 2004, 90, 2312–2316. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, Y.; Ran, J.; Wang, Y. MicroRNA-21 promotes cell metastasis in cervical cancer through modulating epithelial-mesenchymal transition. Oncol. Lett. 2020, 19, 3289–3295. [Google Scholar] [CrossRef]
- Dong, W.; Li, H.; Zhang, Y.; Yang, H.; Guo, M.; Li, L.; Liu, T. Matrix metalloproteinase 2 promotes cell growth and invasion in colorectal cancer. Acta Biochim. Biophys. Sin. 2011, 43, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Dallas, N.A.; Fan, F.; Gray, M.J.; Van Buren, G., 2nd; Lim, S.J.; Xia, L.; Ellis, L.M. Functional significance of vascular endothelial growth factor receptors on gastrointestinal cancer cells. Cancer Metastasis Rev. 2007, 26, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidis, S.; Andreakos, E.; Monaco, C.; Foxwell, B.; Feldmann, M.; Paleolog, E. VEGF expression in human macrophages is NF-kappaB-dependent: Studies using adenoviruses expressing the endogenous NF-kappaB inhibitor IkappaBalpha and a kinase-defective form of the IkappaB kinase 2. J. Cell Sci. 2003, 116 Pt 4, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 2010, 23, 725–732. [Google Scholar]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alotaibi, A.G.; Li, J.V.; Gooderham, N.J. Tumour Necrosis Factor-Alpha (TNF-α)-Induced Metastatic Phenotype in Colorectal Cancer Epithelial Cells: Mechanistic Support for the Role of MicroRNA-21. Cancers 2023, 15, 627. https://doi.org/10.3390/cancers15030627
Alotaibi AG, Li JV, Gooderham NJ. Tumour Necrosis Factor-Alpha (TNF-α)-Induced Metastatic Phenotype in Colorectal Cancer Epithelial Cells: Mechanistic Support for the Role of MicroRNA-21. Cancers. 2023; 15(3):627. https://doi.org/10.3390/cancers15030627
Chicago/Turabian StyleAlotaibi, Aminah G., Jia V. Li, and Nigel J. Gooderham. 2023. "Tumour Necrosis Factor-Alpha (TNF-α)-Induced Metastatic Phenotype in Colorectal Cancer Epithelial Cells: Mechanistic Support for the Role of MicroRNA-21" Cancers 15, no. 3: 627. https://doi.org/10.3390/cancers15030627
APA StyleAlotaibi, A. G., Li, J. V., & Gooderham, N. J. (2023). Tumour Necrosis Factor-Alpha (TNF-α)-Induced Metastatic Phenotype in Colorectal Cancer Epithelial Cells: Mechanistic Support for the Role of MicroRNA-21. Cancers, 15(3), 627. https://doi.org/10.3390/cancers15030627