
Synovial Sarcoma Preclinical Modeling: Integrating Transgenic Mouse Models and Patient-Derived Models for Translational Research



Abstract
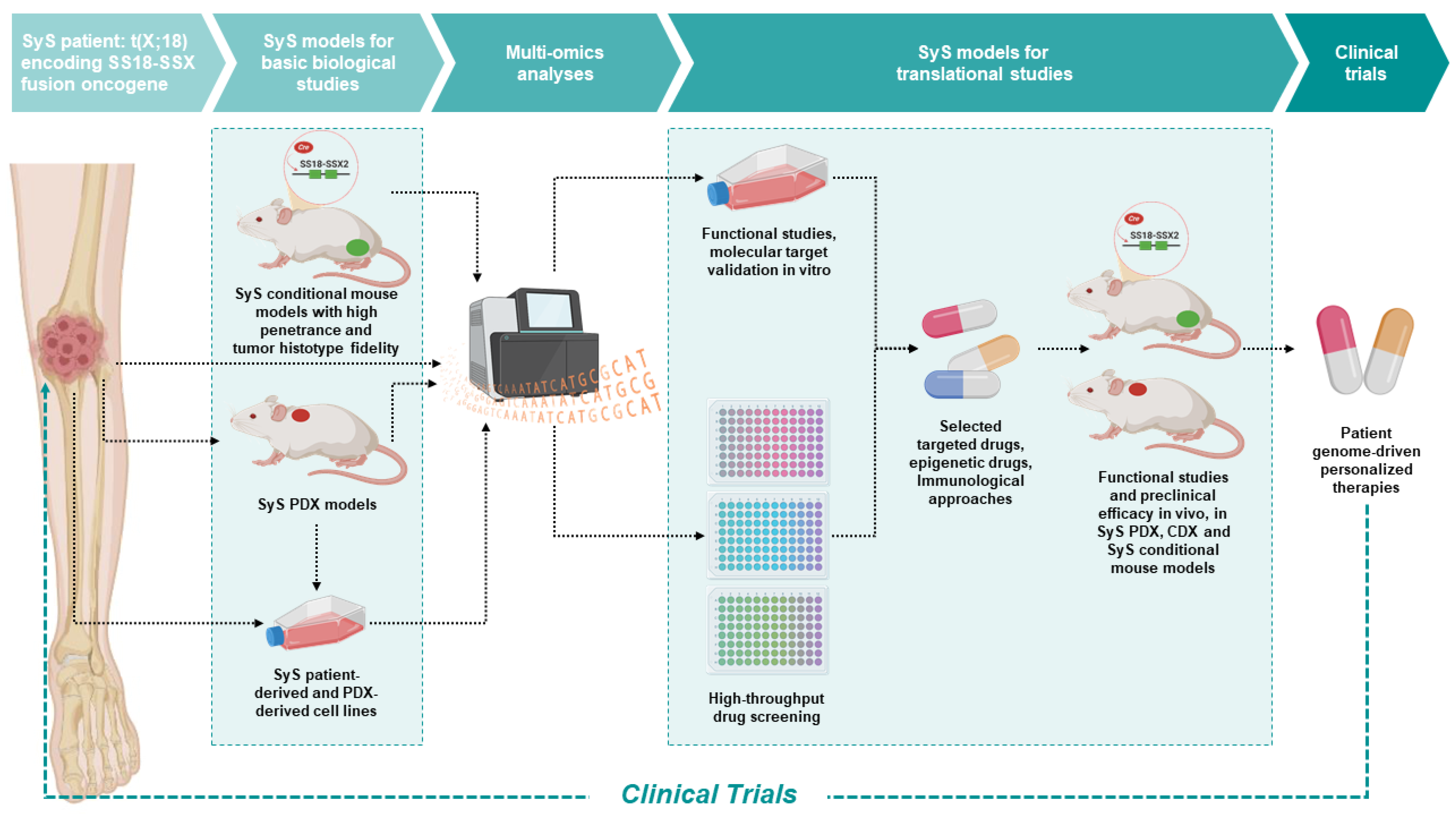
Simple Summary
Abstract
1. Introduction
2. Modeling SyS in the Mouse
3. SyS Conditional Transgenic Mouse Models
3.1. Role of Proto-Oncogenes Explored in Conditional SS18-SSX Transgenic Mouse Models
3.1.1. Bcl-2 Family
3.1.2. WNT/β-Catenin Signaling
3.1.3. Fibroblast Growth Factor Receptor (FGFR) Family and ETV4/ETV5 Transcription Factor
3.2. Role of Tumor Suppressor Genes in Conditional SS18-SSX Transgenic Mouse Models
3.2.1. PTEN and the Phosphatidyl Inositol (PI)3′-Lipid Pathway
3.2.2. SMARCB1 and BAF-Family Complex Dysregulation
3.3. Future Developments and Applications of GEMM in SyS Research
4. SyS Patient-Derived Xenograft (PDX) Models
Innovative Drugs Studied in SyS PDX Models
5. SyS Patient-Derived Cell Lines and Cell-Derived Xenografts (CDX)
6. Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gazendam, A.M.; Popovic, S.; Munir, S.; Parasu, N.; Wilson, D.; Ghert, M. Synovial Sarcoma: A Clinical Review. Curr. Oncol. 2021, 28, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, T.; Nakata, E.; Fujiwara, T.; Hosono, A.; Takihira, S.; Kondo, H.; Ozaki, T. Soft-tissue sarcoma in adolescents and young adults. Int. J. Clin. Oncol. 2022, 28, 1–11. [Google Scholar] [CrossRef]
- Palmerini, E.; Staals, E.L.; Alberghini, M.; Zanella, L.; Ferrari, C.; Benassi, M.S.; Picci, P.; Mercuri, M.; Bacci, G.; Ferrari, S. Synovial sarcoma: Retrospective analysis of 250 patients treated at a single institution. Cancer 2009, 115, 2988–2998. [Google Scholar] [CrossRef]
- Smolle, M.A.; Parry, M.; Jeys, L.; Abudu, S.; Grimer, R. Synovial sarcoma: Do children do better? Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2019, 45, 254–260. [Google Scholar] [CrossRef]
- Aytekin, M.N.; Öztürk, R.; Amer, K.; Yapar, A. Epidemiology; incidence, and survival of synovial sarcoma subtypes: SEER database analysis. J. Orthop. Surg. 2020, 28, 2309499020936009. [Google Scholar] [CrossRef] [PubMed]
- Barrott, J.J.; Illum, B.E.; Jin, H.; Hedberg, M.L.; Wang, Y.; Grossmann, A.; Haldar, M.; Capecchi, M.R.; Jones, K.B. Paracrine osteoprotegerin and β-catenin stabilization support synovial sarcomagenesis in periosteal cells. J. Clin. Investig. 2018, 128, 207–218. [Google Scholar] [CrossRef]
- Fiore, M.; Sambri, A.; Spinnato, P.; Zucchini, R.; Giannini, C.; Caldari, E.; Pirini, M.G.; de Paolis, M. The Biology of Synovial Sarcoma: State-of-the-Art and Future Perspectives. Curr. Treat. Options Oncol. 2021, 22, 109. [Google Scholar] [CrossRef]
- Stacchiotti, S.; van Tine, B.A. Synovial Sarcoma: Current Concepts and Future Perspectives. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 180–187. [Google Scholar] [CrossRef]
- Rhomberg, W. The radiation response of sarcomas by histologic subtypes: A review with special emphasis given to results achieved with razoxane. Sarcoma 2006, 2006, 87367. [Google Scholar] [CrossRef]
- Desar, I.M.E.; Fleuren, E.D.G.; van der Graaf, W.T.A. Systemic Treatment for Adults with Synovial Sarcoma. Curr. Treat. Options Oncol. 2018, 19, 13. [Google Scholar] [CrossRef]
- Tetta, C.; Montrone, G.; Longhi, A.; Rocca, M.; Londero, F.; Parise, G.; Parise, O.; Maessen, J.G.; Miceli, M.; Gelsomino, S. Chemosensitivity of Lung Metastatic High-Grade Synovial Sarcoma. J. Clin. Med. 2021, 10, 5956. [Google Scholar] [CrossRef] [PubMed]
- Vlenterie, M.; Litière, S.; Rizzo, E.; Marréaud, S.; Judson, I.; Gelderblom, H.; Le Cesne, A.; Wardelmann, E.; Messiou, C.; Gronchi, A.; et al. Outcome of chemotherapy in advanced synovial sarcoma patients: Review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; setting a new landmark for studies in this entity. Eur. J. Cancer 2016, 58, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; de Salvo, G.L.; Brennan, B.; van Noesel, M.M.; de Paoli, A.; Casanova, M.; Francotte, N.; Kelsey, A.; Alaggio, R.; Oberlin, O.; et al. Synovial sarcoma in children and adolescents: The European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Hibshoosh, H.; Lattes, R. Immunohistochemical and molecular genetic approaches to soft tissue tumor diagnosis: A primer. Semin. Oncol. 1997, 24, 515–525. [Google Scholar] [PubMed]
- Pelmus, M.; Guillou, L.; Hostein, I.; Sierankowski, G.; Lussan, C.; Coindre, J.-M. Monophasic fibrous and poorly differentiated synovial sarcoma: Immunohistochemical reassessment of 60 t(X;18)(SYT-SSX)-positive cases. Am. J. Surg. Pathol. 2002, 26, 1434–1440. [Google Scholar] [CrossRef]
- Limon, J.; Cin, P.D.; Sandberg, A.A. Translocations involving the X chromosome in solid tumors: Presentation of two sarcomas with t(X;18)(q13;p11). Cancer Genet. Cytogenet. 1986, 23, 87–91. [Google Scholar] [CrossRef]
- Clark, J.; Rocques, P.J.; Crew, A.J.; Gill, S.; Shipley, J.; Chan, A.M.; Gusterson, B.A.; Cooper, C.S. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat. Genet. 1994, 7, 502–508. [Google Scholar] [CrossRef]
- Crew, A.J.; Clark, J.; Fisher, C.; Gill, S.; Grimer, R.; Chand, A.; Shipley, J.; Gusterson, B.A.; Cooper, C.S. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. 1995, 14, 2333–2340. [Google Scholar] [CrossRef]
- de Leeuw, B.; Balemans, M.; Weghuis, D.O.; van Geurts Kessel, A. Identification of two alternative fusion genes, SYT-SSX1 and SYT-SSX2, in t(X;18)(p11.2;q11.2)-positive synovial sarcomas. Hum. Mol. Genet. 1995, 4, 1097–1099. [Google Scholar] [CrossRef]
- Santos, N.R.; de Bruijn, D.R.; van Kessel, A.G. Molecular mechanisms underlying human synovial sarcoma development. Genes Chromosom. Cancer 2001, 30, 1–14. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Mertens, F.; Isaksson, M.; Limon, J.; Gustafson, P.; Skytting, B.; Akerman, M.; Sciot, R.; Cin, P.D.; Samson, I.; et al. Clinical impact of molecular and cytogenetic findings in synovial sarcoma. Genes Chromosom. Cancer 2001, 31, 362–372. [Google Scholar] [CrossRef]
- Kadoch, C.; Williams, R.T.; Calarco, J.P.; Miller, E.L.; Weber, C.M.; Braun, S.M.G.; Pulice, J.L.; Chory, E.J.; Crabtree, G.R. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 2017, 49, 213–222. [Google Scholar] [CrossRef]
- Li, J.; Mulvihill, T.S.; Li, L.; Barrott, J.J.; Nelson, M.L.; Wagner, L.; Lock, I.C.; Pozner, A.; Lambert, S.L.; Ozenberger, B.B.; et al. A Role for SMARCB1 in Synovial Sarcomagenesis Reveals That SS18-SSX Induces Canonical BAF Destruction. Cancer Discov. 2021, 11, 2620–2637. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Poulin, N.M.; Ladanyi, M. Synovial sarcoma: Recent discoveries as a roadmap to new avenues for therapy. Cancer Discov. 2015, 5, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Banito, A.; Li, X.; Laporte, A.N.; Roe, J.-S.; Sanchez-Vega, F.; Huang, C.-H.; Dancsok, A.R.; Hatzi, K.; Chen, C.-C.; Tschaharganeh, D.F.; et al. The SS18-SSX Oncoprotein Hijacks KDM2B-PRC1.1 to Drive Synovial Sarcoma. Cancer cell 2018, 33, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; Decarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef]
- Imle, R.; Kommoss, F.K.F.; Banito, A. Preclinical In Vivo Modeling of Pediatric Sarcoma-Promises and Limitations. J. Clin. Med. 2021, 10, 1578. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Neftel, C.; Shore, M.E.; Weisman, H.R.; Mathewson, N.D.; McBride, M.J.; Haas, B.; Izar, B.; Volorio, A.; Boulay, G.; et al. Opposing immune and genetic mechanisms shape oncogenic programs in synovial sarcoma. Nat. Med. 2021, 27, 289–300. [Google Scholar] [CrossRef]
- Nanni, P.; Landuzzi, L.; Manara, M.C.; Righi, A.; Nicoletti, G.; Cristalli, C.; Pasello, M.; Parra, A.; Carrabotta, M.; Ferracin, M.; et al. Bone sarcoma patient-derived xenografts are faithful and stable preclinical models for molecular and therapeutic investigations. Sci. Rep. 2019, 9, 12174. [Google Scholar] [CrossRef]
- Landuzzi, L.; Palladini, A.; Ceccarelli, C.; Asioli, S.; Nicoletti, G.; Giusti, V.; Ruzzi, F.; Ianzano, M.L.; Scalambra, L.; Laranga, R.; et al. Early stability and late random tumor progression of a HER2-positive primary breast cancer patient-derived xenograft. Sci. Rep. 2021, 11, 1563. [Google Scholar] [CrossRef]
- Nanni, P.; Nicoletti, G.; de Giovanni, C.; Croci, S.; Astolfi, A.; Landuzzi, L.; Di Carlo, E.; Iezzi, M.; Musiani, P.; Lollini, P.-L. Development of rhabdomyosarcoma in HER-2/neu transgenic p53 mutant mice. Cancer Res. 2003, 63, 2728–2732. [Google Scholar] [PubMed]
- Keller, C.; Hansen, M.S.; Coffin, C.M.; Capecchi, M.R. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: Implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 2004, 18, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Dodd, R.D.; Mito, J.K.; Kirsch, D.G. Animal models of soft-tissue sarcoma. Dis. Model. Mech. 2010, 3, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Straessler, K.M.; Jones, K.B.; Hu, H.; Jin, H.; van de Rijn, M.; Capecchi, M.R. Modeling clear cell sarcomagenesis in the mouse: Cell of origin differentiation state impacts tumor characteristics. Cancer Cell 2013, 23, 215–227. [Google Scholar] [CrossRef]
- Goodwin, M.L.; Jin, H.; Straessler, K.; Smith-Fry, K.; Zhu, J.-F.; Monument, M.J.; Grossmann, A.; Randall, R.L.; Capecchi, M.R.; Jones, K.B. Modeling alveolar soft part sarcomagenesis in the mouse: A role for lactate in the tumor microenvironment. Cancer Cell 2014, 26, 851–862. [Google Scholar] [CrossRef] [PubMed]
- de Giovanni, C.; Landuzzi, L.; Palladini, A.; Nicoletti, G.; Nanni, P.; Lollini, P.-L. HER Tyrosine Kinase Family and Rhabdomyosarcoma: Role in Onset and Targeted Therapy. Cells 2021, 10, 1808. [Google Scholar] [CrossRef]
- Haldar, M.; Hancock, J.D.; Coffin, C.M.; Lessnick, S.L.; Capecchi, M.R. A conditional mouse model of synovial sarcoma: Insights into a myogenic origin. Cancer Cell 2007, 11, 375–388. [Google Scholar] [CrossRef]
- Haldar, M.; Hedberg, M.L.; Hockin, M.F.; Capecchi, M.R. A CreER-based random induction strategy for modeling translocation-associated sarcomas in mice. Cancer Res. 2009, 69, 3657–3664. [Google Scholar] [CrossRef]
- Jones, K.B.; Barrott, J.J.; Xie, M.; Haldar, M.; Jin, H.; Zhu, J.-F.; Monument, M.J.; Mosbruger, T.L.; Langer, E.M.; Randall, R.L.; et al. The impact of chromosomal translocation locus and fusion oncogene coding sequence in synovial sarcomagenesis. Oncogene 2016, 35, 5021–5032. [Google Scholar] [CrossRef]
- Fairchild, C.K.; Floros, K.V.; Jacob, S.; Coon, C.M.; Puchalapalli, M.; Hu, B.; Harada, H.; Dozmorov, M.G.; Koblinski, J.E.; Smith, S.C.; et al. Unmasking BCL-2 Addiction in Synovial Sarcoma by Overcoming Low NOXA Cancers 2021, 13, 2310. Cancers 2021, 13, 2310. [Google Scholar] [CrossRef]
- Barrott, J.J.; Zhu, J.-F.; Smith-Fry, K.; Susko, A.M.; Nollner, D.; Burrell, L.D.; Pozner, A.; Capecchi, M.R.; Yap, J.T.; Cannon-Albright, L.A.; et al. The Influential Role of BCL2 Family Members in Synovial Sarcomagenesis. Mol. Cancer Res. MCR 2017, 15, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.B.; Su, L.; Jin, H.; Lenz, C.; Randall, R.L.; Underhill, T.M.; Nielsen, T.O.; Sharma, S.; Capecchi, M.R. SS18-SSX2 and the mitochondrial apoptosis pathway in mouse and human synovial sarcomas. Oncogene 2013, 32, 2365–2371. [Google Scholar] [CrossRef] [PubMed]
- Barham, W.; Frump, A.L.; Sherrill, T.P.; Garcia, C.B.; Saito-Diaz, K.; VanSaun, M.N.; Fingleton, B.; Gleaves, L.; Orton, D.; Capecchi, M.R.; et al. Targeting the Wnt pathway in synovial sarcoma models. Cancer Discov. 2013, 3, 1286–1301. [Google Scholar] [CrossRef]
- Barrott, J.J.; Illum, B.E.; Jin, H.; Zhu, J.-F.; Mosbruger, T.; Monument, M.J.; Smith-Fry, K.; Cable, M.G.; Wang, Y.; Grossmann, A.H.; et al. β-catenin stabilization enhances SS18-SSX2-driven synovial sarcomagenesis and blocks the mesenchymal to epithelial transition. Oncotarget 2015, 6, 22758–22766. [Google Scholar] [CrossRef]
- Garcia, C.B.; Shaffer, C.M.; Alfaro, M.P.; Smith, A.L.; Sun, J.; Zhao, Z.; Young, P.P.; VanSaun, M.N.; Eid, J.E. Reprogramming of mesenchymal stem cells by the synovial sarcoma-associated oncogene SYT-SSX2. Oncogene 2012, 31, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Ishibe, T.; Nakayama, T.; Okamoto, T.; Aoyama, T.; Nishijo, K.; Shibata, K.R.; Shima, Y.; Nagayama, S.; Katagiri, T.; Nakamura, Y.; et al. Disruption of fibroblast growth factor signal pathway inhibits the growth of synovial sarcomas: Potential application of signal inhibitors to molecular target therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 2702–2712. [Google Scholar] [CrossRef] [PubMed]
- DeSalvo, J.; Ban, Y.; Li, L.; Sun, X.; Jiang, Z.; Kerr, D.A.; Khanlari, M.; Boulina, M.; Capecchi, M.R.; Partanen, J.M.; et al. ETV4 and ETV5 drive synovial sarcoma through cell cycle and DUX4 embryonic pathway control. J. Clin. Investig. 2021, 131, e141908. [Google Scholar] [CrossRef]
- Barrott, J.J.; Kafchinski, L.A.; Jin, H.; Potter, J.W.; Kannan, S.D.; Kennedy, R.; Mosbruger, T.; Wang, W.-L.; Tsai, J.-W.; Araujo, D.M.; et al. Modeling synovial sarcoma metastasis in the mouse: PI3′-lipid signaling and inflammation. J. Exp. Med. 2016, 213, 2989–3005. [Google Scholar] [CrossRef]
- Su, L.; Cheng, H.; Sampaio, A.V.; Nielsen, T.O.; Underhill, T.M. EGR1 reactivation by histone deacetylase inhibitors promotes synovial sarcoma cell death through the PTEN tumor suppressor. Oncogene 2010, 29, 4352–4361. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.-W.; Wang, H.-W.; Chen, W.-M.; Chao, T.-C.; Hsieh, Y.-Y.; Hsih, C.-H.; Tzeng, C.-H.; Chen, P.C.-H.; Yen, C.-C. Prevalence and prognostic influence of genomic changes of EGFR pathway markers in synovial sarcoma. J. Surg. Oncol. 2011, 103, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Laporte, A.N.; Barrott, J.J.; Yao, R.J.; Poulin, N.M.; Brodin, B.A.; Jones, K.B.; Underhill, T.M.; Nielsen, T.O. HDAC and Proteasome Inhibitors Synergize to Activate Pro-Apoptotic Factors in Synovial Sarcoma. PLoS ONE 2017, 12, e0169407. [Google Scholar] [CrossRef] [PubMed]
- Laporte, A.N.; Poulin, N.M.; Barrott, J.J.; Wang, X.Q.; Lorzadeh, A.; Werff, R.V.; Jones, K.B.; Underhill, T.M.; Nielsen, T.O. Death by HDAC Inhibition in Synovial Sarcoma Cells. Mol. Cancer Ther. 2017, 16, 2656–2667. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 2018, 7, e41305. [Google Scholar] [CrossRef]
- Jackson, K.L.; Agafonov, R.V.; Carlson, M.W.; Chaturvedi, P.; Cocozziello, D.; Cole, K.; Deibler, R.; Eron, S.J.; Good, A.; Hart, A.A.; et al. Abstract ND09: The discovery and characterization of CFT8634: A potent and selective degrader of BRD9 for the treatment of SMARCB1-perturbed cancers. Cancer Res. 2022, 82, ND09. [Google Scholar] [CrossRef]
- Huang, J.; Chen, M.; Whitley, M.J.; Kuo, H.-C.; Xu, E.S.; Walens, A.; Mowery, Y.M.; van Mater, D.; Eward, W.C.; Cardona, D.M.; et al. Generation and comparison of CRISPR-Cas9 and Cre-mediated genetically engineered mouse models of sarcoma. Nat. Commun. 2017, 8, 15999. [Google Scholar] [CrossRef]
- Vassal, G.; Houghton, P.J.; Pfister, S.M.; Smith, M.A.; Caron, H.N.; Li, X.-N.; Shields, D.J.; Witt, O.; Molenaar, J.J.; Colombetti, S.; et al. International Consensus on Minimum Preclinical Testing Requirements for the Development of Innovative Therapies for Children and Adolescents with Cancer. Mol. Cancer Ther. 2021, 20, 1462–1468. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Maki, R.G.; Jungbluth, A.A.; Gnjatic, S.; Schwartz, G.K.; D’Adamo, D.R.; Keohan, M.L.; Wagner, M.J.; Scheu, K.; Chiu, R.; Ritter, E.; et al. A Pilot Study of Anti-CTLA4 Antibody Ipilimumab in Patients with Synovial Sarcoma. Sarcoma 2013, 2013, 168145. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Rytlewski, J.; Milhem, M.M.; Monga, V. Turning ‘Cold’ tumors ‘Hot’: Immunotherapies in sarcoma. Ann. Transl. Med. 2021, 9, 1039. [Google Scholar] [CrossRef]
- Gyurdieva, A.; Zajic, S.; Chang, Y.-F.; Houseman, E.A.; Zhong, S.; Kim, J.; Nathenson, M.; Faitg, T.; Woessner, M.; Turner, D.C.; et al. Biomarker correlates with response to NY-ESO-1 TCR T cells in patients with synovial sarcoma. Nat. Commun. 2022, 13, 5296. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.; Federico, S.M.; Chen, X.; Shelat, A.A.; Bradley, C.; Gordon, B.; Karlstrom, A.; Twarog, N.R.; Clay, M.R.; Bahrami, A.; et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 2017, 549, 96–100. [Google Scholar] [CrossRef]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.-Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019, 9, 46–63. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]
- Landuzzi, L.; Manara, M.C.; Lollini, P.-L.; Scotlandi, K. Patient Derived Xenografts for Genome-Driven Therapy of Osteosarcoma. Cells 2021, 10, 416. [Google Scholar] [CrossRef]
- Stewart, E.; Federico, S.; Karlstrom, A.; Shelat, A.; Sablauer, A.; Pappo, A.; Dyer, M.A. The Childhood Solid Tumor Network: A new resource for the developmental biology and oncology research communities. Dev. Biol. 2016, 411, 287–293. [Google Scholar] [CrossRef]
- Conte, N.; Mason, J.C.; Halmagyi, C.; Neuhauser, S.; Mosaku, A.; Yordanova, G.; Chatzipli, A.; Begley, D.A.; Krupke, D.M.; Parkinson, H.; et al. PDX Finder: A portal for patient-derived tumor xenograft model discovery. Nucleic Acids Res. 2019, 47, D1073–D1079. [Google Scholar] [CrossRef] [PubMed]
- Meehan, T.F.; Conte, N.; Goldstein, T.; Inghirami, G.; Murakami, M.A.; Brabetz, S.; Gu, Z.; Wiser, J.A.; Dunn, P.; Begley, D.A.; et al. PDX-MI: Minimal Information for Patient-Derived Tumor Xenograft Models. Cancer Res. 2017, 77, e62–e66. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Chao, T.; Ruiqi, C.; Juan, S.; Zhihong, L. Patient-derived xenograft models in musculoskeletal malignancies. J. Transl. Med. 2018, 16, 16. [Google Scholar] [CrossRef] [PubMed]
- Cornillie, J.; Wozniak, A.; Li, H.; Wang, Y.; Boeckx, B.; Gebreyohannes, Y.K.; Wellens, J.; Vanleeuw, U.; Hompes, D.; Stas, M.; et al. Establishment and Characterization of Histologically and Molecularly Stable Soft-tissue Sarcoma Xenograft Models for Biological Studies and Preclinical Drug Testing. Mol. Cancer Ther. 2019, 18, 1168–1178. [Google Scholar] [CrossRef]
- Isfort, I.; Cyra, M.; Elges, S.; Kailayangiri, S.; Altvater, B.; Rossig, C.; Steinestel, K.; Grünewald, I.; Huss, S.; Eßeling, E.; et al. SS18-SSX-Dependent YAP/TAZ Signaling in Synovial Sarcoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3718–3731. [Google Scholar] [CrossRef]
- Kawano, S.; Grassian, A.R.; Tsuda, M.; Knutson, S.K.; Warholic, N.M.; Kuznetsov, G.; Xu, S.; Xiao, Y.; Pollock, R.M.; Smith, J.S.; et al. Preclinical Evidence of Anti-Tumor Activity Induced by EZH2 Inhibition in Human Models of Synovial Sarcoma. PLoS ONE 2016, 11, e0158888. [Google Scholar] [CrossRef]
- Hajdu, S.I.; Lemos, L.B.; Kozakewich, H.; Helson, L.; Beattie, E.J. Growth pattern and differentiation of human soft tissue sarcomas in nude mice. Cancer 1981, 47, 90–98. [Google Scholar] [CrossRef]
- Marín-Jiménez, J.A.; Capasso, A.; Lewis, M.S.; Bagby, S.M.; Hartman, S.J.; Shulman, J.; Navarro, N.M.; Yu, H.; Rivard, C.J.; Wang, X.; et al. Testing Cancer Immunotherapy in a Human Immune System Mouse Model: Correlating Treatment Responses to Human Chimerism, Therapeutic Variables and Immune Cell Phenotypes. Front. Immunol. 2021, 12, 607282. [Google Scholar] [CrossRef]
- Boven, E.; Pinedo, H.M.; van Hattum, A.H.; Scheffer, P.G.; Peters, W.H.; Erkelens, C.A.; Schlüper, H.M.; Kuiper, C.M.; van Ark-Otte, J.; Giaccone, G. Characterization of human soft-tissue sarcoma xenografts for use in secondary drug screening. Br. J. Cancer 1998, 78, 1586–1593. [Google Scholar] [CrossRef]
- Monsma, D.J.; Monks, N.R.; Cherba, D.M.; Dylewski, D.; Eugster, E.; Jahn, H.; Srikanth, S.; Scott, S.B.; Richardson, P.J.; Everts, R.E.; et al. Genomic characterization of explant tumorgraft models derived from fresh patient tumor tissue. J. Transl. Med. 2012, 10, 125. [Google Scholar] [CrossRef]
- Stebbing, J.; Paz, K.; Schwartz, G.K.; Wexler, L.H.; Maki, R.; Pollock, R.E.; Morris, R.; Cohen, R.; Shankar, A.; Blackman, G.; et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer 2014, 120, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Sun, M.; Zhang, L.; Hua, Y.; Li, H.Q.; Ouyang, D.X. Abstract 4609: Establishment of sarcoma PDX models with various subtypes for drug efficacy evaluation. Cancer Res. 2019, 79, 4609. [Google Scholar] [CrossRef]
- Jones, S.E.; Fleuren, E.D.G.; Frankum, J.; Konde, A.; Williamson, C.T.; Krastev, D.B.; Pemberton, H.N.; Campbell, J.; Gulati, A.; Elliott, R.; et al. ATR Is a Therapeutic Target in Synovial Sarcoma. Cancer Res. 2017, 77, 7014–7026. [Google Scholar] [CrossRef] [PubMed]
- Schoffski, P.; Agulnik, M.; Stacchiotti, S.; Davis, L.E.; Villalobos, V.M.; Italiano, A.; George, S.; Cote, G.M.; Blakemore, S.; Clawson, A.; et al. Phase 2 multicenter study of the EZH2 inhibitor tazemetostat in adults with synovial sarcoma (NCT02601950). J. Clin. Oncol. 2017, 35, 11057. [Google Scholar] [CrossRef]
- Yakushiji, T.; Yonemura, K.; Tsuruta, J.; Nishida, K.; Kato, T.; Takagi, K. Capacity for epithelial differentiation in synovial sarcoma: Analysis of a new human cell line. J. Clin. Pathol. 2000, 53, 525–531. [Google Scholar] [CrossRef]
- Kerrison, W.G.J.; Ning, J.; Krasny, L.; Arthur, A.; Guljar, N.; Elms, M.L.; Swain, A.; Jones, R.L.; Thway, K.; Huang, P.H. Characterisation of a Novel Cell Line (ICR-SS-1) Established from a Patient-Derived Xenograft of Synovial Sarcoma. Cells 2022, 11, 2418. [Google Scholar] [CrossRef]
- Hattori, E.; Oyama, R.; Kondo, T. Systematic Review of the Current Status of Human Sarcoma Cell Lines. Cells 2019, 8, 157. [Google Scholar] [CrossRef]
- Naka, N.; Takenaka, S.; Araki, N.; Miwa, T.; Hashimoto, N.; Yoshioka, K.; Joyama, S.; Hamada, K.-I.; Tsukamoto, Y.; Tomita, Y.; et al. Synovial sarcoma is a stem cell malignancy. Stem Cells 2010, 28, 1119–1131. [Google Scholar] [CrossRef]
- Renwick, P.J.; Reeves, B.R.; Cin, P.D.; Fletcher, C.D.; Kempski, H.; Sciot, R.; Kazmierczak, B.; Jani, K.; Sonobe, H.; Knight, J.C. Two categories of synovial sarcoma defined by divergent chromosome translocation breakpoints in Xp11.2, with implications for the histologic sub-classification of synovial sarcoma. Cytogenet. Cell Genet. 1995, 70, 58–63. [Google Scholar] [CrossRef]
- Koelsche, C.; Renner, M.; Hartmann, W.; Brandt, R.; Lehner, B.; Waldburger, N.; Alldinger, I.; Schmitt, T.; Egerer, G.; Penzel, R.; et al. TERT promoter hotspot mutations are recurrent in myxoid liposarcomas but rare in other soft tissue sarcoma entities. J. Exp. Clin. Cancer Res. CR 2014, 33, 33. [Google Scholar] [CrossRef]
- Cassinelli, G.; Bo, L.D.; Favini, E.; Cominetti, D.; Pozzi, S.; Tortoreto, M.; de Cesare, M.; Lecis, D.; Scanziani, E.; Minoli, L.; et al. Supersulfated low-molecular weight heparin synergizes with IGF1R/IR inhibitor to suppress synovial sarcoma growth and metastases. Cancer Lett. 2018, 415, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, C.; Bo, L.D.; Favini, E.; Tortoreto, M.; Beretta, G.L.; Arrighetti, N.; Zaffaroni, N.; Cassinelli, G. Overactive IGF1/Insulin Receptors and NRASQ61R Mutation Drive Mechanisms of Resistance to Pazopanib and Define Rational Combination Strategies to Treat Synovial Sarcoma. Cancers 2019, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Nojima, T.; Wang, Y.S.; Abe, S.; Matsuno, T.; Yamawaki, S.; Nagashima, K. Morphological and cytogenetic studies of a human synovial sarcoma xenotransplanted into nude mice. Acta Pathol. Jpn. 1990, 40, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Nishio, J.; Iwasaki, H.; Ishiguro, M.; Ohjimi, Y.; Fujita, C.; Isayama, T.; Naito, M.; Oda, Y.; Kaneko, Y.; Kikuchi, M. Establishment of a new human synovial sarcoma cell line, FU-SY-1, that expresses c-Met receptor and its ligand hepatocyte growth factor. Int. J. Oncol. 2002, 21, 17–23. [Google Scholar] [CrossRef]
- Soni, E.E.C.; Schlottman, S.; Erkizan, H.V.; Uren, A.; Toretsky, J.A. Loss of SS18-SSX1 inhibits viability and induces apoptosis in synovial sarcoma. Clin. Orthop. Relat. Res. 2014, 472, 874–882. [Google Scholar] [CrossRef]
- Sonobe, H.; Manabe, Y.; Furihata, M.; Iwata, J.; Oka, T.; Ohtsuki, Y.; Mizobuchi, H.; Yamamoto, H.; Kumano, O.; Abe, S. Establishment and characterization of a new human synovial sarcoma cell line, HS-SY-II. Lab. Investig. A J. Tech. Methods Pathol. 1992, 67, 498–505. [Google Scholar]
- Teicher, B.A.; Polley, E.; Kunkel, M.; Evans, D.; Silvers, T.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Selby, M.; et al. Sarcoma Cell Line Screen of Oncology Drugs and Investigational Agents Identifies Patterns Associated with Gene and microRNA Expression. Mol. Cancer Ther. 2015, 14, 2452–2462. [Google Scholar] [CrossRef]
- Sonobe, H.; Takeuchi, T.; Liag, S.-B.; Taguchi, T.; Yuri, K.; Shimizu, K.; Iwata, J.; Furihata, M.; Ohtsuki, Y.; Testa, J.R. A new human synovial sarcoma cell line, HS-SY-3, with a truncated form of hybridSYT/SSX1 gene. Int. J. Cancer 1999, 82, 459–464. [Google Scholar] [CrossRef]
- Raquib, A.R.; Hofvander, J.; Ta, M.; Nielsen, T.O. Expanding the Use of an SS18-SSX Antibody for Molecular Assays in Synovial Sarcoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2022, 30, 531–539. [Google Scholar] [CrossRef]
- Kito, F.; Oyama, R.; Takai, Y.; Sakumoto, M.; Shiozawa, K.; Qiao, Z.; Uehara, T.; Yoshida, A.; Kawai, A.; Kondo, T. Establishment and characterization of the NCC-SS1-C1 synovial sarcoma cell line. Hum. Cell 2018, 31, 167–174. [Google Scholar] [CrossRef]
- Oyama, R.; Kito, F.; Sakumoto, M.; Shiozawa, K.; Toki, S.; Endo, M.; Yoshida, A.; Kawai, A.; Kondo, T. Establishment and proteomic characterization of a novel synovial sarcoma cell line, NCC-SS2-C1. Vitr. Cell. Dev. Biol. Anim. 2018, 54, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, Y.; Noguchi, R.; Tsuchiya, R.; Sei, A.; Sugaya, J.; Iwata, S.; Yoshida, A.; Kawai, A.; Kondo, T. Establishment and characterization of NCC-SS3-C1: A novel patient-derived cell line of synovial sarcoma. Hum. Cell 2020, 33, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, R.; Yoshimatsu, Y.; Noguchi, R.; Ono, T.; Sei, A.; Takeshita, F.; Sugaya, J.; Iwata, S.; Yoshida, A.; Ohtori, S.; et al. Establishment and characterization of NCC-SS4-C1: A novel patient-derived cell line of synovial sarcoma. Hum. Cell 2021, 34, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, Y.; Noguchi, R.; Sin, Y.; Tsuchiya, R.; Ono, T.; Akiyama, T.; Sugaya, J.; Kojima, N.; Yoshida, A.; Kawai, A.; et al. Establishment and characterization of NCC-SS5-C1: A novel patient-derived cell line of synovial sarcoma. Hum. Cell 2022, 35, 1290–1297. [Google Scholar] [CrossRef]
- Berardi, A.C.; Parafioriti, A.; Barisani, D.; Papp, B.; Armiraglio, E.; Martinoli, M.; Dalprà, L.; Santoro, A. A new human cell line, PDSS-26, from poorly differentiated synovial sarcoma, with unique chromosomal anomalies. Cancer Genet. Cytogenet. 2003, 146, 116–124. [Google Scholar] [CrossRef]
- Noguchi, S.-I.; Ueki, T.; Kawauchi, S.; Fukuda, T.; Matsuura, H.; Sonoda, T.; Tsuneyoshi, M. Establishment and characterization of a new synovial sarcoma cell line, SN-SY-1: Special reference to bcl-2 protein and SYT-SSX1 hybrid transcripts. Int. J. Cancer 1997, 72, 995–1002. [Google Scholar] [CrossRef]
- Reeves, B.R.; Smith, S.; Fisher, C.; Warren, W.; Knight, J.; Martin, C.; Chan, A.M.; Gusterson, B.A.; Westbury, G.; Cooper, C.S. Characterization of the translocation between chromosomes X and 18 in human synovial sarcomas. Oncogene 1989, 4, 373–378. [Google Scholar]
- Knight, J.C.; Reeves, B.R.; Kearney, L.; Monaco, A.P.; Lehrach, H.; Cooper, C.S. Localization of the synovial sarcoma t(X;18)(p11.2;q11.2) breakpoint by fluorescence in situ hybridization. Human Mol. Genet. 1992, 1, 633–637. [Google Scholar] [CrossRef]
- Kawai, A.; Naito, N.; Yoshida, A.; Morimoto, Y.; Ouchida, M.; Shimizu, K.; Beppu, Y. Establishment and characterization of a biphasic synovial sarcoma cell line, SYO-1. Cancer Lett. 2004, 204, 105–113. [Google Scholar] [CrossRef]
- Cyra, M.; Schulte, M.; Berthold, R.; Heinst, L.; Jansen, E.-P.; Grünewald, I.; Elges, S.; Larsson, O.; Schliemann, C.; Steinestel, K.; et al. SS18-SSX drives CREB activation in synovial sarcoma. Cell. Oncol. 2022, 45, 399–413. [Google Scholar] [CrossRef]
- Ito, T.; Ouchida, M.; Morimoto, Y.; Yoshida, A.; Jitsumori, Y.; Ozaki, T.; Sonobe, H.; Inoue, H.; Shimizu, K. Significant growth suppression of synovial sarcomas by the histone deacetylase inhibitor FK228 in vitro and in vivo. Cancer Lett. 2005, 224, 311–319. [Google Scholar] [CrossRef]
- Yamasaki, H.; Miyamoto, M.; Yamamoto, Y.; Kondo, T.; Watanabe, T.; Ohta, T. Synovial sarcoma cell lines showed reduced DNA repair activity and sensitivity to a PARP inhibitor. Genes Cells Devoted Mol. Cell. Mech. 2016, 21, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Skytting, B.; Nilsson, G.; Gasbarri, A.; Haslam, K.; Bartolazzi, A.; Brodin, B.; Mandahl, N.; Larsson, O. SYT-SSX is critical for cyclin D1 expression in synovial sarcoma cells: A gain of function of the t(X;18)(p11.2;q11.2) translocation. Cancer Res. 2002, 62, 3861–3867. [Google Scholar] [PubMed]
- Qiao, Z.; Shiozawa, K.; Kondo, T. Proteomic approach toward determining the molecular background of pazopanib resistance in synovial sarcoma. Oncotarget 2017, 8, 109587–109595. [Google Scholar] [CrossRef] [PubMed]
- Avdonkina, N.A.; Danilova, A.B.; Misyurin, V.A.; Prosekina, E.A.; Girdyuk, D.V.; Emelyanova, N.V.; Nekhaeva, T.L.; Gafton, G.I.; Baldueva, I.A. Biological features of tissue and bone sarcomas investigated using an in vitro model of clonal selection. Pathol. Res. Pract. 2021, 217, 153214. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Cooley, C.; Su, L. HDAC2 links ubiquitination to tumor suppression in synovial sarcoma. Mol. Cell. Oncol. 2021, 8, 1914291. [Google Scholar] [CrossRef]


{kind=link}
| Genotype | Tumor Histology or Main Phenotype | Tumor Incidence | Tumor Latency | Main Features and Notes | References |
|---|---|---|---|---|---|
| Rosa26/hSS2 or Rosa26/hSS1 | No tumor | None | Not applicable | Carrying a floxed SS18-SSX2 or SS18-SSX1 allele at the Rosa26 locus Viable, fertile, no tumor formation | [37,39] |
| hSS1 or hSS2 mice +TATCre recombinant protein injection in hind limb | SyS | 40–60% | >1 year median 18.8 months | Induction of SyS after Cre protein administration | [39] |
| Homozygous hSS2 mice +TATCre recombinant protein injection in hind limb | SyS | 100% | 6 months | Dose dependent increase in SyS onset | [23] |
| Myf5–Cre/hSS2 | SyS | 100% | 3–5 months | -Multiple tumors (3–5/mouse) of extremities and rib cage -Monphasic >> biphasic -Myogenin-neg, Cytokeratin-pos, Vimentin-pos -Gene expression signature “SS18-SSX model synovial subset” shared by human and murine SyS is present only in human SyS, not in other STS | [37] |
| Pax3–Cre/hSS2 or Pax7–Cre/hSS2 or Hprt–Cre/hSS2 or Hprt–Cre/hSS1 or Ap2–Cre/hSS2 or Sox9–Cre/hSS2 or Dermo1–Cre/hSS2 or Flk1–Cre/hSS2 or Tie2–Cre/hSS2 or Nestin1–Cre/hSS2 or Prx1Cre/hSS2 | Embryonic lethality | Expression in earlier ectodermic, neural, or mesenchymal precursors, or bone and cartilage precursors, or vascular and hematopoietic precursors, disrupted embryogenesis | [6,37,38,39] | ||
| Col1a1Cre/hSS2 or OcCre/hSS2 | Perinatal lethality | Bone and skeletal defects No tumors | [6] | ||
| Myf6–Cre/hSS2 | Severe myopathy No tumor | Died within 6 months of age | [37] | ||
| Prx1CreERT2hSS2 +tamoxifen | SyS | 100% | 9 months | Perimandibular SyS | [6] |
| OsxCreERT hSS2 +tamoxifen | No tumor | No tumors or skeletal defects up to 1 year | [6] | ||
| Rosa26CreER/hSS2 | SyS | 100% | 5–14 months | 3 tumors/mouse with a distinct anatomical distribution compared to Myf5–Cre/hSS2 mice | [38] |
| Myf5–Cre/hSS1 or Rosa26CreER/hSS1 | SyS | 80–50% | >1 year | Low multiplicity | [39] |
| Genotype | Tumor Histology or Main Phenotype | Tumor Incidence | Tumor Latency | Main Features and Notes | References |
|---|---|---|---|---|---|
| hSS2/TATCre recombinant protein injection in hind limb+ Bcl2 overexpressing (Bcl-2OE) mice | SyS | 80% | Median 9 months | Enhanced synovial sarcomagenesis, increased incidence, and reduced tumor latency compared to hSS2 mice/TATCre | [41] |
| Myf5–Cre/hSS2 + Bcl2 overexpression | SyS | 100% | Earlier onset | Significantly reduced tumor latency | [41] |
| Myf5–Cre/hSS2, Bcl2fl/fl (Bcl2 deleted) | SyS, monophasic subtype only | 80% | 5–9 months | Slightly later onset and lower multiplicity compared to Myf5 –Cre/hSS2 mice | [41] |
| SSM2+/B-CATfl+/+/Myf5–Cre+ | Strong inhibition of synovial sarcomagenesis by β-catenin silencing | 20% | NA | Strong β-catenin nuclear signal in the few developing tumors, indicating incomplete silencing | [43] |
| Rosa26hSS2/wt; Ctnnb1ex3fl/wt +AdCre injection | Increased synovial sarcomagenesis; poorly differentiated SyS subtype | 90% | 3 months | Beta-catenin stabilization, increased SyS sarcomagenesis No metastasis | [44] |
| Myf5–Cre; Ctnnb1ex3fl | Embryonic lethality | [44] | |||
| Prx1CreERT2 hSS2 Ctnnb1ex3fl +tamoxifen | SyS in the forelimbs | 100% | 3 months | [6] | |
| OsxCreERT Ctnnb1ex3fl +tamoxifen | SyS | 100% | NA | Osteopetrosis | [6] |
| hSS2 Myf5–Cre/Fgfr1,2,3fl/fl (SMF1,2,3.HO) and each single Fgfr knockout (FGFR silencing) | Significantly reduced SyS incidence and multiplicity | 10–35% | Observed at 10 weeks | FGFR homozygous (HO) silencing. Implication of mechanisms related to ETV4 and ETV5 through DUX4 embryonic pathway | [47] |
| Genotype | Tumor Histology or Main Phenotype | Tumor Incidence | Tumor Latency | Main Features and Notes | References |
|---|---|---|---|---|---|
| hSS1 or hSS2/PTENfllfl +TATCre injection in the hind limb (PTEN silencing) | Increased SyS incidence and acquisition of metastatic potential to the lung, metastatic SyS | 100% | <1 year (8–10 months) | -TATCre induction of homozygous silencing of PTEN alone induced no tumors -40% incidence of lung macrometastses -70% incidence of lung micrometastases ->90% incidence of lung disseminated tumor cells | [48,51] |
| hSS2/SMARCB1fl/fl +TATCre injection (SMARCB1 silencing) | Increased sarcomagenesis, but development of epithelioid sarcoma or mesenchymal rhabdoid tumor, not SyS | 100% | 3 months | BAF-family complexes perturbation | [23] |
| Number of Established * SyS PDX Models | Mouse Strain | Rate of Engraftment (PDX/Implanted Tumors) | PDX Model ID and Annotations | References |
|---|---|---|---|---|
| 6 | athymic nu/nu mice | 100% (6/6) | Histological evaluation | [77] |
| 2 | athymic NMRI-nu/nu mice | NA | PDX ID: S.Lt, S.To Histological evaluation | [79] |
| 1 | athymic nu/nu mice | 100% (1/1) | 0.91 Pearson correlation coefficient between the originating patient tumor and the PDX, based on Affymetrix gene expression data | [80] |
| 1 | athymic nu/nu mice | 50% (1/2) | PDX ID: 1152, SS18-SSX fusion High drug sensitivity to Ifosfamide and trabectedin, intermediate sensitivity to gemcitabine and pazopanib | [81] |
| 2 | athymic nu/nu mice | 100% (2/2) | PDX ID: CTG-0771, SS18-SSX2 fusion PDX ID: CTG-1169, SS18-SSX1 fusion | [64] |
| 1 | NSG, athymic nu/nu mice | 50% (1/2) | Pediatric SyS, time to passage 8 months | [65] |
| 1 | athymic NMRI-nu/nu mice | 12% (1/8) | PDX ID: UZLX-STS7 SS18-SSX1 fusion, Poorly differentiated subtype, over 48 passages, growth rate 1 month | [74] |
| International Repositories and Internet Links § | Number of SyS PDX Models | PDX Model ID, Annotations | References |
|---|---|---|---|
| Champions Oncology Model Cohorts (championsoncology.com) (https://www.championsoncology.com/resource-library/model-cohorts, (accessed on 28 November 2022)) | 3 | CTG-0771, SS18-SSX2 fusion CTG-1169, SS18-SSX1 fusion CTG-0331, SS18-SSX2 fusion | [64,76] |
| Xenosarc Platform (Leuven, Belgium) XenoSarc platform—Laboratory of Experimental Oncology (kuleuven.be, (accessed on 28 November 2022)) (https://gbiomed.kuleuven.be/english/research/50488876/50488902/xenosarc, (accessed on 28 November 2022)) | 1 | UZLX –STS7, SS18-SSX1 fusion | [74,75] |
| Crown Bioscience Patient-Derived Xenograft—PDX Models|Crown Bioscience (https://www.crownbio.com/model-systems/in-vivo/pdx-models, (accessed on 28 November 2022)) | 3 | SA10159 SA10162 SA10175 [SA13412 previously diagnosed as SyS but later diagnosed as primitive neuroectodermal tumor (PNET)] | [82,83] |
| NCI Patient-Derived Models Repository (PDMR) PDCM Finder—Search (cancermodels.org, (accessed on 28 November 2022)) (https://www.cancermodels.org/data/search?q=Synovial%20Sarcoma, (accessed on 28 November 2022)) Including Childhood Solid Tumor Network www.stjude.org/CSTN/, (accessed on 28 November 2022) (http://www.stjude.org/CSTN, (accessed on 28 November 2022)) | 12 from 4 sources (JAX, SJCRH, WUSTL, PDMR) | PDMR/119177-322-R1 PDMR/197587-005-T PDMR/761936-265-R PDMR/571681-099-R PDMR/957923-259-R WUSTL/WUSTL SHIM9 WUSTL/WUSTL SHIM11 WUSTL/WUSTL SHIM12 SJCRH/SJSS049190_X1 SJCRH/SJSS063828_X1 JAX/J000104314 (SS18-SSX1 fusion, monophasic subtype) JAX/TM01634 (SS18-SSX1 fusion) | [40,65] |
| Cell Line | Fusion Gene | Histology | Tumorigenic Ability in Mice and Annotations | References and Banks |
|---|---|---|---|---|
| A2243 | SS18-SSX2 | Biphasic | NA | [18] |
| ASKA-SS | SS18-SSX1 | Biphasic | In BALB/c nu/nu, 1000–1 × 107 cells sc, tumor incidence was 100% within 5 months | [88] Cell Engineering Division-CELL BANK-(RIKEN BRC) (https://cell.brc.riken.jp/en/, (accessed on 28 November 2022)) |
| CME-1 | SS18-SSX2 | Monophasic | In SCID mice, 20 × 106 cells im | [89,90,91,92] |
| Fuji | SS18-SSX2 | Monophasic | In Balb/C-nu, 1 × 107 cells sc 50% Matrigel 200 mm3 at 25 days after cell injection | [18,93] |
| FU-SY-1 | SS18-SSX1 | Monophasic | Not tumorigenic | [94] |
| GUSS-1 | SS18-SSX1 | Biphasic | NA | [95] |
| GUSS-2 | SS18-SSX1 | Monophasic | NA | [95] |
| GUSS-3 | SS18-SSX1 | Biphasic | NA | [95] |
| GUSS-3b | SS18-SSX1 | Biphasic | NA, deriving from the same patient of GUSS-3 after neoadjuvant chemotherapy and radiation | [95] |
| HS-SY-II | SS18-SSX1 | Monophasic | In Balb/C-nu, 1 × 107 cells sc 50% Matrigel 170 mm3 at 35 days after cell injection | [25,96,97] Cell Engineering Division-CELL BANK-(RIKEN BRC) (https://cell.brc.riken.jp/en/, (accessed on 28 November 2022)) |
| HS-SY-3 | SS18-SSX1 truncated | Monophasic | Not tumorigenic in nude mice | [98] |
| ICR-SS-1 | SS18-SSX1 | Monophasic | NA | [86] Not included in Cellosaurus |
| KU-SS-1 | SS18-SSX2 | Monophasic | In SICD mice, 8 × 107 cells sc, tumor latency 16 weeks. Derived from a PDX at the third in vivo passage | [85] |
| MoJo | SS18-SSX1 | Monophasic | In SCID mice, 20 × 106 cells im, tumor growth within 60 days from cell injection Resistant to pazopanib both in vitro and in vivo Harbor the NRAS Q61R mutation | [39,92,99] |
| NCC-SS1-C1 | SS18-SSX1 | Poorly differentiated | NA | [100] |
| NCC-SS2-C1 | SS18-SSX2 | Poorly differentiated | NA | [101] |
| NCC-SS3-C1 | SS18-SSX1 | Monophasic | NA | [102] |
| NCC-SS4-C1 | SS18-SSX1 | Monophasic | No in Balb/C-nu, 1 × 106 cells sc 50% Matrigel | [103] |
| NCC-SS5-C1 | SS18-SSX1 | Poorly differentiated | NA | [104] |
| PDSS-26 | SS18-SSX1 | Poorly differentiated, small cell variant | NA | [105] |
| SCS214 | SS18-SSX2 | NA | NA | Cellosaurus SCS214 (CVCL_WU91) |
| SN-SY-1 | SS18-SSX1 | Monophasic | In Balb/C-nu, 1.3 × 107 cells sc 33% positive after 23 weeks from cell injection | [106] |
| SS.PDX | SS18-SSX1 | Monophasic | NA | [40] Not included in Cellosaurus |
| SS255 | SS18-SSX2 | Monophasic | NA | [18,107,108] |
| SYO-1 | SS18-SSX2 | Biphasic | Yes, 5 × 106 sc In NSG or SCID mice or 105 cells/mouse inBALB/c nu/nu SYO-1 cells harbor mutation in CTNNB1 (G34L) with nuclear accumulation of Beta-catenin | [24,25,92,109,110,111] |
| YaFuSS | SS18-SSX1 | Monophasic | NA | [25,46,112] |
| Yamato-SS | SS18-SSX1 | Biphasic | In BALB/c nu/nu 1000–1 × 105 cells sc, tumor incidence was 100% within 5 months; 1 × 107 cells sc tumor latency 2 weeks | [88] Cell Engineering Division-CELL BANK-(RIKEN BRC) (https://cell.brc.riken.jp/en/, (accessed on 28 November 2022)) |
| 1273/99 | SS18-SSX2 | NA | NA | [90,113,114] |
| 716 SS MNV | SS18-SSX | NA | NA | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landuzzi, L.; Ruzzi, F.; Lollini, P.-L.; Scotlandi, K. Synovial Sarcoma Preclinical Modeling: Integrating Transgenic Mouse Models and Patient-Derived Models for Translational Research. Cancers 2023, 15, 588. https://doi.org/10.3390/cancers15030588
Landuzzi L, Ruzzi F, Lollini P-L, Scotlandi K. Synovial Sarcoma Preclinical Modeling: Integrating Transgenic Mouse Models and Patient-Derived Models for Translational Research. Cancers. 2023; 15(3):588. https://doi.org/10.3390/cancers15030588
Chicago/Turabian StyleLanduzzi, Lorena, Francesca Ruzzi, Pier-Luigi Lollini, and Katia Scotlandi. 2023. "Synovial Sarcoma Preclinical Modeling: Integrating Transgenic Mouse Models and Patient-Derived Models for Translational Research" Cancers 15, no. 3: 588. https://doi.org/10.3390/cancers15030588
APA StyleLanduzzi, L., Ruzzi, F., Lollini, P.-L., & Scotlandi, K. (2023). Synovial Sarcoma Preclinical Modeling: Integrating Transgenic Mouse Models and Patient-Derived Models for Translational Research. Cancers, 15(3), 588. https://doi.org/10.3390/cancers15030588