
Augmented Concentration of Isopentyl-Deoxynyboquinone in Tumors Selectively Kills NAD(P)H Quinone Oxidoreductase 1-Positive Cancer Cells through Programmed Necrotic and Apoptotic Mechanisms

 and
and 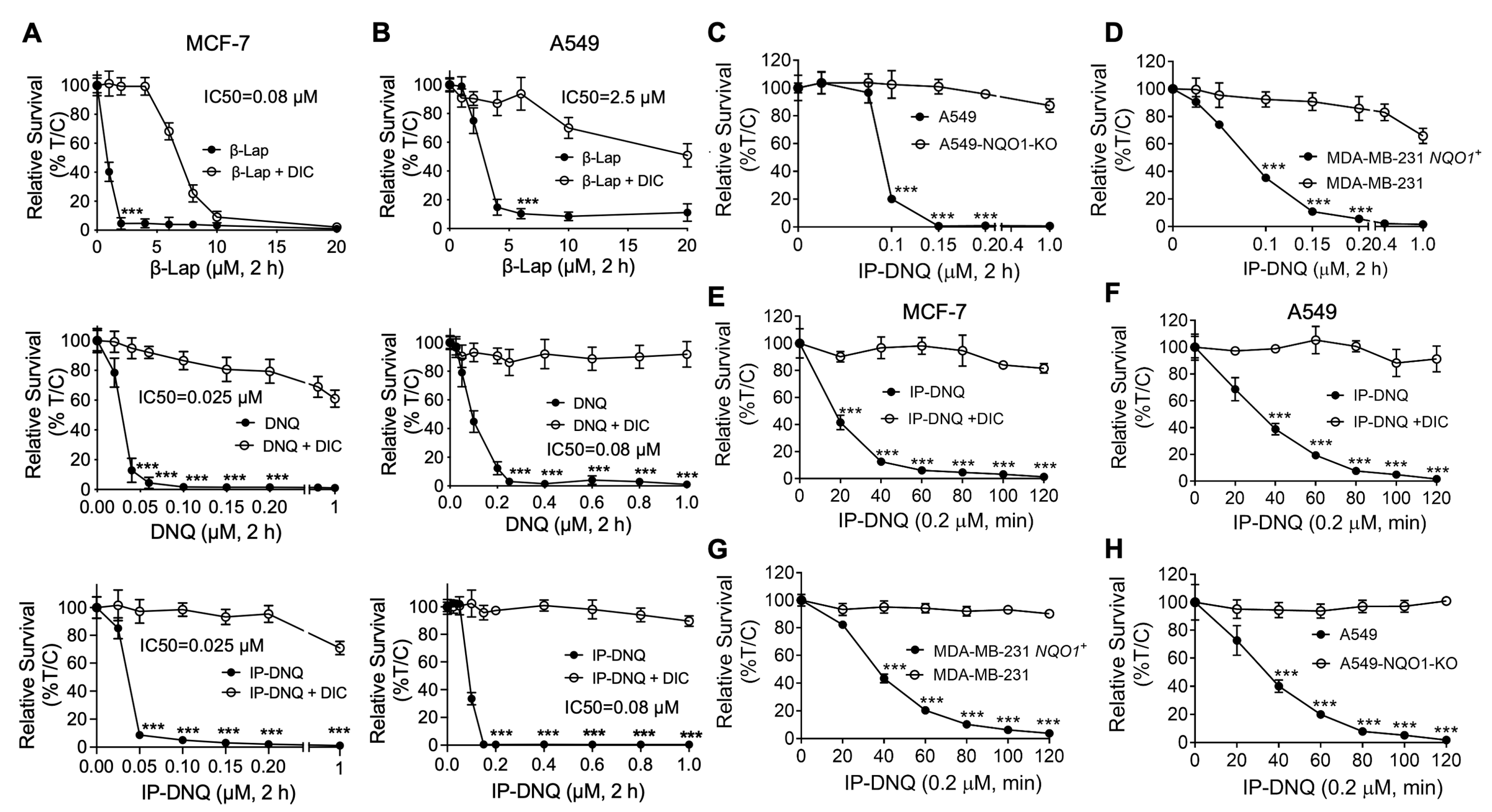{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Antibodies
2.3. Reagents and Chemicals
2.4. Relative Survival Assays (DNA Assay)
2.5. O2 Consumption Rate (OCR) and Extracellular Acidification Rates (ECAR) Assessments
2.6. Western Blotting
2.7. H2O2, ATP, and NAD+ Quantification
2.8. Immunofluorescence Assay
2.9. Alkaline Comet Assay
2.10. Caspase Activity Assay for Apoptosis Detection
2.11. Annexin-V FITC/PI Assay
2.12. In Vivo Antitumor Efficacy and Pharmacokinetic (PK) and Pharmacodynamic (PD) Studies
2.13. Statistical Analysis
3. Results
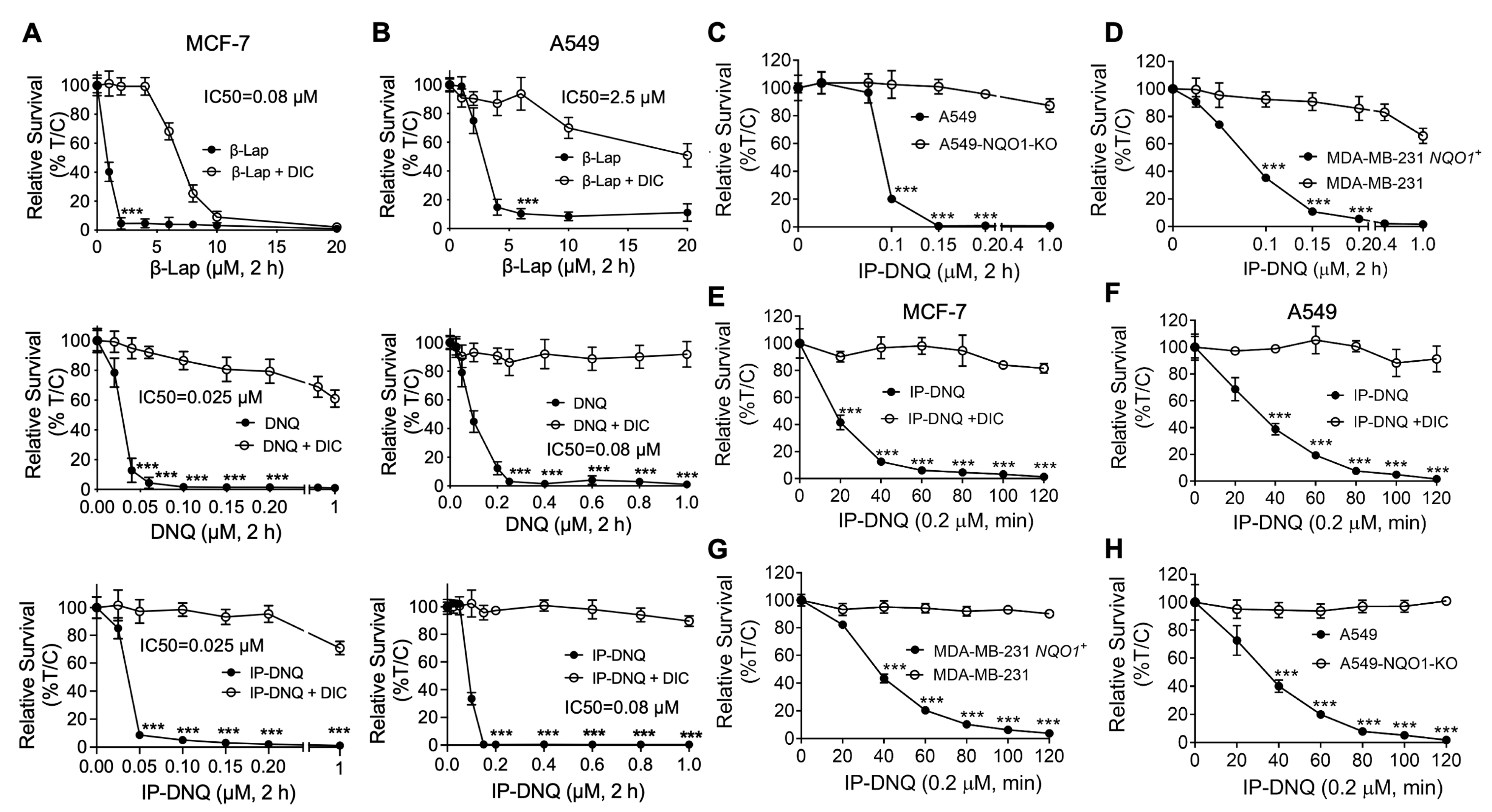
3.1. IP-DNQ Exerts Potent Cytotoxicity in Various Types of NQO1+ Cancer Cells
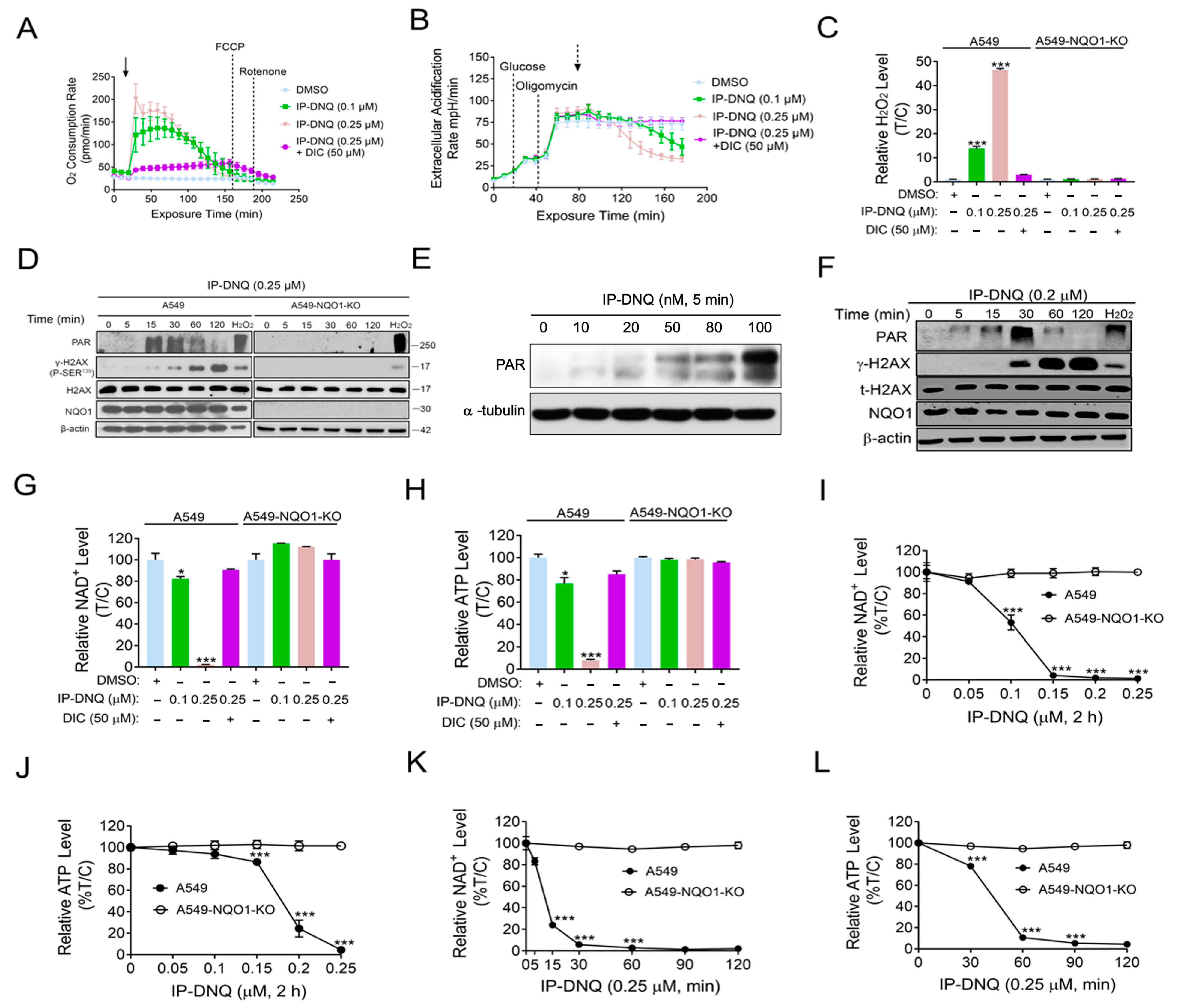
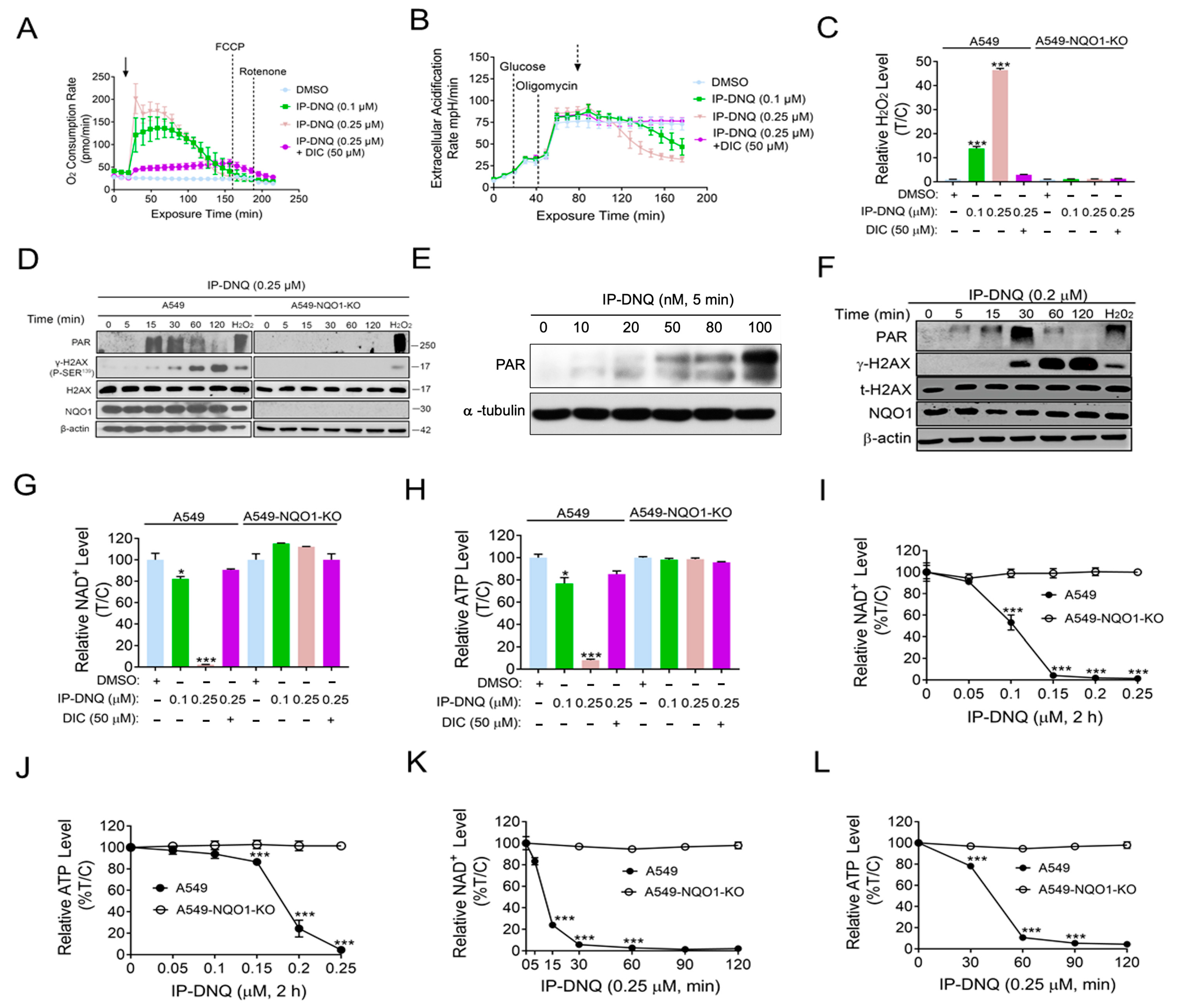
3.2. IP-DNQ Induces NQO1-Dependent ROS Formation, PARP1 Hyperactivation, and NAD+/ATP Loss
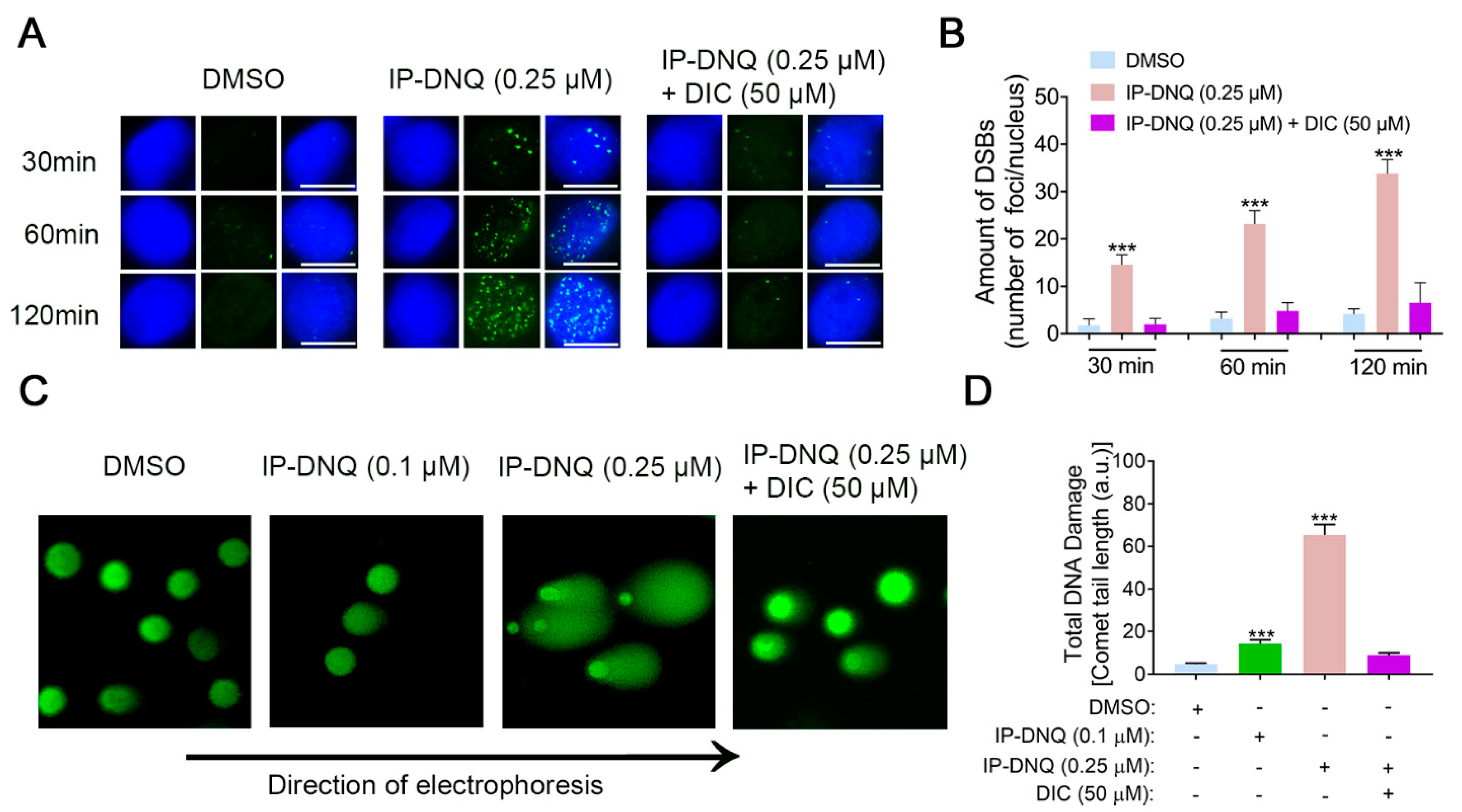
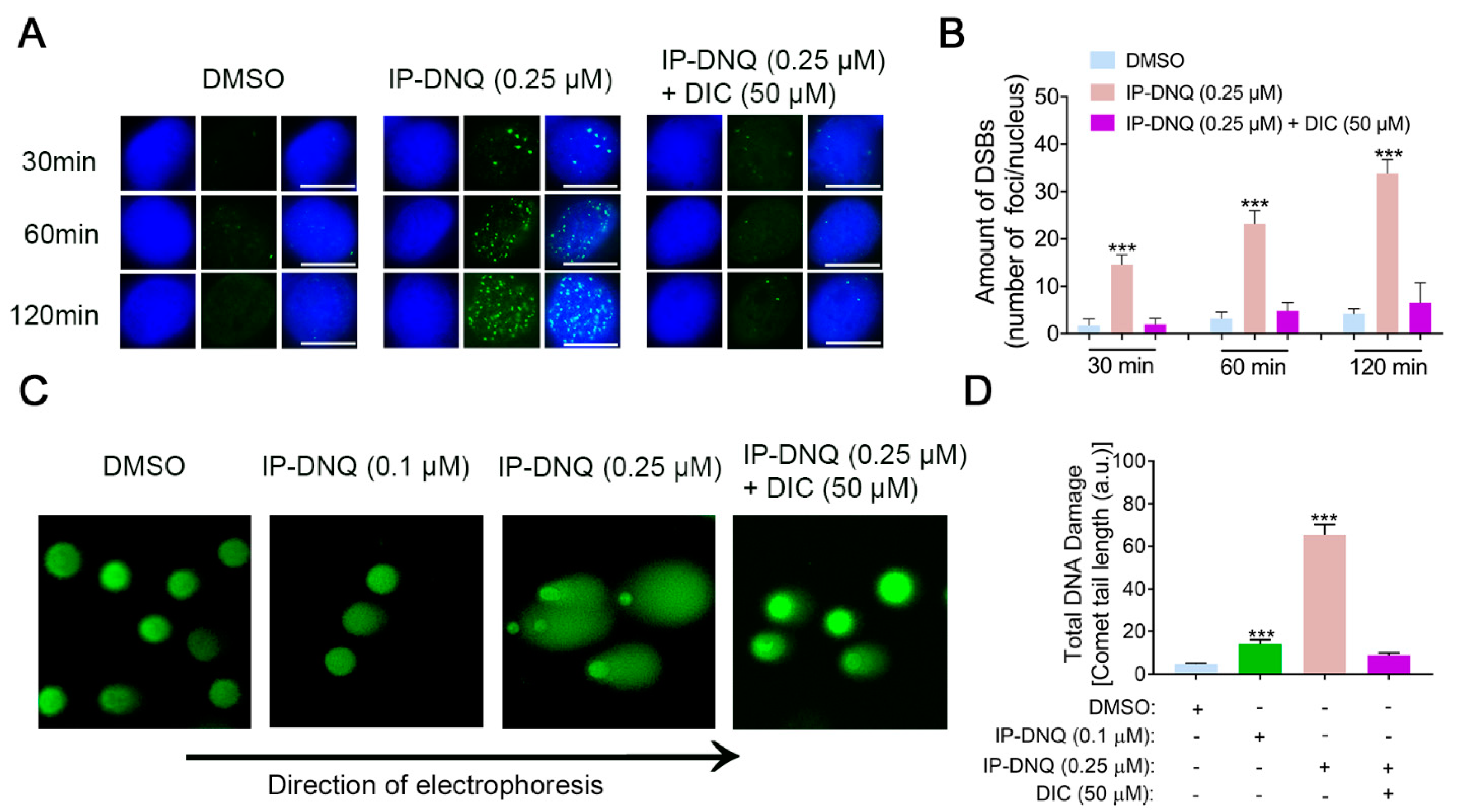
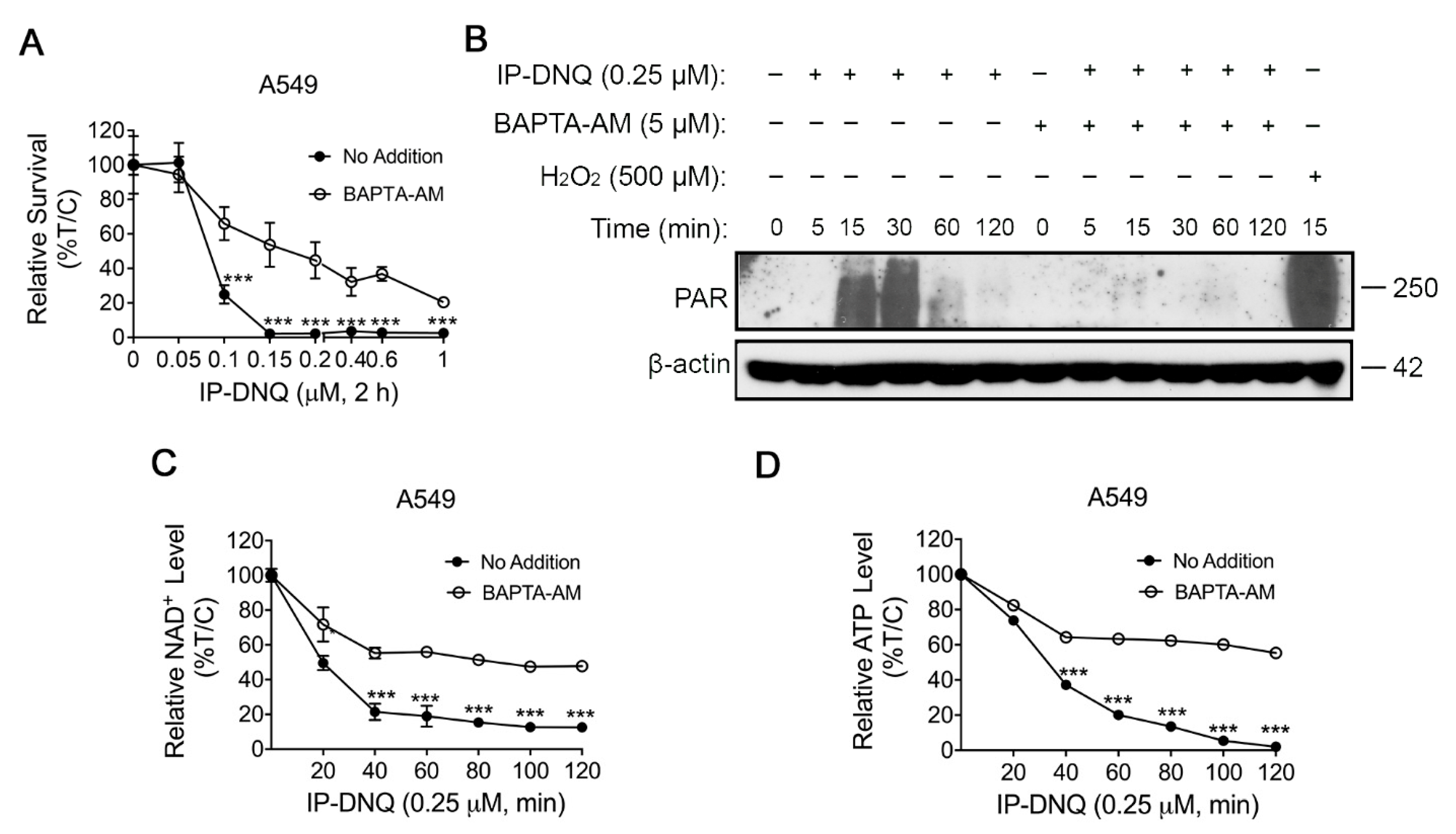
3.3. IP-DNQ Induces NQO1-Dependent DNA Damage, and Ca2+ Plays a Pivotal Role in IP-DNQ-Induced Cytotoxicity
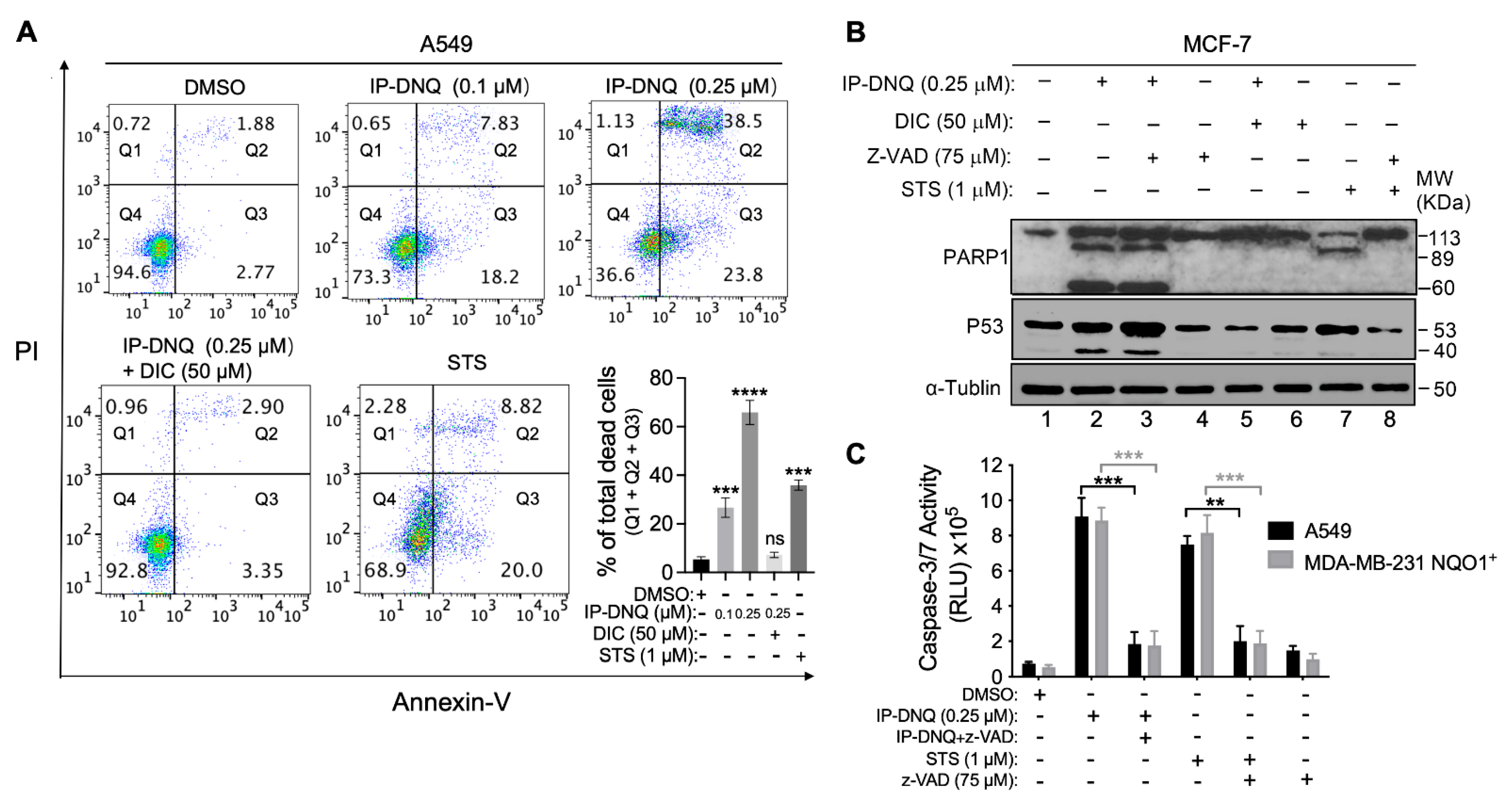
3.4. IP-DNQ Initiates Both Apoptosis and Programmed Necrosis in NQO1+ Cancer Cells
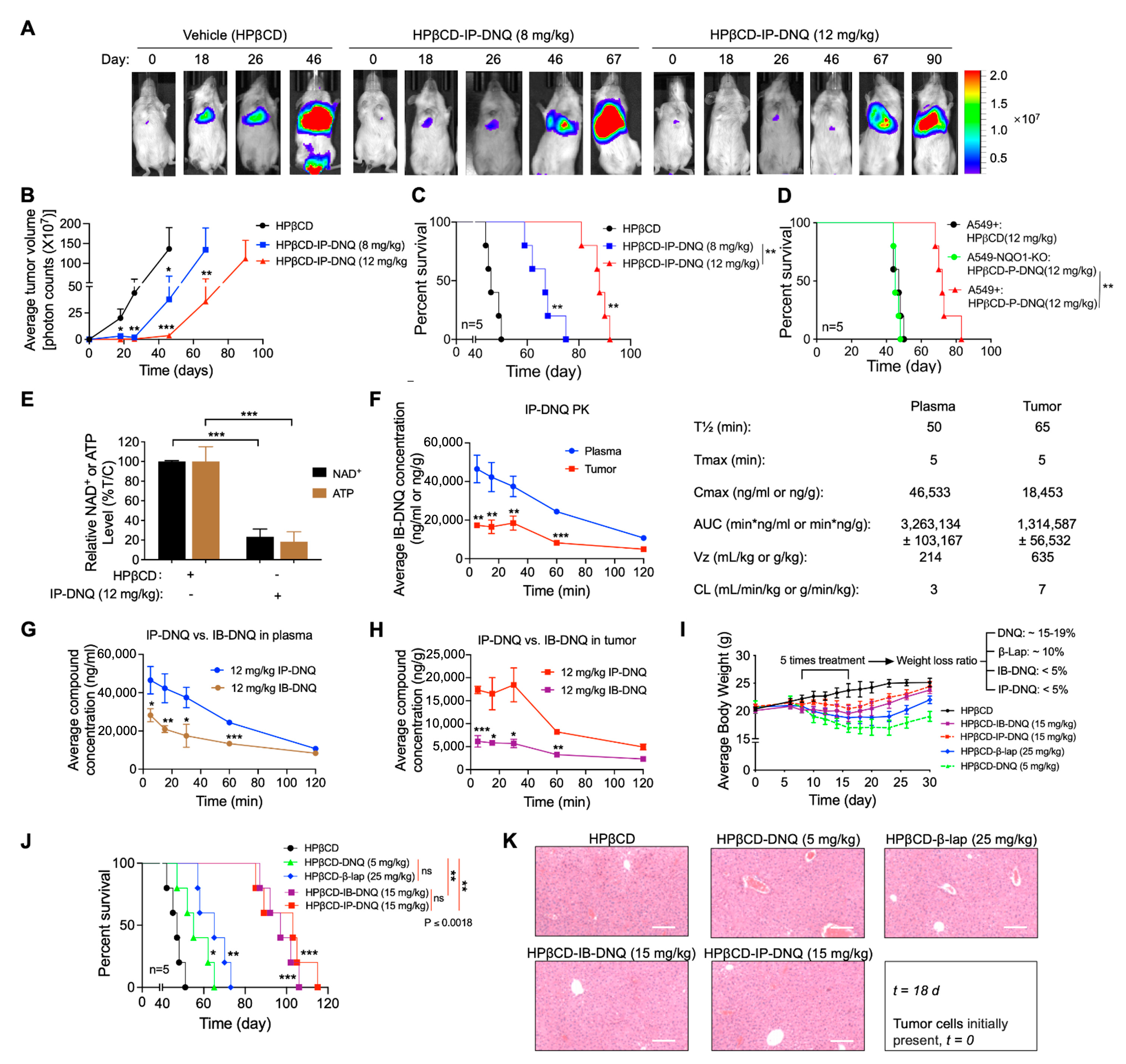
3.5. Antitumor Efficacy of IP-DNQ in Orthotopic NSCLC Xenografts, Pharmacokinetics (PK) and Tissue Pharmacodynamics (PD)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Sun, X.; Lu, W.; Xu, L.; Shi, J.; Yang, S.; Zhou, M.; Su, F.; Lin, F.; Cao, F. Synthesis of novel, DNA binding heterocyclic dehydroabietylamine derivatives as potential antiproliferative and apoptosis-inducing agents. Drug Deliv. 2020, 27, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Puyang, X.; Jeremy Wu, Z.; Seiler, R.; Furman, C.; Oo, H.Z.; Seiler, M.; Irwin, S.; Subramanian, V.; Julie Joshi, J.; et al. Evasion of immunosurveillance by genomic alterations of PPARγ/RXRα in bladder cancer. Nat. Commun. 2017, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.J.; Lim, J.W.; Kim, H. Docosahexaenoic Acid Induces Expression of NAD(P)H: Quinone Oxidoreductase and Heme Oxygenase-1 through Activation of Nrf2 in Cerulein-Stimulated Pancreatic Acinar Cells. Antioxidants 2020, 9, 1084. [Google Scholar] [CrossRef] [PubMed]
- Bey, E.A.; Bentle, M.S.; Reinicke, K.E.; Dong, Y.; Yang, C.R.; Girard, L.; Minna, J.D.; Bornmann, W.G.; Gao, J.; Boothman, D.A. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc. Natl. Acad. Sci. USA 2007, 104, 11832–11837. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Li, L.S.; Spruell, C.; Xiao, L.; Chakrabarti, G.; Bey, E.A.; Reinicke, K.E.; Srougi, M.C.; Moore, Z.; Dong, Y.; et al. Tumor-selective, futile redox cycle-induced bystander effects elicited by NQO1 bioactivatable radiosensitizing drugs in triple-negative breast cancers. Antioxid. Redox Signal. 2014, 21, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Bey, E.A.; Li, L.S.; Kabbani, W.; Yan, J.; Xie, X.J.; Hsieh, J.T.; Gao, J.; Boothman, D.A. Prostate cancer radiosensitization through poly(ADP-Ribose) polymerase-1 hyperactivation. Cancer Res. 2010, 70, 8088–8096. [Google Scholar] [CrossRef]
- Huang, X.; Motea, E.A.; Moore, Z.R.; Yao, J.; Dong, Y.; Chakrabarti, G.; Kilgore, J.A.; Silvers, M.A.; Patidar, P.L.; Cholka, A.; et al. Leveraging an NQO1 Bioactivatable Drug for Tumor-Selective Use of Poly(ADP-ribose) Polymerase Inhibitors. Cancer Cell 2016, 30, 940–952. [Google Scholar] [CrossRef]
- Li, L.S.; Bey, E.A.; Dong, Y.; Meng, J.; Patra, B.; Yan, J.; Xie, X.J.; Brekken, R.A.; Barnett, C.C.; Bornmann, W.G.; et al. Modulating endogenous NQO1 levels identifies key regulatory mechanisms of action of β-lapachone for pancreatic cancer therapy. Clin. Cancer Res. 2011, 17, 275–285. [Google Scholar] [CrossRef]
- Kim, D.W.; Cho, J.Y. NQO1 is Required for β-Lapachone-Mediated Downregulation of Breast-Cancer Stem-Cell Activity. Int. J. Mol. Sci. 2018, 19, 3813. [Google Scholar] [CrossRef] [PubMed]
- Silvers, M.A.; Deja, S.; Singh, N.; Egnatchik, R.A.; Sudderth, J.; Luo, X.; Beg, M.S.; Burgess, S.C.; DeBerardinis, R.J.; Boothman, D.A.; et al. The NQO1 bioactivatable drug, β-lapachone, alters the redox state of NQO1+ pancreatic cancer cells, causing perturbation in central carbon metabolism. J. Biol. Chem. 2017, 292, 18203–18216. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, X.; Xu, M.; Piao, J.; Zhang, Y.; Lin, Z.; Chen, L. β-lapachone suppresses tumour progression by inhibiting epithelial-to-mesenchymal transition in NQO1-positive breast cancers. Sci. Rep. 2017, 7, 2681. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Huang, X.; Silvers, M.A.; Gerber, D.E.; Bolluyt, J.; Sarode, V.; Fattah, F.; Deberardinis, R.J.; Merritt, M.E.; Xie, X.J.; et al. Using a novel NQO1 bioactivatable drug, beta-lapachone (ARQ761), to enhance chemotherapeutic effects by metabolic modulation in pancreatic cancer. J. Surg. Oncol. 2017, 116, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, G.; Silvers, M.A.; Ilcheva, M.; Liu, Y.; Moore, Z.R.; Luo, X.; Gao, J.; Anderson, G.; Liu, L.; Sarode, V.; et al. Tumor-selective use of DNA base excision repair inhibition in pancreatic cancer using the NQO1 bioactivatable drug, β-lapachone. Sci. Rep. 2015, 5, 17066. [Google Scholar] [CrossRef] [PubMed]
- Bey, E.A.; Reinicke, K.E.; Srougi, M.C.; Varnes, M.; Anderson, V.E.; Pink, J.J.; Li, L.S.; Patel, M.; Cao, L.; Moore, Z.; et al. Catalase abrogates β-lapachone-induced PARP1 hyperactivation-directed programmed necrosis in NQO1-positive breast cancers. Mol. Cancer Ther. 2013, 12, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef]
- Huang, X.; Dong, Y.; Bey, E.A.; Kilgore, J.A.; Bair, J.S.; Li, L.S.; Patel, M.; Parkinson, E.I.; Wang, Y.; Williams, N.S.; et al. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis. Cancer Res. 2012, 72, 3038–3047. [Google Scholar] [CrossRef]
- Parkinson, E.I.; Hergenrother, P.J. Deoxynyboquinones as NQO1-Activated Cancer Therapeutics. Acc. Chem. Res. 2015, 48, 2715–2723. [Google Scholar] [CrossRef]
- Gerber, D.E.; Beg, M.S.; Fattah, F.; Frankel, A.E.; Fatunde, O.; Arriaga, Y.; Dowell, J.E.; Bisen, A.; Leff, R.D.; Meek, C.C.; et al. Phase 1 study of ARQ 761, a β-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. Br. J. Cancer 2018, 119, 928–936. [Google Scholar] [CrossRef]
- Su, X.; Wang, J.; Jiang, L.; Chen, Y.; Lu, T.; Mendonca, M.S.; Huang, X. PCNA inhibition enhances the cytotoxicity of β-lapachone in NQO1-Positive cancer cells by augmentation of oxidative stress-induced DNA damage. Cancer Lett. 2021, 519, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Motea, E.A.; Huang, X.; Singh, N.; Kilgore, J.A.; Williams, N.S.; Xie, X.J.; Gerber, D.E.; Beg, M.S.; Bey, E.A.; Boothman, D.A. NQO1-dependent, Tumor-selective Radiosensitization of Non-small Cell Lung Cancers. Clin. Cancer Res. 2019, 25, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- Li, L.S.; Reddy, S.; Lin, Z.H.; Liu, S.; Park, H.; Chun, S.G.; Bornmann, W.G.; Thibodeaux, J.; Yan, J.; Chakrabarti, G.; et al. NQO1-Mediated Tumor-Selective Lethality and Radiosensitization for Head and Neck Cancer. Mol. Cancer Ther. 2016, 15, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Shukla, K.; Singh, N.; Lewis, J.E.; Tsang, A.W.; Boothman, D.A.; Kemp, M.L.; Furdui, C.M. MTHFD2 Blockade Enhances the Efficacy of β-Lapachone Chemotherapy With Ionizing Radiation in Head and Neck Squamous Cell Cancer. Front. Oncol. 2020, 10, 536377. [Google Scholar] [CrossRef] [PubMed]
- Moore, Z.; Chakrabarti, G.; Luo, X.; Ali, A.; Hu, Z.; Fattah, F.J.; Vemireddy, R.; DeBerardinis, R.J.; Brekken, R.A.; Boothman, D.A. NAMPT inhibition sensitizes pancreatic adenocarcinoma cells to tumor-selective, PAR-independent metabolic catastrophe and cell death induced by β-lapachone. Cell Death Dis. 2015, 6, e1599. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Li, Q.R.; Cheng, X.F.; Wang, G.J.; Hao, H.P. NAMPT inhibition synergizes with NQO1-targeting agents in inducing apoptotic cell death in non-small cell lung cancer cells. Chin. J. Nat. Med. 2016, 14, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, E.I.; Bair, J.S.; Cismesia, M.; Hergenrother, P.J. Efficient NQO1 substrates are potent and selective anticancer agents. ACS Chem. Biol. 2013, 8, 2173–2183. [Google Scholar] [CrossRef]
- Motea, E.A.; Fattah, F.J.; Xiao, L.; Girard, L.; Rommel, A.; Morales, J.C.; Patidar, P.; Zhou, Y.; Porter, A.; Xie, Y.; et al. Kub5-Hera (RPRD1B) Deficiency Promotes “BRCAness” and Vulnerability to PARP Inhibition in BRCA-proficient Breast Cancers. Clin. Cancer Res. 2018, 24, 6459–6470. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Bentle, M.S.; Reinicke, K.E.; Bey, E.A.; Spitz, D.R.; Boothman, D.A. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J. Biol. Chem. 2006, 281, 33684–33696. [Google Scholar] [CrossRef]
- Lundberg, A.P.; Francis, J.M.; Pajak, M.; Parkinson, E.I.; Wycislo, K.L.; Rosol, T.J.; Brown, M.E.; London, C.A.; Dirikolu, L.; Hergenrother, P.J.; et al. Pharmacokinetics and derivation of an anticancer dosing regimen for the novel anti-cancer agent isobutyl-deoxynyboquinone (IB-DNQ), a NQO1 bioactivatable molecule, in the domestic felid species. Investig. New Drugs 2017, 35, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Nilsson, M.; Goldman, J.; Reck, M.; Nakagawa, K.; Kato, T.; Ares, L.P.; Frimodt-Moller, B.; Wolff, K.; Visseren-Grul, C.; et al. Dual EGFR-VEGF Pathway Inhibition: A Promising Strategy for Patients With EGFR-Mutant NSCLC. J. Thorac. Oncol. 2021, 16, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Danielson, L.; Ernster, L. Lack of relationship between mitochondrial oxidative phosphorylation and the dicoumarol-sensitive flavoenzyme ‘DT diaphorase’or ‘vitamin K reductase’. Nature 1962, 194, 155–157. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Jin, T.; Men, J.; Lin, Z.; Qi, P.; Piao, Y.; Yan, G. NQO1 protein expression predicts poor prognosis of non-small cell lung cancers. BMC Cancer 2015, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.; Wu, Q.; Cui, X.; Lin, Z.; Liu, S.; Chen, L. Clinical implications of high NQO1 expression in breast cancers. J. Exp. Clin. Cancer Res. 2014, 33, 14. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Wei, Y.; Jiang, T.; Wang, S. Correlation of Nrf2, NQO1, MRP1, cmyc and p53 in colorectal cancer and their relationships to clinicopathologic features and survival. Int. J. Clin. Exp. Pathol. 2014, 7, 1124–1131. [Google Scholar] [PubMed]
- Mendonca, M.S.; Howard, K.L.; Farrington, D.L.; Desmond, L.A.; Temples, T.M.; Mayhugh, B.M.; Pink, J.J.; Boothman, D.A. Delayed apoptotic responses associated with radiation-induced neoplastic transformation of human hybrid cells. Cancer Res. 1999, 59, 3972–3979. [Google Scholar] [PubMed]
- Blanco, E.; Bey, E.A.; Khemtong, C.; Yang, S.G.; Setti-Guthi, J.; Chen, H.; Kessinger, C.W.; Carnevale, K.A.; Bornmann, W.G.; Boothman, D.A.; et al. Beta-lapachone micellar nanotherapeutics for non-small cell lung cancer therapy. Cancer Res. 2010, 70, 3896–3904. [Google Scholar] [CrossRef]
- Dong, Y.; Chin, S.F.; Blanco, E.; Bey, E.A.; Kabbani, W.; Xie, X.J.; Bornmann, W.G.; Boothman, D.A.; Gao, J. Intratumoral delivery of beta-lapachone via polymer implants for prostate cancer therapy. Clin. Cancer Res. 2009, 15, 131–139. [Google Scholar] [CrossRef]
- Lundberg, A.P.; Boudreau, M.W.; Selting, K.A.; Chatkewitz, L.E.; Samuelson, J.; Francis, J.M.; Parkinson, E.I.; Barger, A.M.; Hergenrother, P.J.; Fan, T.M. Utilizing feline oral squamous cell carcinoma patients to develop NQO1-targeted therapy. Neoplasia 2021, 23, 811–822. [Google Scholar] [CrossRef]
- Ma, X.; Huang, X.; Moore, Z.; Huang, G.; Kilgore, J.A.; Wang, Y.; Hammer, S.; Williams, N.S.; Boothman, D.A.; Gao, J. Esterase-activatable beta-lapachone prodrug micelles for NQO1-targeted lung cancer therapy. J. Control. Release 2015, 200, 201–211. [Google Scholar] [CrossRef]
- Ma, X.; Huang, X.; Huang, G.; Li, L.; Wang, Y.; Luo, X.; Boothman, D.A.; Gao, J. Prodrug strategy to achieve lyophilizable, high drug loading micelle formulations through diester derivatives of beta-Lapachone. Adv. Healthc. Mater. 2014, 3, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Cortazzo, J.A.; Lichtman, A.D. Methemoglobinemia: A review and recommendations for management. J. Cardiothorac. Vasc. Anesth. 2014, 28, 1043–1047. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Su, X.; Jiang, L.; Boudreau, M.W.; Chatkewitz, L.E.; Kilgore, J.A.; Zahid, K.R.; Williams, N.S.; Chen, Y.; Liu, S.; et al. Augmented Concentration of Isopentyl-Deoxynyboquinone in Tumors Selectively Kills NAD(P)H Quinone Oxidoreductase 1-Positive Cancer Cells through Programmed Necrotic and Apoptotic Mechanisms. Cancers 2023, 15, 5844. https://doi.org/10.3390/cancers15245844
Wang J, Su X, Jiang L, Boudreau MW, Chatkewitz LE, Kilgore JA, Zahid KR, Williams NS, Chen Y, Liu S, et al. Augmented Concentration of Isopentyl-Deoxynyboquinone in Tumors Selectively Kills NAD(P)H Quinone Oxidoreductase 1-Positive Cancer Cells through Programmed Necrotic and Apoptotic Mechanisms. Cancers. 2023; 15(24):5844. https://doi.org/10.3390/cancers15245844
Chicago/Turabian StyleWang, Jiangwei, Xiaolin Su, Lingxiang Jiang, Matthew W. Boudreau, Lindsay E. Chatkewitz, Jessica A. Kilgore, Kashif Rafiq Zahid, Noelle S. Williams, Yaomin Chen, Shaohui Liu, and et al. 2023. "Augmented Concentration of Isopentyl-Deoxynyboquinone in Tumors Selectively Kills NAD(P)H Quinone Oxidoreductase 1-Positive Cancer Cells through Programmed Necrotic and Apoptotic Mechanisms" Cancers 15, no. 24: 5844. https://doi.org/10.3390/cancers15245844
APA StyleWang, J., Su, X., Jiang, L., Boudreau, M. W., Chatkewitz, L. E., Kilgore, J. A., Zahid, K. R., Williams, N. S., Chen, Y., Liu, S., Hergenrother, P. J., & Huang, X. (2023). Augmented Concentration of Isopentyl-Deoxynyboquinone in Tumors Selectively Kills NAD(P)H Quinone Oxidoreductase 1-Positive Cancer Cells through Programmed Necrotic and Apoptotic Mechanisms. Cancers, 15(24), 5844. https://doi.org/10.3390/cancers15245844