

Stem Cell Theory of Cancer: Clinical Implications of Epigenomic versus Genomic Biomarkers in Cancer Care

 ,
, 
Abstract
:Simple Summary
Abstract
A good decision is based on knowledge not numbers.Plato
1. Introduction
2. Brief History
3. Origin of Cancer
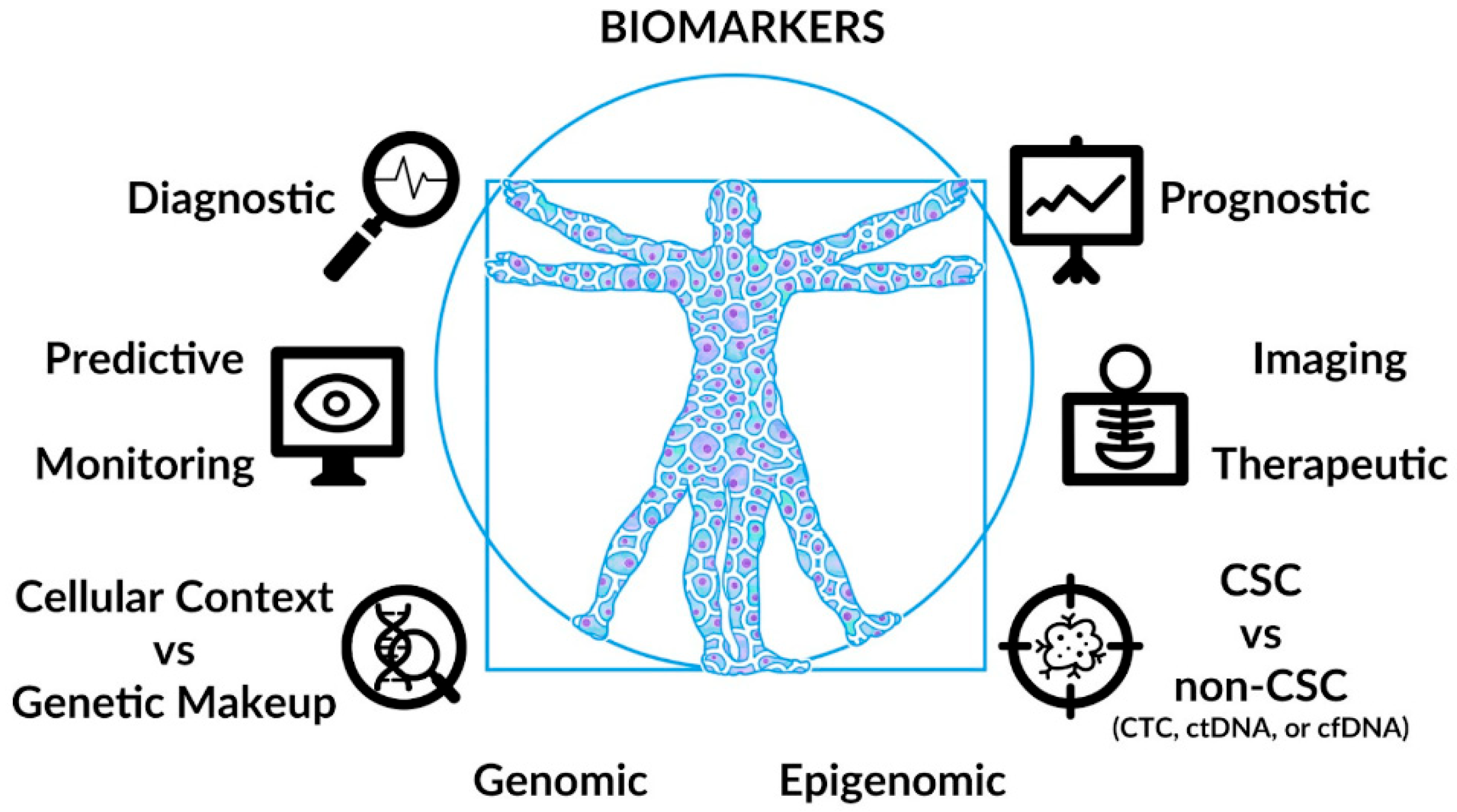
4. Diagnostic/Prognostic Biomarkers
4.1. Primary Tumors
4.2. Mixed Tumors
4.2.1. Testicular Cancer
4.2.2. Breast Cancer
4.2.3. Lung Cancer
4.2.4. Osteosarcoma
4.2.5. Additional Solid Tumors
4.3. Tumor Subtypes
4.3.1. Breast Cancer
4.3.2. Prostate Cancer
4.3.3. Bladder Cancer
4.3.4. Kidney Cancer
5. Predictive/Therapeutic Biomarkers
5.1. Monitoring
5.2. Imaging
5.3. Therapeutics
5.3.1. MiRNA
5.3.2. HDACi
6. Clinical Implications
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bence Jones, H. Papers on chemical pathology. Lancet 1847, 50, 88–92. [Google Scholar] [CrossRef]
- Kamel, H.F.M.; Al-Amodi, H.S.B. Cancer Biomarkers in Role of Biomarkers in Medicine; Wang, M., Witzmann, F.A., Eds.; IntechOpen: London, UK, 2006. [Google Scholar] [CrossRef]
- Gold, P.; Freedman, S.O. Demonstration of tumor-specific antigens in human colonic carcinomata by immunological tolerance and absorption techniques. J. Exp. Med. 1965, 121, 439–462. [Google Scholar] [CrossRef]
- Masopust, J.; Kithier, K.; Radl, J.; Koutecky, J.; Kotal, L. Occurrence of fetoprotein in patients with neoplasms and non-neoplastic diseases. Int. J. Cancer 1968, 3, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Papsidero, L.D.; Wang, M.C.; Valenzuela, L.A.; Murphy, G.P.; Chu, T.M. A prostate antigen in sera of prostatic cancer patients. Cancer Res. 1980, 40, 2428–2432. [Google Scholar] [PubMed]
- Bast, R.C., Jr.; Feeney, M.; Lazarus, H.; Nadler, L.M.; Colvin, R.B.; Knapp, R.C. Reactivity of a monoclonal antibody with human ovarian carcinoma. J. Clin. Investig. 1981, 68, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M. Origin of Cancers: Clinical Perspectives and Implications of a Stem-Cell Theory of Cancer. In Cancer Treatment and Research; Rosen, S.T., Ed.; Springer: New York, NY, USA, 2010; Volume 154. [Google Scholar]
- Tu, S.M. Story of Hydra: Portrait of Cancer as a Stem-Cell Disease; Nova Medicine & Health: New York, NY, USA, 2019; pp. 43–53. [Google Scholar]
- Tu, S.M.; Bilen, M.A.; Tannir, N.M. Pillar and pitfall of cancer research. Cancer Med. 2014, 3, 1035–1037. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Tu, S.M.; Singh, S.; Arnaoutakis, K.; Malapati, S.; Bhatti, S.A.; Joon, A.Y.; Atiq, O.T.; Posters, L.L. Stem cell theory of cancer: Implications for translational research from bedside to bench. Cancers 2022, 14, 3345. [Google Scholar] [CrossRef]
- Levine, A.J.; Jenkins, N.A.; Copeland, N.G. The roles of initiating truncal mutations in human cancers: The order of mutations and tumor cell type matters. Cancer Cell 2019, 35, 10–15. [Google Scholar] [CrossRef]
- Vezzoni, L.; Parmiani, G. Limitations of the cancer stem cell theory. Cytotechnology 2008, 58, 3–9. [Google Scholar] [CrossRef]
- Bartram, I.; Jeschke, J.M. Do cancer stem cells exist? A pilot study combining a systematic review with the hierarchy-of-hypotheses approach. PLoS ONE 2019, 14, e0225898. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, V.; Kulkarni, Y.; Felix, K.; Azad, N.; Iyer, A.K.V.; Yakisich, J.S. Alternative models of cancer stem cells: The Stemness phenotype model, 10 years later. World J. Stem Cells 2021, 13, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.H.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.L.; Hognas, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Bennett, H.M.; Stephenson, W.; Rose, C.M.; Darmanis, S. Single-cell proteomics enabled by next-generation sequencing or mass spectrometry. Nat. Methods 2023, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Lim, A.; Odegaard, J.I.; Honeycutt, J.D.; Kawano, S.; Hsieh, M.H.; Beachy, P.A. Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nat. Cell Biol. 2014, 16, 469–478. [Google Scholar] [CrossRef]
- Tu, S.M.; Lin, S.H.; Logothetis, C.J. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol. 2002, 3, 508–513. [Google Scholar] [CrossRef]
- Tu, S.M.; Bilen, M.A.; Hess, K.R.; Broaddus, R.R.; Kopetz, S.; Wei, C.; Pagliaro, L.C.; Karam, J.A.; Ward, J.F.; Wood, C.G.; et al. Intratumoral heterogeneity: Role of differentiation in a potentially lethal phenotype of testicular cancer. Cancer 2016, 122, 1836–1843. [Google Scholar] [CrossRef]
- Tu, S.M.; Campbell, M.; Shah, A.; Logothetis, C.J. Application of a successful germ cell tumor paradigm to the challenges of common adult solid cancers. J. Cell Sci. Ther. 2021, 12, 301. [Google Scholar]
- McCart-Reed, A.E.; Kutasovic, J.; Nones, K.; Saunus, J.M.; Da Silva, L.; Newell, F.; Kazakoff, S.; Melville, L.; Jayanthan, J.; Vargas, A.C.; et al. Mixed ductal-lobular carcinomas: Evidence for progression from ductal to lobular morphology. J. Pathol. 2018, 244, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; McCutcheon, J.N.; Kallakury, B.; Chahine, J.J.; Pratt, D.; Raffeld, M.; Chen, Y.; Wang, C.; Giaccone, G. Combined small cell carcinoma of the lung: Is it a single entity? J. Thorac. Oncol. 2017, 13, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.; Zaccaria, S.; Cannon, M.V.; Cam, M.; Gross, A.C.; Raphael, B.J.; Roberts, R.D. Structural complex osteosarcoma genomes exhibit limited heterogeneity within individual tumors and across evolutionary time. Cancer Res. Commun. 2023, 3, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.; Chang, B.; Barsky, S.H. Monoclonal origins of malignant mixed tumors (carcinosarcomas). Evidence for a divergent histogenesis. Am. J. Surg. Pathol. 1996, 20, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Boerman, R.; Anderl, K.; Herath, J.; Borell, T.; Johnson, N.; SchaefferKlein, J.; Kirchof, A.; Raap, A.K.; Scheithauer, B.W.; Jenkins, R.B. The glial and mesenchymal elements of gliosarcomas share similar genetic alterations. J. Neuropathol. Exp. Neurol. 1996, 55, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Actor, B.; Cobbers, J.M.; Buschges, R.; Wolter, M.; Knobbe, C.B.; Lichter, P.; Reifenberger, G.; Weber, R.G. Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes. Chromosomes Cancer 2002, 34, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Zhang, S.; Lopez-Beltran, A.; Shah, R.B.; Montironi, R.; Tan, P.H.; Wang, M.; Baldridge, L.A.; MacLennan, G.T.; Cheng, L. Lymphoepithelioma-like carcinoma of the urinary bladder: Clinicopathologic, immunohistochemical, and molecular features. Am. J. Surg. Pathol. 2011, 35, 474–483. [Google Scholar] [CrossRef]
- Verbiest, A.; Van Hoef, V.; Rodriguez-Antona, C.; Gracia-Donas, J.; Grana-Castro, O.; Albersen, M.; Baldewijns, M.; Laenen, A.; Roussel, E.; Schoffski, P.; et al. MicroRNA expression profiles in molecular subtypes of clear-cell renal cell carcinoma are associated with clinical outcome and repression of specific mRNA targets. PLoS ONE 2020, 15, e0238809. [Google Scholar] [CrossRef]
- Voss, M.H.; Kuo, F.; Chen, D.; Marker, M.; Patel, P.; Redzematovic, A.; Riaz, N.; Chan, T.A.; Choueiri, T.K.; Hsieh, J.J.D.; et al. Integrated biomarker analysis for 412 renal cell cancer patients treated on the phase 3 COMPARZ trial: Correlating common mutation events in PBRM1 and BAP1 with angiogenesis expression signatures and outcomes on tyrosine kinase inhibitor therapy. J. Clin. Oncol. 2017, 35, 4523. [Google Scholar] [CrossRef]
- Conway, E.; Rossi, F.; Fernandez-Perez, D.; Ponzo, E.; Ferrari, K.J.; Zanotti, M.; Manganaro, D.; Rodighierio, S.; Tamburri, S.; Pasini, D. BAP1 enhances Polycomb repression by counteracting widespread H2AK119ub1 deposition and chromatin condensation. Mol. Cell 2021, 81, 3526–3541. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc.Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Wicha, M.S. Breast cancer stem cells: We have got them surrounded. Clin. Cancer Res. 2013, 19, 511–513. [Google Scholar] [CrossRef] [PubMed]
- McDougall, A.R.A.; Tolcos, M.; Hooper, S.B.; Cole, T.J.; Wallace, M.J. Trop2: From development to disease. Dev. Dyn. 2015, 244, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- de Wit, S.; van Dalum, G.; Lenferink, A.T.M.; Tibbe, A.G.J.; Hiltermann, T.J.N.; Groen, H.J.M.; van Rijn, C.J.M.; Terstappen, L.W.M.M. The detection of EpCAM+ and EpCAM− circulating tumor cells. Sci. Rep. 2015, 5, 12270. [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, A.; Leversha, M.A.; Satagopan, J.M.; Zhou, Q.; Al-Ahmadie, H.A.; Fine, S.W.; Eastham, J.A.; Scardino, P.T.; Scher, H.I.; Tickoo, S.K.; et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res. 2009, 69, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, A.; Graff, R.E.; Bauer, S.R.; Pitt, M.J.; Lis, R.T.; Stack, E.C.; Martin, N.E.; Kunz, L.; Penney, K.L.; Ligon, A.H.; et al. The TMPRSS2-ERG rearrangement, ERG expression, and prostate cancer outcome: A cohort study and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1497–1509. [Google Scholar] [CrossRef]
- Van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; da Costa, R.M.G.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 130, 4492–4505. [Google Scholar] [CrossRef]
- Tang, F.; Xu, D.; Wang, S.; Wong, C.K.; Martinez-Fundichely, A.; Lee, C.J.; Cohen, S.; Park, J.; Hill, C.E.; Eng, K.; et al. Chromatin profiles classify castration-resistant prostate cancers suggesting therapeutic targets. Science 2022, 376, eabe1505. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2018, 174, 1033. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, A.; de Reynies, A.; Allory, Y.; Sjodahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. The consensus molecular classification of muscle-invasive bladder cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Mejean, A.; Ravaud, A.; Thezenas, S.; Colas, S.; Beauval, J.B.; Bensalah, K.; Geoffrois, L.; Thiery-Vuillemin, A.; Cormier, L.; Lang, H.; et al. Sunitinib alone or after nephrectomy in metastatic renal-cell carcinoma. N. Engl. J. Med. 2018, 379, 417–427. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.G.; Umbreit, E.C.; Holland, L.C.; Gu, C.; Tannir, N.M.; Matin, S.F.; Karam, J.A.; Culp, S.H.; Wood, C.G. Optimizing patient selection for cytoreductive nephrectomy based on outcomes in the contemporary era of systemic therapy. Cancer 2020, 126, 3950–3960. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nichol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell 2018, 173, 581–594. [Google Scholar] [CrossRef]
- Barata, P.C.; Koshkin, V.S.; Funchain, P.; Sohal, D.; Pritchard, A.; Klek, S.; Adamowicz, T.; Gopalakrishnan, D.; Garcia, J.; Rini, B.; et al. Next-generation sequencing (NGS) of cell-free circulating tumor DNA and tumor tissue in patients with advanced urothelial cancer: A pilot assessment of concordance. Ann. Oncol. 2017, 28, 2458–2463. [Google Scholar] [CrossRef]
- Chae, Y.K.; Davis, A.A.; Jain, S.; Santa-Maria, C.; Flaum, L.; Beaubier, N.; Platanias, L.C.; Gradishar, W.; Giles, F.J.; Cristofanilli, M. Concordance of genomic alterations by next-generation sequencing in tumor tissue versus circulating tumor DNA in breast cancer. Mol. Cancer Ther. 2017, 16, 1412–1420. [Google Scholar] [CrossRef]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat. Commun. 2018, 9, 1691. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kalyan, A.; Taxter, T.J.; Wehbe, F. Concordance of mutations identified using circulating tumor DNA (ctDNA) compared with tissue based next generation sequencing (NGS) in gastrointestinal malignancies: A single institution experience. J. Clin. Oncol. 2017, 35 (Suppl. S15), e23023. [Google Scholar] [CrossRef]
- Hahn, A.W.; Nussenzveig, R.H.; Pal, S.K.; Agarwal, N. Blood- and tissue-based tumor genomics: A battle royale or match made in heaven? Ann. Oncol. 2017, 28, 2333–2335. [Google Scholar] [CrossRef]
- Mo, S.; Ye, L.; Wang, D.; Han, L.; Zhou, S.; Wang, H.; Dai, W.; Wang, Y.; Luo, W.; Wang, R.; et al. Early detection of molecular residual disease and risk stratification for stage I to III colorectal cancer via circulating tumor DNA methylation. JAMA Oncol. 2023, 9, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Schuster, T.; Geiger, H. Septins in stem cells. Front. Cell Dev. Biol. 2021, 9, 801507. [Google Scholar] [CrossRef] [PubMed]
- Francois, L.; Boskovic, P.; Knerr, J.; He, W.; Sigismondo, G.; Schwan, C.; More, T.H.; Schlotter, M.; Conway, M.E.; Krijgsveld, J.; et al. BCAT1 redox function maintains mitotic fidelity. Cell Rep. 2022, 41, 111524. [Google Scholar] [CrossRef]
- Chen, S.; Chen, B.; Su, G.; Chen, J.; Guo, D.; Yin, Q.; Wang, W.; Zhao, Z.; Zhang, L.; Shi, J.; et al. Branched-chain amino acid aminotransferase-1 regulates self-renewal and pluripotency of mouse embryonic stem cells through Ras signaling. Stem Cell Res. 2020, 49, 102097. [Google Scholar] [CrossRef]
- Li, Z.; Li, Z.P.; Li, R.Y.; Zhu, H.; Liu, X.; Guo, X.L.; Mu, L.L.; Cai, J.J.; Bai, F.; Chen, G.Q.; et al. Leukaemic alterations of IKZF1 primes stemness and malignancy prograns in human lymphocytes. Cell Death Dis. 2018, 9, 526. [Google Scholar] [CrossRef]
- Miao, R.; Huang, D.; Zhao, K.; Li, Y.; Zhang, X.; Cheng, Y.; Guo, N. VAV3 regulates glioblastoma cell proliferation, migration, invasion and cancer stem-like cell self-renewal. Mol. Med. Rep. 2023, 27, 94. [Google Scholar] [CrossRef]
- Han, D.; Choi, M.R.; Jung, K.H.; Kim, N.; Kim, S.K.; Chai, J.C.; Lee, Y.S.; Chai, Y.G. Global transcriptome profiling of genes that are differentially regulated during differentiation of mouse embryonic neural stem cells into astrocytes. J. Mol. Neurosci. 2015, 55, 109–125. [Google Scholar] [CrossRef]
- Taylor, M.S.; Wu, C.; Fridy, P.C.; Zhang, S.J.; Senussi, Y.; Wolters, J.C.; Cajuso, T.; Cheng, W.C.; Heaps, J.D.; Miller, B.D.; et al. Ultrasensitive detection of circulating LINE-1 ORF1p as a specific multi-cancer biomarker. Cancer Discov. 2023. [Google Scholar] [CrossRef]
- MacLennan, M.; Garcia-Canadas, M.; Reichmann, J.; Khazina, E.; Wagner, G.; Playfoot, C.J.; Salvador-Palomeque, C.; Mann, A.R.; Preressini, P.; Sanchez, L.; et al. Mobilization of LINE-1 retrotransposons is restricted by Tex19.1 in mouse embryonic stem cells. eLife 2017, 6, e26152. [Google Scholar] [CrossRef] [PubMed]
- Walker-Samuel, S.; Ramasawmy, R.; Torrealdea, F.; Rega, M.; Rajkumar, V.; Johnson, S.P.; Richardson, S.; Goncalves, M.; Parkes, H.G.; Arstad, E.; et al. In vivo imaging of glucose uptake and metabolism in tumors. Nat. Med. 2013, 19, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Kang, J.; Kim, T.I.; Joo, C.G. MRI assessment of glutamine uptake correlates with the distribution of glutamine transporters and cancer stem cell markers. Sci. Rep. 2022, 12, 5511. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. P32 (C1QBP) and cancer cell metabolism: Is the Warburg effect a lot of hot air? Mol. Cell. Biol. 2010, 30, 1300–1302. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Balakrishnan, A.; Bok, R.A.; Anderton, B.; Larson, P.E.Z.; Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Goga, A. 13C-pyruvate imaging reveals alterations in glycolysis that precede c-myc-induced tumor formation and regression. Cell Metab. 2011, 14, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Gjyrezi, A.; Galletti, G.; Zhang, J.; Bareja, R.; Halima, A.; Worroll, D.; Nanus, D.; Tagawa, S.; Beltran, H.; Giannakakou, P. Transferrin receptor identifies a distinct pool of circulating tumor cells from metastatic prostate cancer patients with unique molecular profiles enriched in AR variants. Cancer Res. 2019, 79 (Suppl. S13), 451. [Google Scholar] [CrossRef]
- De Kouchkovsky, I.; Behr, S.; Zhang, L.; Foye, A.; Vanbrocklin, H.; Small, E.J.; Feng, F.Y.; Friedlander, T.W.; Li, P.; Frey, N.; et al. Feasibility study of gallium-68 citrate PET as a bone-tropic imaging biomarker in, mCRPC. J. Clin. Oncol. 2021, 39 (Suppl. S6), 31. [Google Scholar] [CrossRef]
- Lambert, L.A.; Mitchell, S.L. Molecular evolution of the transferrin receptor/glutamate carboxypeptidase II family. J. Mol. Evol. 2007, 64, 113–128. [Google Scholar] [CrossRef]
- Toloudi, M.; Apostolou, P.; Chatziioannou, M.; Kourtidou, E.; Vlachou, I.; Mimikakou, G.; Papasotiriou, I. How prostate-specific membrane antigen level may be correlated with stemness in prostate cancer stem cell-like cell populations? J. Cancer Res. Ther. 2014, 10, 133–141. [Google Scholar]
- Kasimir-Bauer, S.; Keup, C.; Hoffmann, O.; Hauch, S.; Kimmig, R.; Bittner, A.K. Circulating tumor cells expressing the prostate specific membrane antigen indicate worse outcome in primary, non-metastatic triple-negative breast cancer. Front. Oncol. 2020, 10, 1658. [Google Scholar] [CrossRef]
- Calais, J.; Czermin, J.; Fendler, W.P.; Elashoff, D.; Nickols, N.G. Randomized prospective phase III trial of 68Ga-PSMA-11 PET/CT molecular imaging for prostate cancer salvage radiotherapy planning (PSMA-SRT). BMC Cancer 2019, 19, 18. [Google Scholar]
- Hope, T.A.; Eiber, M.; Armstrong, W.R.; Juarez, R.; Murthy, V.; Lawhn-Heath, C.; Behr, S.C.; Zhang, L.; Barbato, F.; Ceci, F.; et al. Diagnostic accuracy of 68Ga-PSMA-11 PET for pelvic nodal metastasis detection prior to radical prostatectomy and pelvic lymph node dissection: A multicenter prospective phase 3 imaging trial. JAMA Onol. 2021, 7, 1635–1642. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Abba, M.; Novak, D.; Moniuszko, M.; Allgayer, H. Function and significance of MicroRNAs in benign and malignant human stem cells. Semin. Cancer Biol. 2015, 35, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Filipow, S.; Laczmanski, L. Blood circulating miRNAs as cancer biomarkers for diagnosis and surgical treatment response. Front. Genet. 2019, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.D.; Ross, S.A. Evidence for dietary regulation of microRNA expression in cancer cells. Nutr. Rev. 2008, 66, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Misso, G.; Di Martino, M.T.; De Rosa, G.; Farooqi, A.A.; Lombardi, A.; Campani, V.; Zarone, M.R.; Gulla, A.; Tagliaferri, P.; Tassone, P.; et al. MiR-34: A new weapon against cancer? Mol. Ther. Nucleic Acids 2014, 3, e194. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.S.; Davidson, L.A.; Chapkin, R.S. Mechanistic insights into the role of microRNAs in cancer: Influence of nutrient crosstalk. Front. Genet. 2012, 3, 305. [Google Scholar] [CrossRef]
- Bao, B.; Ahmad, A.; Kong, D.; Ali, S.; Azmi, A.S.; Li, Y.; Banerjee, S.; Padhye, S.; Sarkar, F.H. Hypoxia induced aggressiveness of prostate cancer cells is linked with deregulated expression of VEGF, IL-6, and miRNAs that are attenuated by CDF. PLoS ONE 2012, 7, e43726. [Google Scholar] [CrossRef]
- Li, Y.; VandenBoom, I.I.T.G.; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and Let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar] [CrossRef] [PubMed]
- Chiyomaru, T.; Yamamura, S.; Fukuhara, S.; Yoshino, H.; Kinoshita, T.; Majid, S.; Saini, S.; Chang, I.; Tanaka, Y.; Enokida, H.; et al. Genistein inhibits prostate cancer cell growth by targeting miR-34a and oncogenic HOTAIR. PLoS ONE 2013, 8, e70372. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Ai, W.; Banik, N.L.; Ray, S.K. Overexpression of miR-7-1 increases efficacy of green tea polyphenols for induction of apoptosis in human malignant neuroblastoma SH-SY5Y and SK-N-DZ cells. Neurochem. Res. 2013, 38, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.S.; Schwartz, S.L.; Zhao, C.; Davidson, L.A.; Zhou, B.; Lupton, J.R.; Ivanov, I.; Chapkin, R.S. Integrated microRNA and mRNA expression profiling in a rat colon carcinogenesis model: Effect of a chemo-protective diet. Physiol. Genom. 2011, 43, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2016, 36, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Ho, E.; Williams, D.E.; Dashwood, R.H. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin. Epigenet. 2011, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Williams, M.; Sharma, H.; Chaudry, A.; Bellamy, P. A double-blind, placebo-controlled randomized trial evaluating the effect of a polyphenol-rich whole food supplement on PSA progression in men with prostate cancer—The UK NCRN Pomi-T study. Prostate Cancer Prostatic Dis. 2014, 17, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Capodice, J.L.; Gorroochurn, P.; Cammack, A.S.; Goluboff, E.; McKiernan, J.M.; Benson, M.C.; Stone, B.A.; Katz, A.E. Zyflamend in men with high-grade prostatic intraepithelial neoplasia: Results of a phase I clinical trial. J. Soc. Integr. Oncol. 2009, 7, 43–51. [Google Scholar]
- Park, W.; Amin, A.R.M.R.; Chen, Z.G.; Shin, D.M. New perspectives of curcumin in cancer prevention. Cancer Prev. Res. 2013, 6, 387–400. [Google Scholar] [CrossRef]
- Hartojo, W.; Silvers, A.L.; Thomas, D.G.; Seder, C.W.; Lin, L.; Rao, H.; Wang, Z.; Greenson, J.K.; Giordano, T.J.; Orringer, M.B.; et al. Curcumin promotes apoptosis, increases chemosensitivity, and inhibits nuclear factor kappaB in esophageal adenocarcinoma. Transl. Oncol. 2010, 3, 99–108. [Google Scholar] [CrossRef]
- Sandur, S.K.; Deorukhkar, A.; Pandey, M.K.; Pabon, A.M.; Shentu, S.; Guha, S.; Aggarwal, B.B.; Krishnan, S. Curcumin modulates the radiosensitivity of colorectal cancer cells by suppressing constitutive and inducible NF-kB activity. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Kim, C.G.; Lim, Y.; Shin, S.Y.; Lee, Y.H. Curcumin down-regulates the multidrug-resistance mdr1b gene by inhibiting the PI3K/Akt/NF kappa B pathway. Cancer Lett. 2007, 259, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Chearwae, W.; Shukla, S.; Limtrakul, P.; Ambudkar, S.V. Modulation of the function of the multidrug resistance-linked ATP-binding cassette transporter ABCG2 by the cancer preventive agent curcumin. Mol. Cancer Ther. 2006, 5, 1995–2006. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Kanwar, S.S.; Patel, B.B.; Nautiyal, J.; Sarkar, F.H.; Majumdar, A.P.N. Elimination of cancer stem-like cells by the combination of curcumin and FOLFOX. Transl. Oncol. 2009, 2, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.; Yeh, A.; Naftalovich, R.; Vhoi, T.H.; Chan, M.M. Curcumin inhibits the side population phenotype of the rat glioma cell line: Towards targeting of cancer stem cells with phytochemicals. Cancer Lett. 2010, 293, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Liu, Y.; Li, H.; Li, P.K.; Fuchs, J.; Shibata, H.; Iwabuchi, Y.; Lin, J. Targeting colon cancer stem cells using a new curcumin analogue, GO-Y030. Br. J. Cancer 2011, 105, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Straub, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer stem cells—Origins and biomarkers: Perspectives for targeted personalized therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Saito, S.; Ku, C.C.; Wuputra, K.; Pan, J.B.; Lin, C.S.; Lin, Y.C.; Wu, D.C.; Yokoyama, K.K. Biomarkers of cancer stem cells for experimental research and clinical application. J. Pers. Med. 2022, 12, 715. [Google Scholar] [CrossRef]
- Bilen, M.A.; Lin, S.H.; Tang, D.G.; Parikh, K.; Lee, M.H.; Yeung, S.C.J.; Tu, S.M. Maintenance therapy containing metformin and/or zyflamend for advanced prostate cancer: A case series. Case Rep. Oncol. Med. 2015, 2015, 471861. [Google Scholar] [CrossRef]
- Tu, S.M.; Aydin, A.M.; Maraboyina, S.; Chen, Z.; Singh, S.; Gokden, N.; Langford, T. Stem cell origin of cancer: Implications of oncogenesis recapitulating embryogenesis in cancer care. Cancers 2023, 15, 2516. [Google Scholar] [CrossRef] [PubMed]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A patient-derived, pan-cancer EMT signature identifies global molecular alterations and immune target enrichment following epithelial-to-mesenchymal transition. Clin. Cancer Res. 2015, 22, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development, and treatment: Beyond immune evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.M.; Xia, W.; Hsu, Y.H.; Chan, L.C.; Yu, W.H.; Cha, J.H.; Chen, C.T.; Liao, H.W.; Kuo, C.W.; Khoo, K.H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [PubMed]
- Siddarth, S.; Goutam, K.; Das, S.; Nayak, A.; Nayak, D.; Sethy, C.; Wyatt, M.D.; Kundu, C.N. Nectin-4 is a breast cancer stem cell marker that induces WNT/beta-catenin signaling via Pi3k/Akt axis. Int. J. Biochem. Cell Biol. 2017, 89, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Hoimes, C.J.; Flaig, T.W.; Milowsky, M.I.; Friedlander, T.W.; Bilen, M.A.; Gupta, S.; Srinivas, S.; Merchan, J.R.; McKay, R.R.; Petrylak, D.P.; et al. Enfortumab vedotin plus pembrolizumab in previously untreated advanced urothelial cancer. J. Clin. Oncol. 2023, 41, 22–31. [Google Scholar] [CrossRef]

{kind=link}
| Subtypes (Incidence) | Biology | Treatment |
|---|---|---|
| Luminal-papillary (35%) | FGFR alterations | Low response to NAC |
| Luminal-infiltrated (19%) | EMT, PD-L1 | CPI, resistance to NAC |
| Luminal (6%) | ||
| Basal-squamous (35%) | PD-L1 | CPI, NAC |
| Neuronal (5%) | NAC (EP) |
| ITH | SCNA | |
|---|---|---|
| LOW | HIGH | No surgery, may not respond to CPI or TKI |
| Increased metastatic potential | ||
| HIGH | May benefit from surgery + CPI (PDL1+) | |
| Delayed metastatic potential | ||
| LOW | LOW | Benefit from surgery + TKI (angiogenesis+) |
| Decreased metastatic potential |
| ccrcc | 1 | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| PD | 22% | 3% | 0% | 27% | |
| PR/CR | 41% | 53% | 70% | 21% | p = 0.005 |
| PFS (mos) | 13 | 19 | 24 | 8 | p = 0.0003 |
| OS (mos) | 24 | 35 | 50 | 14 | p = 0.0002 |
| ccrcc4 and ccrcc1 = ITH LOW, SCNA HIGH: No surgery | |||||
| ccrcc3 = ITH LOW, SCNA LOW: Benefit from surgery | |||||
| ccrcc2 = ITH HIGH: benefit from surgery + TKI/CPI | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, S.-M.; Chen, J.Z.; Singh, S.R.; Aydin, A.M.; Gokden, N.; Tam, N.N.C.; Leung, Y.-K.; Langford, T.; Ho, S.-M. Stem Cell Theory of Cancer: Clinical Implications of Epigenomic versus Genomic Biomarkers in Cancer Care. Cancers 2023, 15, 5533. https://doi.org/10.3390/cancers15235533
Tu S-M, Chen JZ, Singh SR, Aydin AM, Gokden N, Tam NNC, Leung Y-K, Langford T, Ho S-M. Stem Cell Theory of Cancer: Clinical Implications of Epigenomic versus Genomic Biomarkers in Cancer Care. Cancers. 2023; 15(23):5533. https://doi.org/10.3390/cancers15235533
Chicago/Turabian StyleTu, Shi-Ming, Jim Zhongning Chen, Sunny R. Singh, Ahmet Murat Aydin, Neriman Gokden, Neville Ngai Chung Tam, Yuet-Kin Leung, Timothy Langford, and Shuk-Mei Ho. 2023. "Stem Cell Theory of Cancer: Clinical Implications of Epigenomic versus Genomic Biomarkers in Cancer Care" Cancers 15, no. 23: 5533. https://doi.org/10.3390/cancers15235533
APA StyleTu, S.-M., Chen, J. Z., Singh, S. R., Aydin, A. M., Gokden, N., Tam, N. N. C., Leung, Y.-K., Langford, T., & Ho, S.-M. (2023). Stem Cell Theory of Cancer: Clinical Implications of Epigenomic versus Genomic Biomarkers in Cancer Care. Cancers, 15(23), 5533. https://doi.org/10.3390/cancers15235533