Thinking (Metastasis) outside the (Primary Tumor) Box

 , , , , and
, , , , and 
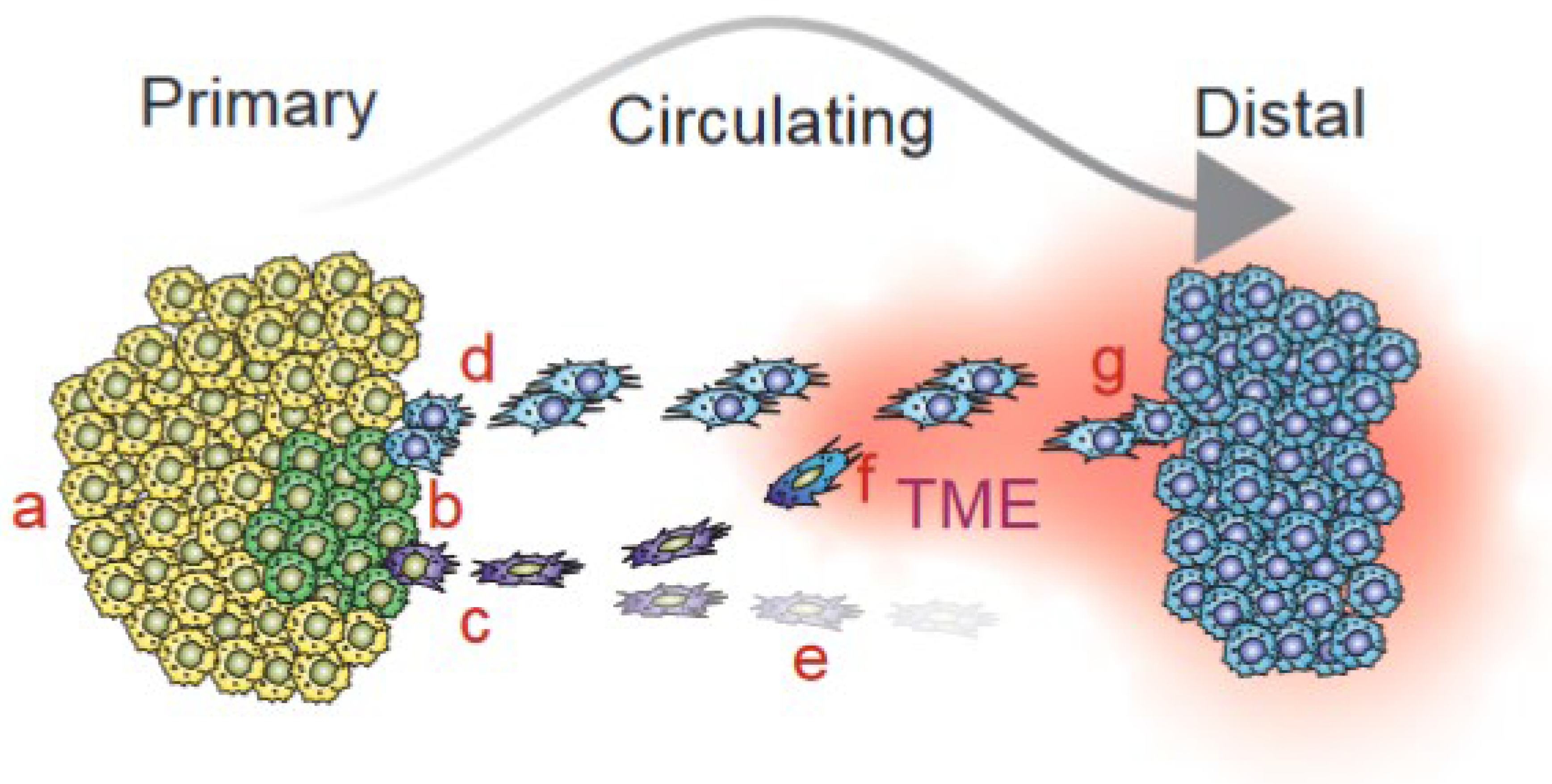
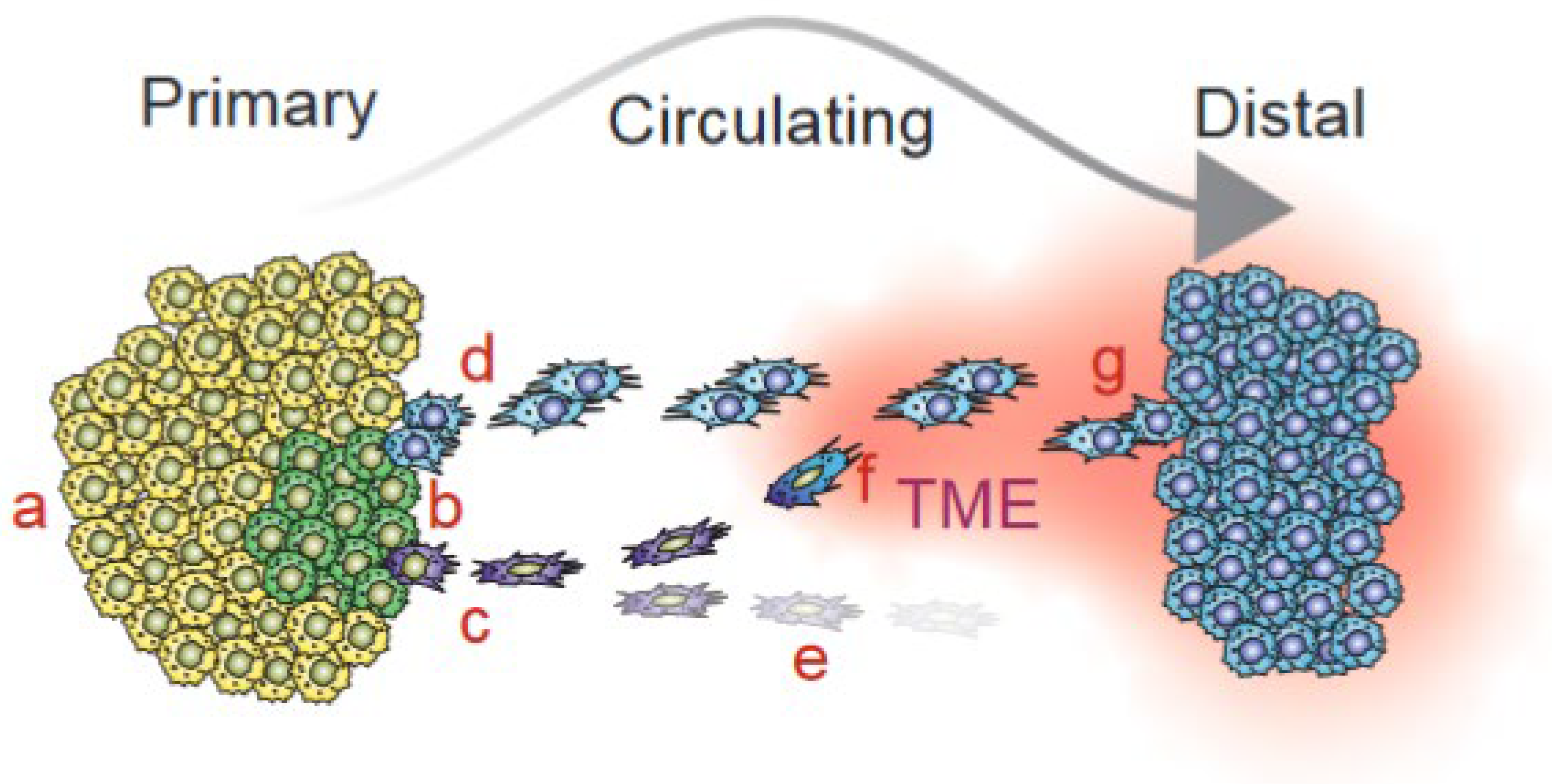{kind=link}
Acknowledgments
Conflicts of Interest
References
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Scheid, A.D.; Beadnell, T.C.; Welch, D.R. Roles of mitochondria in the hallmarks of metastasis. Br. J. Cancer 2021, 124, 124–135. [Google Scholar] [CrossRef]
- Zacksenhaus, E.; Egan, S.E. Progression to Metastasis of Solid Cancer. Cancers 2021, 13, 717. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Bernards, R.; Weinberg, R.A. A progression puzzle. Nature 2002, 418, 823. [Google Scholar] [CrossRef]
- Siegel, M.B.; He, X.; Hoadley, K.A.; Hoyle, A.; Pearce, J.B.; Garrett, A.L.; Kumar, S.; Moylan, V.J.; Brady, C.M.; Van Swearingen, A.E.; et al. Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J. Clin. Investig. 2018, 128, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Savas, P.; Teo, Z.L.; Lefevre, C.; Flensburg, C.; Caramia, F.; Alsop, K.; Mansour, M.; Francis, P.A.; Thorne, H.A.; Silva, M.J.; et al. The Subclonal Architecture of Metastatic Breast Cancer: Results from a Prospective Community-Based Rapid Autopsy Program “CASCADE”. PLoS Med. 2016, 13, e1002204. [Google Scholar] [CrossRef]
- Paul, M.R.; Pan, T.C.; Pant, D.K.; Shih, N.N.; Chen, Y.; Harvey, K.L.; Solomon, A.; Lieberman, D.; Morrissette, J.J.; Soucier-Ernst, D.; et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J. Clin. Investig. 2020, 130, 4252–4265. [Google Scholar] [CrossRef]
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533. [Google Scholar] [CrossRef]
- Hakem, A.; Sanchez-Sweatman, O.; You-Ten, A.; Duncan, G.; Wakeham, A.; Khokha, R.; Mak, T.W. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005, 19, 1974–1979. [Google Scholar] [CrossRef] [PubMed]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat. Commun. 2018, 9, 4782. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563–575.e511. [Google Scholar] [CrossRef] [PubMed]
- Hessey, S.; Fessas, P.; Zaccaria, S.; Jamal-Hanjani, M.; Swanton, C. Insights into the metastatic cascade through research autopsies. Trends. Cancer 2023, 9, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, F.; Movasati, A.; Brunner, S.R.; Nguyen, L.; Priestley, P.; Cuppen, E.; Van Hoeck, A. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature 2023, 618, 333–341. [Google Scholar] [CrossRef]
- Morrissy, A.S.; Cavalli, F.M.G.; Remke, M.; Ramaswamy, V.; Shih, D.J.H.; Holgado, B.L.; Farooq, H.; Donovan, L.K.; Garzia, L.; Agnihotri, S.; et al. Spatial heterogeneity in medulloblastoma. Nat. Genet. 2017, 49, 780–788. [Google Scholar] [CrossRef]
- Rossi, D.; Khiabanian, H.; Spina, V.; Ciardullo, C.; Bruscaggin, A.; Fama, R.; Rasi, S.; Monti, S.; Deambrogi, C.; De Paoli, L.; et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014, 123, 2139–2147. [Google Scholar] [CrossRef]
- Jiang, Z.; Deng, T.; Jones, R.; Li, H.; Herschkowitz, J.I.; Liu, J.C.; Weigman, V.J.; Tsao, M.S.; Lane, T.F.; Perou, C.M.; et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J. Clin. Investig. 2010, 120, 3296–3309. [Google Scholar] [CrossRef]
- Jiang, Z.; Ju, Y.; Ali, A.; Chung, P.E.D.; Skowron, P.; Wang, D.Y.; Shrestha, M.; Li, H.; Liu, J.C.; Vorobieva, I.; et al. Distinct shared and compartment-enriched oncogenic networks drive primary versus metastatic breast cancer. Nat. Commun. 2023, 14, 4313. [Google Scholar] [CrossRef]
- Jiang, Z.; Jones, R.; Liu, J.C.; Deng, T.; Robinson, T.; Chung, P.E.; Wang, S.; Herschkowitz, J.I.; Egan, S.E.; Perou, C.M.; et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle 2011, 10, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Zacksenhaus, E.; Shrestha, M.; Liu, J.C.; Vorobieva, I.; Chung, P.E.D.; Ju, Y.; Nir, U.; Jiang, Z. Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis. Trends Cancer 2017, 3, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, H.; Schroer, S.A.; Voisin, V.; Ju, Y.; Pacal, M.; Erdmann, N.; Shi, W.; Chung, P.E.D.; Deng, T.; et al. Hypophosphorylated pRb knock-in mice exhibit hallmarks of aging and vitamin C-preventable diabetes. EMBO J. 2022, 41, e106825. [Google Scholar] [CrossRef]
- Ciavarra, G.; Zacksenhaus, E. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J. Cell Biol. 2010, 191, 291–301. [Google Scholar] [CrossRef]
- Robinson, T.J.; Liu, J.C.; Vizeacoumar, F.; Sun, T.; Maclean, N.; Egan, S.E.; Schimmer, A.D.; Datti, A.; Zacksenhaus, E. RB1 status in triple negative breast cancer cells dictates response to radiation treatment and selective therapeutic drugs. PLoS ONE 2013, 8, e78641. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, M.; Wang, D.Y.; Ben-David, Y.; Zacksenhaus, E. CDK4/6 inhibitors and the pRB-E2F1 axis suppress PVR and PD-L1 expression in triple-negative breast cancer. Oncogenesis 2023, 12, 29. [Google Scholar] [CrossRef]
- Jones, R.A.; Robinson, T.J.; Liu, J.C.; Shrestha, M.; Voisin, V.; Ju, Y.; Chung, P.E.; Pellecchia, G.; Fell, V.L.; Bae, S.; et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Investig. 2016, 126, 3739–3757. [Google Scholar] [CrossRef]
- Liu, J.C.; Granieri, L.; Shrestha, M.; Wang, D.Y.; Vorobieva, I.; Rubie, E.A.; Jones, R.; Ju, Y.; Pellecchia, G.; Jiang, Z.; et al. Identification of CDC25 as a Common Therapeutic Target for Triple-Negative Breast Cancer. Cell Rep. 2018, 23, 112–126. [Google Scholar] [CrossRef]
- Ho-Yen, C.M.; Jones, J.L.; Kermorgant, S. The clinical and functional significance of c-Met in breast cancer: A review. Breast Cancer Res. 2015, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Smith, M.J. RAS GTPase signalling to alternative effector pathways. Biochem. Soc. Trans. 2020, 48, 2241–2252. [Google Scholar] [CrossRef]
- Liu, W.; Gajendran, B.; Sample, K.M.; Wang, C.; Hu, A.; Chen, B.; Li, Y.; Zacksenhaus, E.; Ben-David, Y. A critical ETV4/Twist1/Vimentin axis in Ha-RAS-induced aggressive breast cancer. Cancer Gene Ther. 2022, 29, 1590–1599. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Andrechek, E.R.; Cardiff, R.D.; Chang, J.T.; Gatza, M.L.; Acharya, C.R.; Potti, A.; Nevins, J.R. Genetic heterogeneity of Myc-induced mammary tumors reflecting diverse phenotypes including metastatic potential. Proc. Natl. Acad. Sci. USA 2009, 106, 16387–16392. [Google Scholar] [CrossRef]
- Gatza, M.L.; Lucas, J.E.; Barry, W.T.; Kim, J.W.; Wang, Q.; Crawford, M.D.; Datto, M.B.; Kelley, M.; Mathey-Prevot, B.; Potti, A.; et al. A pathway-based classification of human breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 6994–6999. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Gendoo, D.M.A.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. A subgroup of microRNAs defines PTEN-deficient, triple-negative breast cancer patients with poorest prognosis and alterations in RB1, MYC, and Wnt signaling. Breast Cancer Res. 2019, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Jiang, Z.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. Molecular stratification within triple-negative breast cancer subtypes. Sci. Rep. 2019, 9, 19107. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Massague, J. Roles of TGFbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Seoane, J.; Gomis, R.R. TGF-beta Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef]
- Tian, M.; Schiemann, W.P. The TGF-beta paradox in human cancer: An update. Future Oncol. 2009, 5, 259–271. [Google Scholar] [CrossRef]
- Ali, R.; Wendt, M.K. The paradoxical functions of EGFR during breast cancer progression. Signal Transduct. Target. Ther. 2017, 2, 16042. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Broz, M.L.; Ranger, J.J.; Ozcelik, J.; Ahn, R.; Zuo, D.; Ursini-Siegel, J.; Hallett, M.T.; Krummel, M.; Muller, W.J. STAT3 Establishes an Immunosuppressive Microenvironment during the Early Stages of Breast Carcinogenesis to Promote Tumor Growth and Metastasis. Cancer Res. 2016, 76, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal 2020, 18, 33. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Huang, B.; Lu, M.; Jolly, M.K.; Tsarfaty, I.; Onuchic, J.; Ben-Jacob, E. The three-way switch operation of Rac1/RhoA GTPase-based circuit controlling amoeboid-hybrid-mesenchymal transition. Sci. Rep. 2014, 4, 6449. [Google Scholar] [CrossRef]
- Graziani, V.; Rodriguez-Hernandez, I.; Maiques, O.; Sanz-Moreno, V. The amoeboid state as part of the epithelial-to-mesenchymal transition programme. Trends. Cell Biol. 2022, 32, 228–242. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e684. [Google Scholar] [CrossRef]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e221. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Mizuno, M.; Khaledian, B.; Maeda, M.; Hayashi, T.; Mizuno, S.; Munetsuna, E.; Watanabe, T.; Kono, S.; Okada, S.; Suzuki, M.; et al. Adipsin-Dependent Secretion of Hepatocyte Growth Factor Regulates the Adipocyte-Cancer Stem Cell Interaction. Cancers 2021, 13, 4238. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tian, G.; Dajac, M.; Doty, A.; Wang, S.; Lee, J.H.; Rahman, M.; Huang, J.; Reynolds, B.A.; Sarkisian, M.R.; et al. Slow-Cycling Cells in Glioblastoma: A Specific Population in the Cellular Mosaic of Cancer Stem Cells. Cancers 2022, 14, 1126. [Google Scholar] [CrossRef] [PubMed]
- Haist, M.; Stege, H.; Ebner, R.; Fleischer, M.I.; Loquai, C.; Grabbe, S. The Role of Treatment Sequencing with Immune-Checkpoint Inhibitors and BRAF/MEK Inhibitors for Response and Survival of Patients with BRAFV600-Mutant Metastatic Melanoma-A Retrospective, Real-World Cohort Study. Cancers 2022, 14, 2082. [Google Scholar] [CrossRef]
- Gul, D.; Schweitzer, A.; Khamis, A.; Knauer, S.K.; Ding, G.B.; Freudelsperger, L.; Karampinis, I.; Strieth, S.; Hagemann, J.; Stauber, R.H. Impact of Secretion-Active Osteoblast-Specific Factor 2 in Promoting Progression and Metastasis of Head and Neck Cancer. Cancers 2022, 14, 2337. [Google Scholar] [CrossRef]
- Lin, C.Y.; Song, X.; Ke, Y.; Raha, A.; Wu, Y.; Wasi, M.; Wang, L.; Geng, F.; You, L. Yoda1 Enhanced Low-Magnitude High-Frequency Vibration on Osteocytes in Regulation of MDA-MB-231 Breast Cancer Cell Migration. Cancers 2022, 14, 3395. [Google Scholar] [CrossRef] [PubMed]
- Ormseth, B.; Onuma, A.; Zhang, H.; Tsung, A. The Hepatic Pre-Metastatic Niche. Cancers 2022, 14, 3731. [Google Scholar] [CrossRef] [PubMed]
- Goyette, M.A.; Cote, J.F. AXL Receptor Tyrosine Kinase as a Promising Therapeutic Target Directing Multiple Aspects of Cancer Progression and Metastasis. Cancers 2022, 14, 466. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Patel, P.S.; Hakem, R. BRCA1 and Metastasis: Outcome of Defective DNA Repair. Cancers 2021, 14, 108. [Google Scholar] [CrossRef]
- Gaebe, K.; Li, A.Y.; Das, S. Clinical Biomarkers for Early Identification of Patients with Intracranial Metastatic Disease. Cancers 2021, 13, 5973. [Google Scholar] [CrossRef]
- Terceiro, L.E.L.; Edechi, C.A.; Ikeogu, N.M.; Nickel, B.E.; Hombach-Klonisch, S.; Sharif, T.; Leygue, E.; Myal, Y. The Breast Tumor Microenvironment: A Key Player in Metastatic Spread. Cancers 2021, 13, 4798. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Lam, S.; Degenhard, T.; Garzia, L.; Guiot, M.C.; Petrecca, K.; Siegel, P.M. The Underlying Biology and Therapeutic Vulnerabilities of Leptomeningeal Metastases in Adult Solid Cancers. Cancers 2021, 13, 732. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Dedhar, S. New Perspectives on the Role of Integrin-Linked Kinase (ILK) Signaling in Cancer Metastasis. Cancers 2022, 14, 3209. [Google Scholar] [CrossRef]
- Bonnelye, E.; Juarez, P. Targeting Bone Metastasis in Cancers. Cancers 2021, 13, 4490. [Google Scholar] [CrossRef]
- Makino, T.; Izumi, K.; Iwamoto, H.; Mizokami, A. Treatment Strategies for High-Risk Localized and Locally Advanced and Oligometastatic Prostate Cancer. Cancers 2021, 13, 4470. [Google Scholar] [CrossRef]
- Care, A.; Del Bufalo, D.; Facchiano, A. Editorial on Special Issue “Advances and Novel Treatment Options in Metastatic Melanoma”. Cancers 2022, 14, 707. [Google Scholar] [CrossRef]
- Hoeppner, J.; Bronsert, P. Metastasis and Tumor Cell Migration of Solid Tumors. Cancers 2021, 13, 5576. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Z.; Ju, Y.-J.; Ali, A.; Chung, P.E.D.; Wang, D.-Y.; Liu, J.C.; Li, H.; Vorobieva, I.; Mwewa, E.; Ghanbari-Azarnier, R.; et al. Thinking (Metastasis) outside the (Primary Tumor) Box. Cancers 2023, 15, 5315. https://doi.org/10.3390/cancers15225315
Jiang Z, Ju Y-J, Ali A, Chung PED, Wang D-Y, Liu JC, Li H, Vorobieva I, Mwewa E, Ghanbari-Azarnier R, et al. Thinking (Metastasis) outside the (Primary Tumor) Box. Cancers. 2023; 15(22):5315. https://doi.org/10.3390/cancers15225315
Chicago/Turabian StyleJiang, Zhe, Young-Jun Ju, Amjad Ali, Philip E. D. Chung, Dong-Yu Wang, Jeff C. Liu, Huiqin Li, Ioulia Vorobieva, Ethel Mwewa, Ronak Ghanbari-Azarnier, and et al. 2023. "Thinking (Metastasis) outside the (Primary Tumor) Box" Cancers 15, no. 22: 5315. https://doi.org/10.3390/cancers15225315
APA StyleJiang, Z., Ju, Y.-J., Ali, A., Chung, P. E. D., Wang, D.-Y., Liu, J. C., Li, H., Vorobieva, I., Mwewa, E., Ghanbari-Azarnier, R., Shrestha, M., Ben-David, Y., & Zacksenhaus, E. (2023). Thinking (Metastasis) outside the (Primary Tumor) Box. Cancers, 15(22), 5315. https://doi.org/10.3390/cancers15225315