
Microbial-Related Metabolites May Be Involved in Eight Major Biological Processes and Represent Potential Diagnostic Markers in Gastric Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Process and Analysis of Metabolomics in GC and NC Tissues
2.2.1. Metabolite Extraction from Tissues
2.2.2. Untargeted Metabolome Analysis by LC-MS/MS
2.2.3. Data Processing and Metabolite Identification for the Metabolome Analysis
2.2.4. Metabolite Annotation, Screening, and Differential Metabolite Analysis
2.3. Sequencing and Analysis of the Microbiome in GC and NC Tissues
2.3.1. 16S rRNA Sequencing and Data Processing
2.3.2. Analysis of Microbial Diversity Differences and Differential Bacteria through 16S rRNA Sequencing
2.4. Microbe-Metabolite Correlation Analysis in GC and NC Tissues
2.4.1. The Overall Correlation Analysis of the Microbes and Metabolites
2.4.2. Coexistence Analysis of Differential Microbes and Metabolites
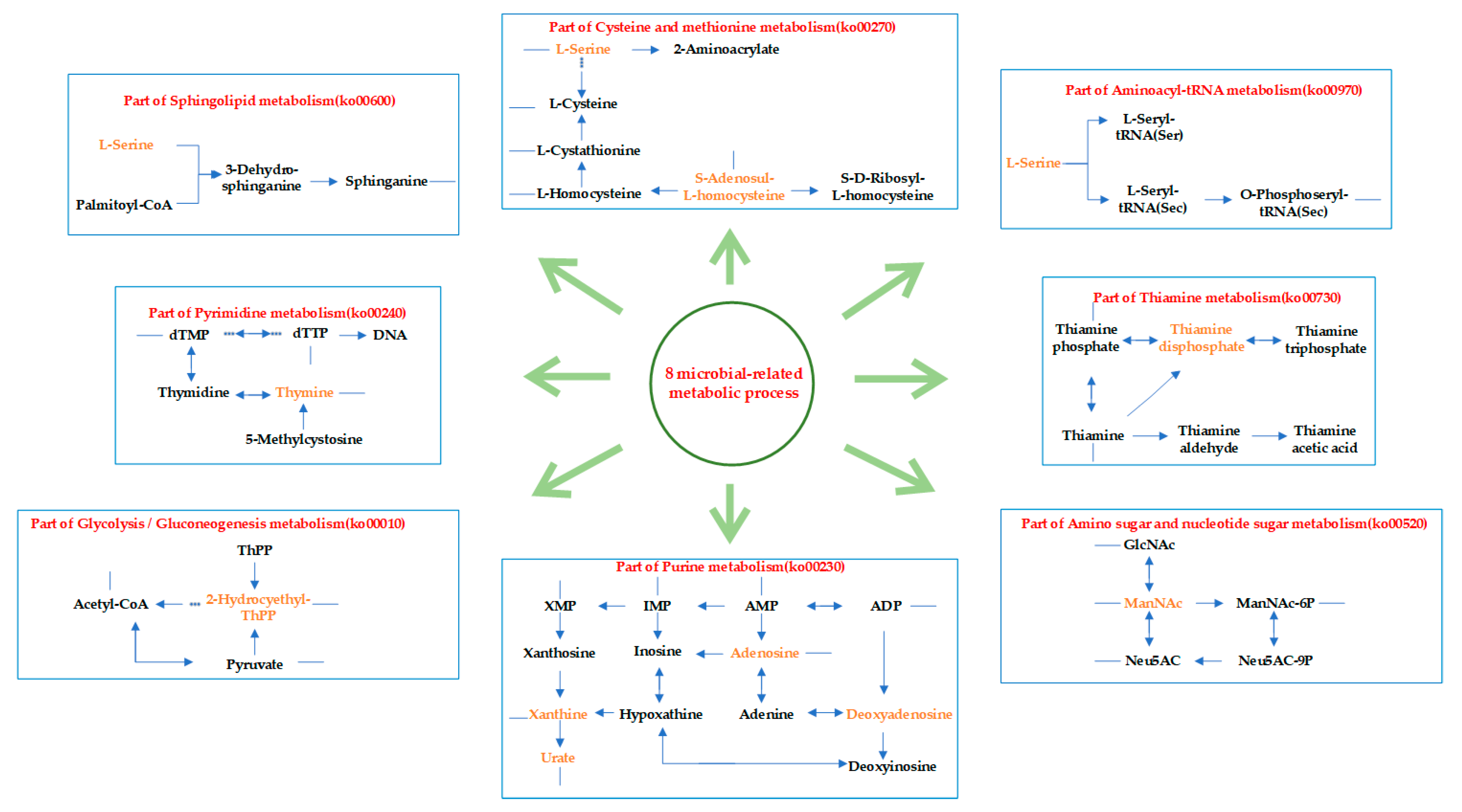
2.4.3. Functional Enrichment Analysis of the Microbial-Related Metabolites
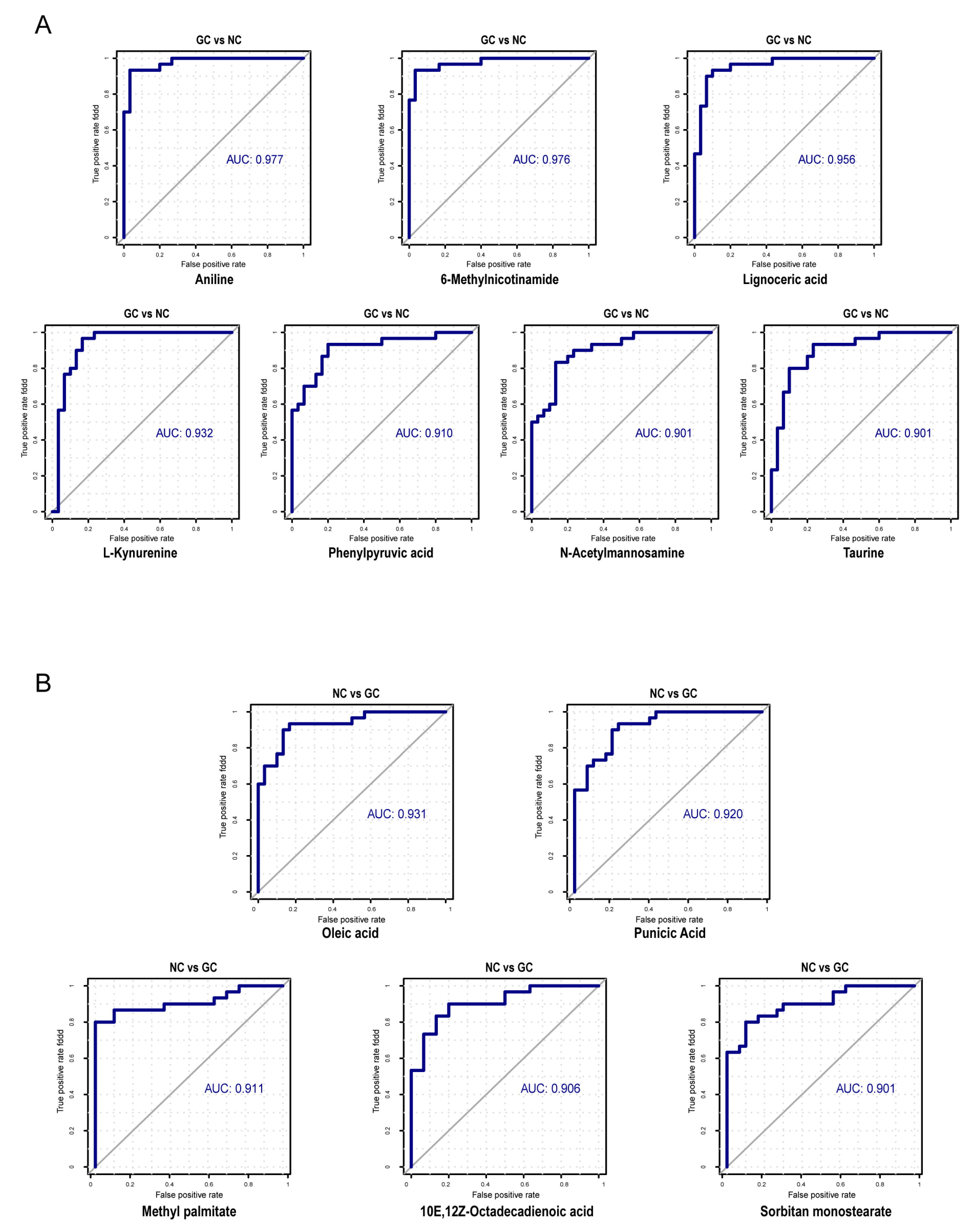
2.5. Diagnostic Efficacy Analysis of Differential Microbial-Related Metabolites between GC and NC Tissues
2.6. Statistical Analysis
3. Results
3.1. General Sample Information
3.2. The Metabolic Characteristics in the GC Tissues and the NC Tissues
3.2.1. The Overall Distribution of Metabolites in the GC Tissues and the NC Tissues
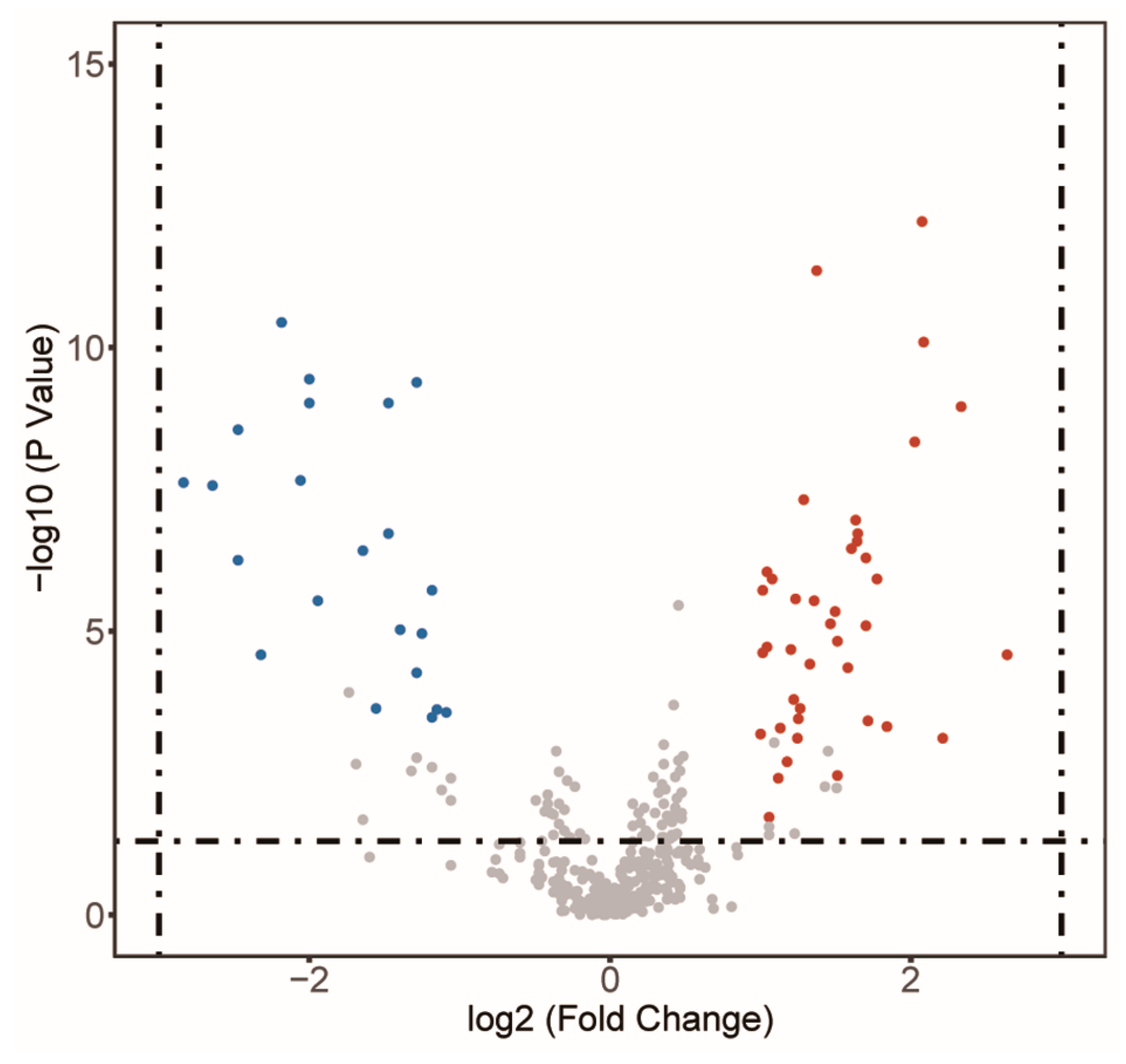
3.2.2. Differential Metabolites between the GC Tissues and the NC Tissues
3.3. The Microbial Characteristics in GC Tissues and NC Tissues
3.3.1. The Overall Microbe Composition and Microbial Diversity in GC Tissues and NC Tissues
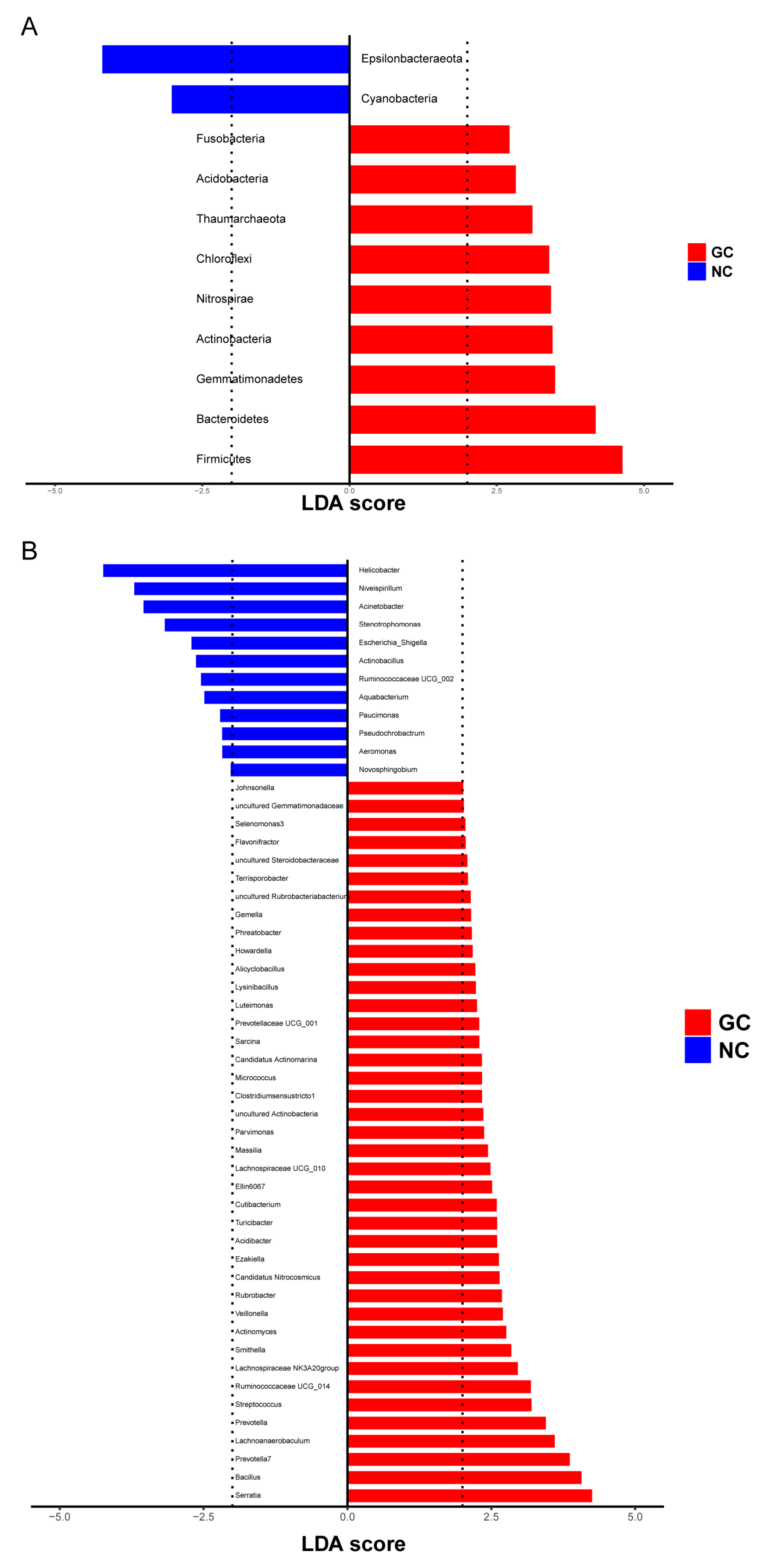
3.3.2. Differential Microbes in GC Tissues and NC Tissues
3.4. Correlation between Metabolites and Microbes in GC Tissues and NC Tissues
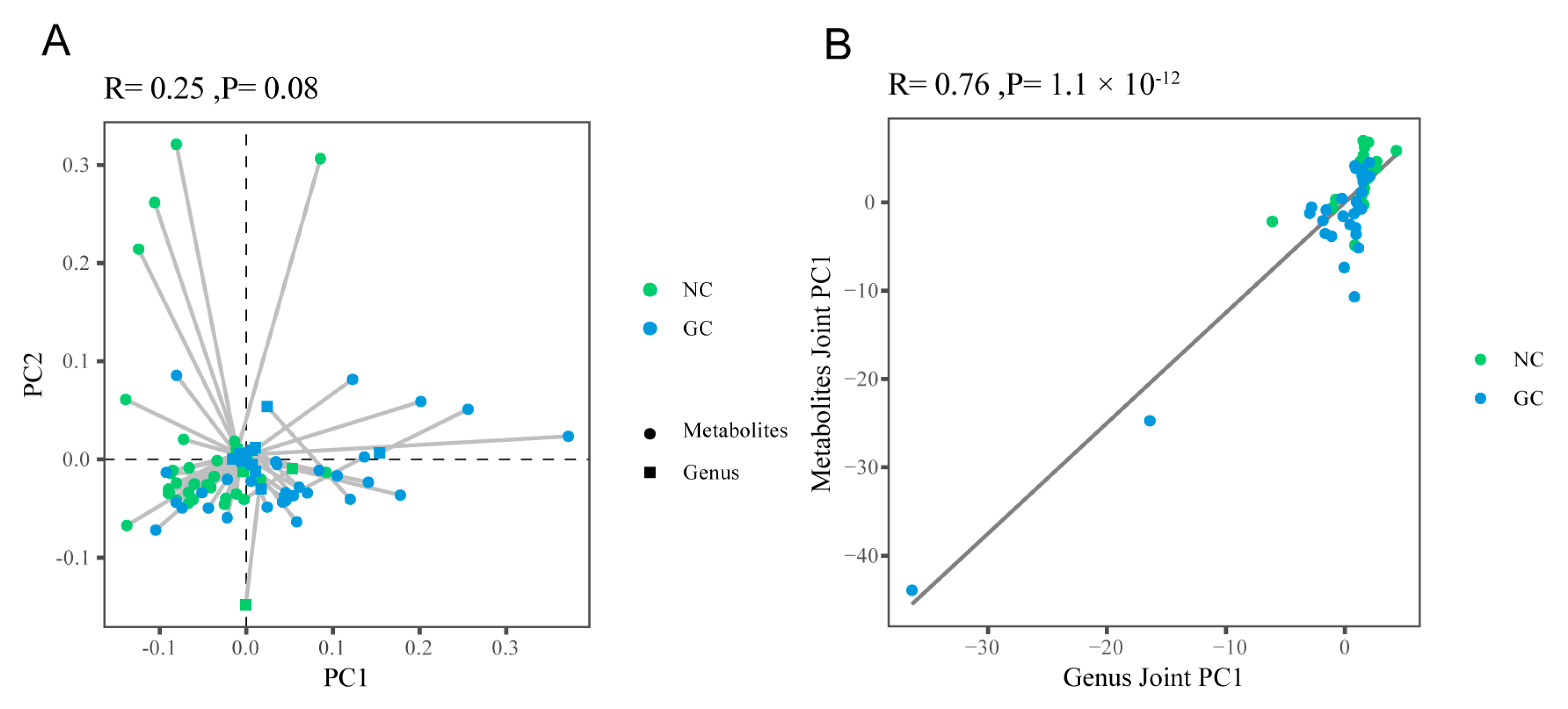
3.4.1. The Overall Correlation between Metabolites and Microbes
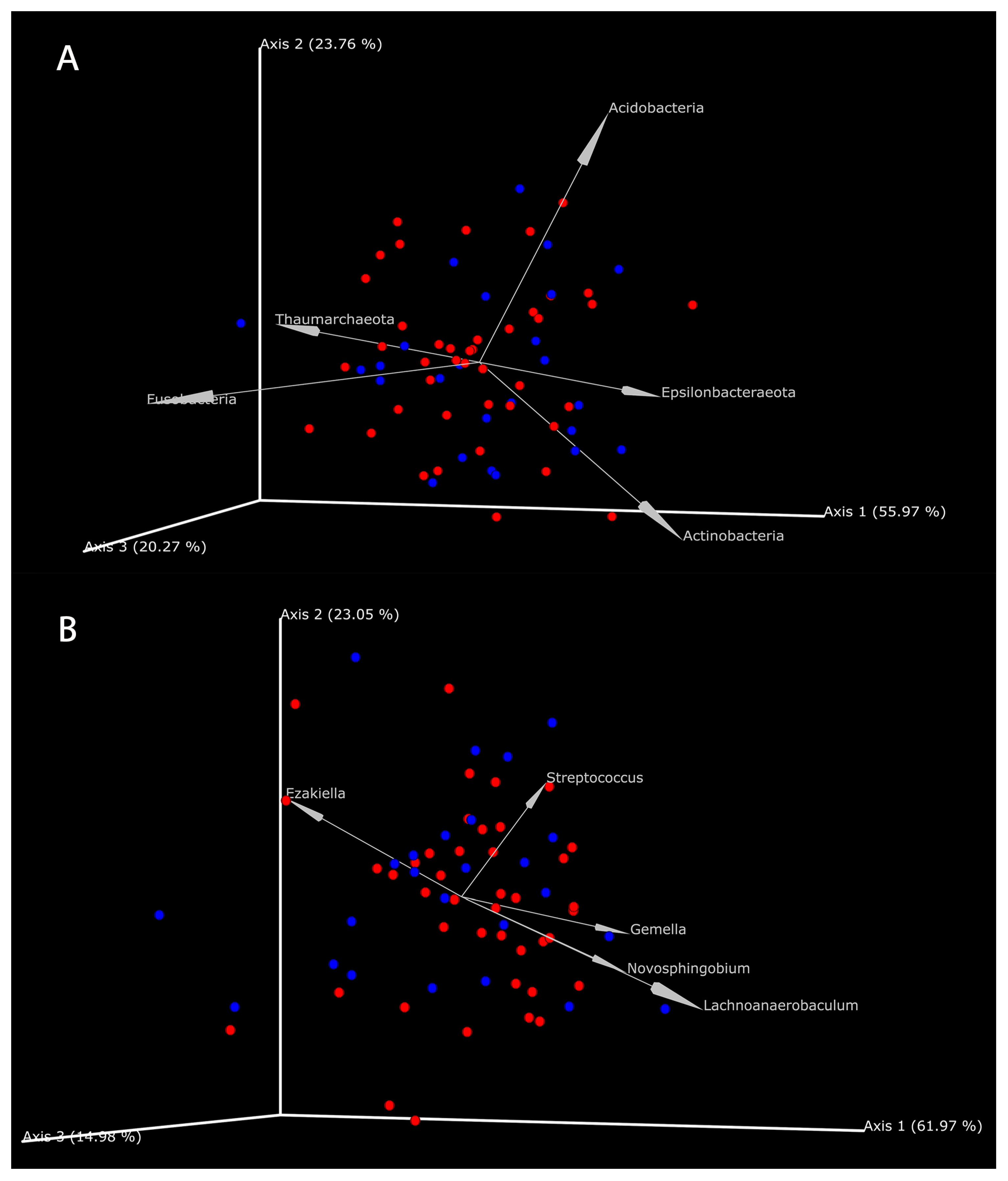
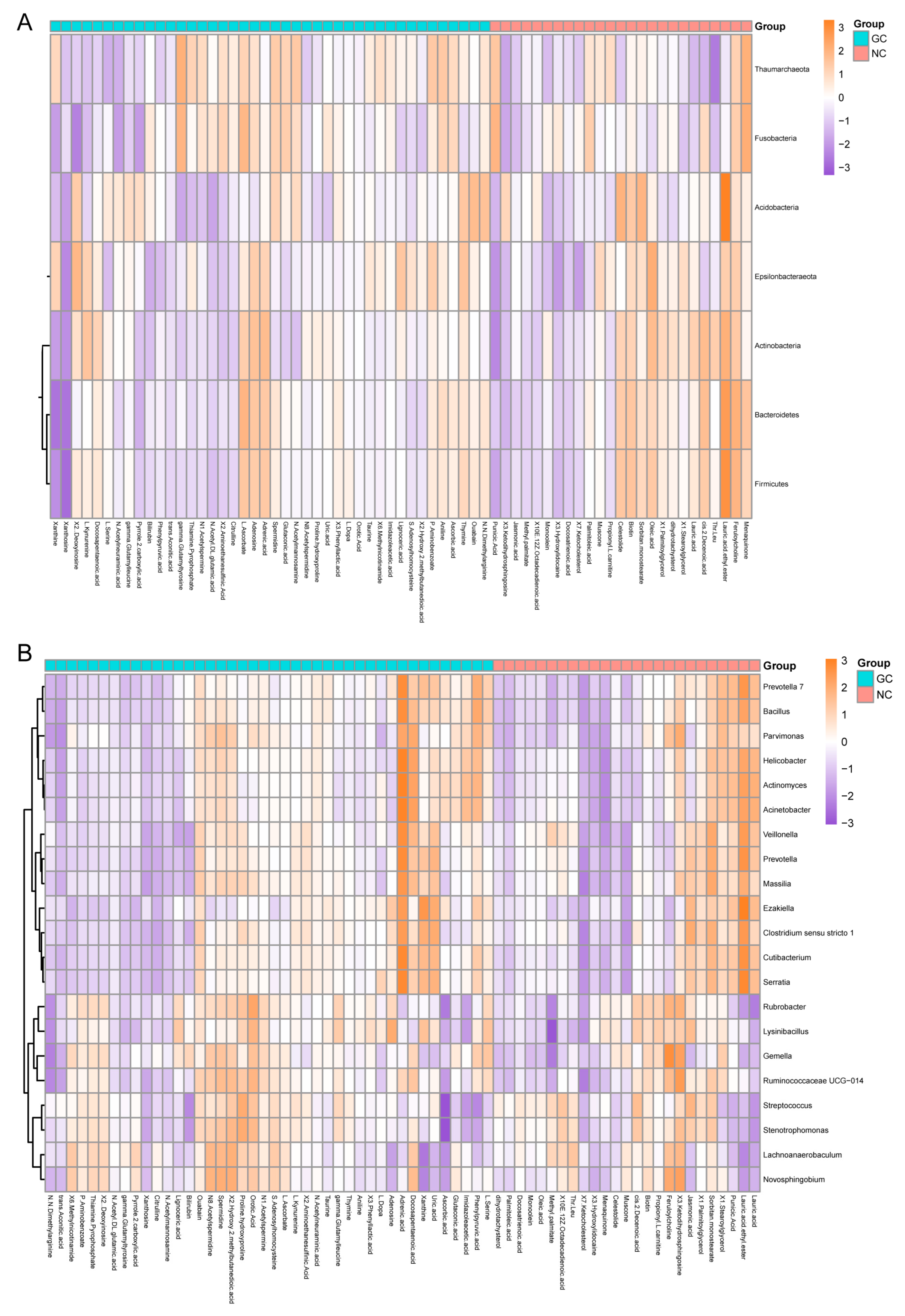
3.4.2. The Coexistence of Metabolites and Microbes in GC Tissues and NC Tissues
3.4.3. Functional Enrichment of Microbial-Related Metabolites in GC Tissues and NC Tissues
3.5. Efficacy of Microbial-Related Differential Metabolites in the Diagnosis of GC
4. Discussion
4.1. Differential Metabolites between GC Tissues and NC Tissues Participated in the Process of Sugar, Amino Acid, Nucleotide, and Lipid Metabolism
4.2. Metabolic Functions of Microbial-Related Metabolites
4.3. The Diagnostic Potential of Microbial-Related Metabolites in GC
5. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cavadas, B.; Camacho, R.; Ferreira, J.C.; Ferreira, R.M.; Figueiredo, C.; Brazma, A.; Fonseca, N.A.; Pereira, L. Gastric Microbiome Diversities in Gastric Cancer Patients from Europe and Asia Mimic the Human Population Structure and Are Partly Driven by Microbiome Quantitative Trait Loci. Microorganisms 2020, 8, 1196. [Google Scholar] [CrossRef] [PubMed]
- Dadgar, N.; Edlukudige Keshava, V.; Raj, M.S.; Wagner, P.L. The Influence of the Microbiome on Immunotherapy for Gastroesophageal Cancer. Cancers 2023, 15, 4426. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.; Hsu, R.; Rafizadeh, D.L.; Wang, L.; Bowlus, C.L.; Kumar, N.; Mishra, J.; Timilsina, S.; Ridgway, W.M.; Gershwin, M.E.; et al. The gut ecosystem and immune tolerance. J. Autoimmun. 2023, 103114. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Sethi, V.; Vitiello, G.A.; Saxena, D.; Miller, G.; Dudeja, V. The Role of the Microbiome in Immunologic Development and its Implication for Pancreatic Cancer Immunotherapy. Gastroenterology 2019, 156, 2097–2115.e2. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, X.; Zeng, R.; Wu, Q.; Sun, H.; Wu, W.; Zhang, X.; Sun, G.; Yan, B.; Wu, L.; et al. Changes of the Gastric Mucosal Microbiome Associated with Histological Stages of Gastric Carcinogenesis. Front. Microbiol. 2020, 11, 997. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.F.; Zou, K.; Wu, G.N.; Jin, Z.J.; Xiang, C.J.; Xu, S.; Wang, Y.H.; Wu, X.Y.; Chen, C.; Xu, Z.; et al. A Comparison of Tumor-Associated and Non-Tumor-Associated Gastric Microbiota in Gastric Cancer Patients. Dig. Dis. Sci. 2020, 66, 1673–1682. [Google Scholar] [CrossRef]
- Liu, X.; Shao, L.; Liu, X.; Ji, F.; Mei, Y.; Cheng, Y.; Liu, F.; Yan, C.; Li, L.; Ling, Z. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine 2019, 40, 336–348. [Google Scholar] [CrossRef]
- Castaño-Rodríguez, N.; Goh, K.L.; Fock, K.M.; Mitchell, H.M.; Kaakoush, N.O. Dysbiosis of the microbiome in gastric carcinogenesis. Sci. Rep. 2017, 7, 15957. [Google Scholar] [CrossRef]
- Chen, X.H.; Wang, A.; Chu, A.N.; Gong, Y.H.; Yuan, Y. Mucosa-associated microbiome in gastric cancer compared with non-cancer tissues. Front. Microbiol. 2019, 10, 1261. [Google Scholar] [CrossRef]
- Kumar, S.; Metz, D.C.; Ellenberg, S.; Kaplan, D.E.; Goldberg, D.S. Risk Factors and Incidence of Gastric Cancer After Detection of Helicobacter pylori Infection: A Large Cohort Study. Gastroenterology 2020, 158, 527–536.e7. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, E.M.; Oien, K.; El-Nujumi, A.; Gillen, D.; Wirz, A.; Dahill, S.; Williams, C.; Ardill, J.E.; McColl, K.E. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology 1997, 113, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, X.; Liu, X.; Ling, Z.; Ji, F. Role of the Gastric Microbiome in Gastric Cancer: From Carcinogenesis to Treatment. Front. Microbiol. 2021, 12, 641322. [Google Scholar] [CrossRef]
- Hirata, S.I.; Kunisawa, J. Gut microbiome, metabolome, and allergic diseases. Allergol. Int. Off. J. Jpn. Soc. Allergol. 2017, 66, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sarwar, K.A.; Lasky-Su, J.; Kelly, R.S.; Litonjua, A.A.; Weiss, S.T. Metabolome-Microbiome Crosstalk and Human Disease. Metabolites 2020, 10, 181. [Google Scholar] [CrossRef]
- Yang, Y.; Misra, B.B.; Liang, L.; Bi, D.; Weng, W.; Wu, W.; Cai, S.; Qin, H.; Goel, A.; Li, X.; et al. Integrated microbiome and metabolome analysis reveals a novel interplay between commensal bacteria and metabolites in colorectal cancer. Theranostics 2019, 9, 4101–4114. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Malczewski, A.B.; Navarro, S.; Coward, J.I.; Ketheesan, N. Microbiome-derived metabolome as a potential predictor of response to cancer immunotherapy. J. Immunother. Cancer 2020, 8, e001383. [Google Scholar] [CrossRef]
- Anand, S.; Kaur, H.; Mande, S.S. Comparative In silico Analysis of Butyrate Production Pathways in Gut Commensals and Pathogens. Front. Microbiol. 2016, 7, 1945. [Google Scholar] [CrossRef]
- Russo, E.; Giudici, F.; Fiorindi, C.; Ficari, F.; Scaringi, S.; Amedei, A. Immunomodulating Activity and Therapeutic Effects of Short Chain Fatty Acids and Tryptophan Post-biotics in Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 2754. [Google Scholar] [CrossRef] [PubMed]
- Marcobal, A.; Kashyap, P.C.; Nelson, T.A.; Aronov, P.A.; Donia, M.S.; Spormann, A.; Fischbach, M.A.; Sonnenburg, J.L. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 2013, 7, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Melnik, A.V.; Vrbanac, A.; Fu, T.; Patras, K.A.; Christy, M.P.; Bodai, Z.; Belda-Ferre, P.; Tripathi, A.; Chung, L.K.; et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 2020, 579, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Mars, R.A.T.; Yang, Y.; Ward, T.; Houtti, M.; Priya, S.; Lekatz, H.R.; Tang, X.; Sun, Z.; Kalari, K.R.; Korem, T.; et al. Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome. Cell 2020, 182, 1460–1473.e1417. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Usyk, M.; Sollecito, C.C.; Qiu, Y.; Williams-Nguyen, J.; Hua, S.; Gradissimo, A.; Wang, T.; Xue, X.; Kurland, I.J.; et al. Altered Gut Microbiota and Host Metabolite Profiles in HIV-infected Women. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2019, 71, 2345–2353. [Google Scholar] [CrossRef]
- Sagar, N.M.; Duboc, H.; Kay, G.L.; Alam, M.T.; Wicaksono, A.N.; Covington, J.A.; Quince, C.; Kokkorou, M.; Svolos, V.; Palmieri, L.J.; et al. The pathophysiology of bile acid diarrhoea: Differences in the colonic microbiome, metabolome and bile acids. Sci. Rep. 2020, 10, 20436. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef]
- Li, B.; Shu, X.; Jiang, H.; Shi, C.; Qi, L.; Zhu, L.; Zhou, J.; Tang, M.; Hu, A. Plasma metabolome identifies potential biomarkers of gastric precancerous lesions and gastric cancer risk. Metabolomics Off. J. Metabolomic Soc. 2023, 19, 73. [Google Scholar] [CrossRef]
- Lei, C.; Gong, D.; Zhuang, B.; Zhang, Z. Alterations in the gastric microbiota and metabolites in gastric cancer: An update review. Front. Oncol. 2022, 12, 960281. [Google Scholar] [CrossRef]
- Morton, J.T.; Aksenov, A.A.; Nothias, L.F.; Foulds, J.R.; Quinn, R.A.; Badri, M.H.; Swenson, T.L.; Van Goethem, M.W.; Northen, T.R.; Vazquez-Baeza, Y.; et al. Learning representations of microbe-metabolite interactions. Nat. Methods 2019, 16, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; O’Maille, G.; Smith, C.A.; Brandon, T.R.; Uritboonthai, W.; Qin, C.; Trauger, S.A.; Siuzdak, G. Solvent-dependent metabolite distribution, clustering, and protein extraction for serum profiling with mass spectrometry. Anal. Chem. 2006, 78, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Ni, Y.; Yu, G.; Chen, H.; Deng, Y.; Wells, P.M.; Steves, C.J.; Ju, F.; Fu, J. M2IA: A Web Server for Microbiome and Metabolome Integrative Analysis. Bioinformatics 2020, 36, 3493–3498. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Jung, J.; Jung, Y.; Bang, E.J.; Cho, S.I.; Jang, Y.J.; Kwak, J.M.; Ryu, D.H.; Park, S.; Hwang, G.S. Noninvasive diagnosis and evaluation of curative surgery for gastric cancer by using NMR-based metabolomic profiling. Ann. Surg. Oncol. 2014, 21, 736–742. [Google Scholar] [CrossRef]
- Song, H.; Wang, L.; Liu, H.L.; Wu, X.B.; Wang, H.S.; Liu, Z.H.; Li, Y.; Diao, D.C.; Chen, H.L.; Peng, J.S. Tissue metabolomic fingerprinting reveals metabolic disorders associated with human gastric cancer morbidity. Oncol. Rep. 2011, 26, 431–438. [Google Scholar] [CrossRef]
- Wu, H.; Xue, R.; Tang, Z.; Deng, C.; Liu, T.; Zeng, H.; Sun, Y.; Shen, X. Metabolomic investigation of gastric cancer tissue using gas chromatography/mass spectrometry. Anal. Bioanal. Chem. 2010, 396, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, J.S.; Li, J.J.; Peng, D.N.; Wang, X.Y.; Chen, T.L.; Qiu, Y.P.; Chen, P.P.; Li, W.J.; Xu, L.Y.; et al. A combined proteomics and metabolomics profiling of gastric cardia cancer reveals characteristic dysregulations in glucose metabolism. Mol. Cell. Proteom. MCP 2010, 9, 2617–2628. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Zhou, L. Gastric cancer: Metabolic and metabolomics perspectives (Review). Int. J. Oncol. 2017, 51, 5–17. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Deng, P.; Liu, C.; Li, D.; Jie, H.; Zhang, H.; Zhou, Z.; Zhao, Y.L. Tissue metabolic profiling of human gastric cancer assessed by (1)H NMR. BMC Cancer 2016, 16, 371. [Google Scholar] [CrossRef]
- Hur, H.; Paik, M.J.; Xuan, Y.; Nguyen, D.T.; Ham, I.H.; Yun, J.; Cho, Y.K.; Lee, G.; Han, S.U. Quantitative measurement of organic acids in tissues from gastric cancer patients indicates increased glucose metabolism in gastric cancer. PLoS ONE 2014, 9, e98581. [Google Scholar] [CrossRef]
- Cui, P.; Huang, C.; Guo, J.; Wang, Q.; Liu, Z.; Zhuo, H.; Lin, D. Metabolic Profiling of Tumors, Sera, and Skeletal Muscles from an Orthotopic Murine Model of Gastric Cancer Associated-Cachexia. J. Proteome Res. 2019, 18, 1880–1892. [Google Scholar] [CrossRef]
- Huang, S.; Guo, Y.; Li, Z.; Zhang, Y.; Zhou, T.; You, W.; Pan, K.; Li, W. A systematic review of metabolomic profiling of gastric cancer and esophageal cancer. Cancer Biol. Med. 2020, 17, 181–198. [Google Scholar] [CrossRef]
- Yang, T.; Luo, P.; Li, Y.; Hua, R.; Yin, P.; Xu, G. A serum metabolomics study of gastric cancer based on pseudotargeted liquid chromatography-mass spectrometry approach. Se Pu = Chin. J. Chromatogr. 2014, 32, 126–132. [Google Scholar] [CrossRef]
- Abbassi-Ghadi, N.; Kumar, S.; Huang, J.; Goldin, R.; Takats, Z.; Hanna, G.B. Metabolomic profiling of oesophago-gastric cancer: A systematic review. Eur. J. Cancer 2013, 49, 3625–3637. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Aa, J.; Xu, J.; Sun, M.; Qian, S.; Cheng, L.; Yang, S.; Shi, R. Metabolomic phenotype of gastric cancer and precancerous stages based on gas chromatography time-of-flight mass spectrometry. J. Gastroenterol. Hepatol. 2011, 26, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.D.; Tang, H.Q.; Zhang, Q.; Fan, J.; Hong, J.; Gu, J.Z.; Chen, J.L. Prediction of gastric cancer metastasis through urinary metabolomic investigation using GC/MS. World J. Gastroenterol. 2011, 17, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Ravegnini, G.; Fosso, B.; Saverio, V.D.; Sammarini, G.; Zanotti, F.; Rossi, G.; Ricci, M.; D’Amico, F.; Valori, G.; Ioli, A.; et al. Gastric Adenocarcinomas and Signet-Ring Cell Carcinoma: Unraveling Gastric Cancer Complexity through Microbiome Analysis-Deepening Heterogeneity for a Personalized Therapy. Int. J. Mol. Sci. 2020, 21, 9735. [Google Scholar] [CrossRef]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Wang, M.; Yang, G.; Tian, Y.; Zhang, Q.; Liu, Z.; Xin, Y. The role of the gut microbiota in gastric cancer: The immunoregulation and immunotherapy. Front. Immunol. 2023, 14, 1183331. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillère, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef]
- Hong, Y.; Li, B.; Zheng, N.; Wu, G.; Ma, J.; Tao, X.; Chen, L.; Zhong, J.; Sheng, L.; Li, H. Integrated Metagenomic and Metabolomic Analyses of the Effect of Astragalus Polysaccharides on Alleviating High-Fat Diet-Induced Metabolic Disorders. Front. Pharmacol. 2020, 11, 833. [Google Scholar] [CrossRef]
- Zhang, G.; Gao, F. Quantitative analysis of correlation between AT and GC biases among bacterial genomes. PLoS ONE 2017, 12, e0171408. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Goswami, A.; Dutta, C. Association of purine asymmetry, strand-biased gene distribution and PolC within Firmicutes and beyond: A new appraisal. BMC Genom. 2014, 15, 430. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.W.; Hao, A.; Satyshur, K.A.; Keck, J.L.; Wang, J.D. Molecular Mechanism of Regulation of the Purine Salvage Enzyme XPRT by the Alarmones pppGpp, ppGpp, and pGpp. J. Mol. Biol. 2020, 432, 4108–4126. [Google Scholar] [CrossRef]
- Mukai, T.; Vargas-Rodriguez, O.; Englert, M.; Tripp, H.J.; Ivanova, N.N.; Rubin, E.M.; Kyrpides, N.C.; Söll, D. Transfer RNAs with novel cloverleaf structures. Nucleic Acids Res. 2017, 45, 2776–2785. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Haorah, J.; Chen, S.C.; Wang, X.; Kolar, C.; Lawson, T.A.; Mirvish, S.S. Nitrosation of glycine ethyl ester and ethyl diazoacetate to give the alkylating agent and mutagen ethyl chloro(hydroximino)acetate. Chem. Res. Toxicol. 2004, 17, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Shephard, S.E.; Hegi, M.E.; Lutz, W.K. In-vitro assays to detect alkylating and mutagenic activities of dietary components nitrosated in situ. In Relevance of N-Nitroso Compounds to Human Cancer; Bartsch, H., Ed.; IARC Scientific Publications: Lyon, France, 1987; pp. 232–236. [Google Scholar]
- Xiang, Z.; Li, J.; Song, S.; Wang, J.; Cai, W.; Hu, W.; Ji, J.; Zhu, Z.; Zang, L.; Yan, R.; et al. A positive feedback between IDO1 metabolite and COL12A1 via MAPK pathway to promote gastric cancer metastasis. J. Exp. Clin. Cancer Res. CR 2019, 38, 314. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, P.; Rodríguez, M.C.; Sánchez-Yagüe, J. Identification of potential erythrocyte phospholipid fatty acid biomarkers of advanced lung adenocarcinoma, squamous cell lung carcinoma, and small cell lung cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 5687–5698. [Google Scholar] [CrossRef]
- Cha, D.; Liu, M.; Zeng, Z.; Cheng, D.; Zhan, G. Analysis of fatty acids in lung tissues using gas chromatography-mass spectrometry preceded by derivatization-solid-phase microextraction with a novel fiber. Anal. Chim. Acta 2006, 572, 47–54. [Google Scholar] [CrossRef]
- Mun, C.W.; Cho, J.Y.; Shin, W.J.; Choi, K.S.; Eun, C.K.; Cha, S.S.; Lee, J.; Yang, Y.I.; Nam, S.H.; Kim, J.; et al. Ex vivo proton MR spectroscopy (1H-MRS) for evaluation of human gastric carcinoma. Magn. Reson. Imaging 2004, 22, 861–870. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Rao, B.; Deng, L. Gut flora profiling and fecal metabolite composition of colorectal cancer patients and healthy individuals. Exp. Ther. Med. 2017, 13, 2848–2854. [Google Scholar] [CrossRef]
- Bimro, E.T.; Hovav, R.; Nyska, A.; Glazer, T.A.; Madar, Z. High oleic peanuts improve parameters leading to fatty liver development and change the microbiota in mice intestine. Food Nutr. Res. 2020, 64. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | FC | VIP | |
|---|---|---|---|
| 25 NC metabolites | Monoolein | 0.04 | 1.76 |
| Palmitoleic acid | 0.07 | 1.65 | |
| 1-Palmitoylglycerol | 0.11 | 1.56 | |
| Menaquinone | 0.14 | 1.66 | |
| dihydrotachysterol | 0.16 | 1.66 | |
| Sorbitan monostearate | 0.18 | 1.71 | |
| cis-2-Decenoic acid | 0.18 | 1.71 | |
| 3-Hydroxylidocaine | 0.2 | 1.12 | |
| Oleic acid | 0.22 | 1.85 | |
| Lauric acid | 0.24 | 1.67 | |
| Punicic Acid | 0.25 | 1.96 | |
| Methyl palmitate | 0.25 | 1.81 | |
| Muscone | 0.26 | 1.44 | |
| 7-Ketocholesterol | 0.32 | 1.36 | |
| Lauric acid ethyl ester | 0.34 | 1.19 | |
| 3-Ketodihydrosphingosine | 0.36 | 1.7 | |
| Propionyl-L-carnitine | 0.36 | 1.67 | |
| 1-Stearoylglycerol | 0.38 | 1.26 | |
| 10E,12Z-Octadecadienoic acid | 0.41 | 1.4 | |
| Docosatrienoic acid | 0.41 | 1.33 | |
| Thr-Leu | 0.42 | 1.75 | |
| Jasmonic acid | 0.44 | 1.34 | |
| Feruloylcholine | 0.44 | 1.23 | |
| Biotin | 0.45 | 1.14 | |
| Celestolide | 0.47 | 1.24 | |
| 42 GC metabolites | 2′-Deoxyinosine | 2 | 1.1 |
| N-Acetylmannosamine | 2.02 | 1.67 | |
| N8-Acetylspermidine | 2.02 | 1.55 | |
| Docosapentaenoic acid | 2.06 | 1.6 | |
| N-Acetylneuraminic acid | 2.06 | 1.38 | |
| Thymine | 2.08 | 1.09 | |
| Adrenic acid | 2.11 | 1.85 | |
| 2-Aminoethanesulfinic Acid | 2.17 | 1.22 | |
| Xanthine | 2.19 | 1.26 | |
| Orotic Acid | 2.26 | 1.22 | |
| Spermidine | 2.3 | 1.36 | |
| Ouabain | 2.33 | 1.13 | |
| N-Acetyl-DL-glutamic acid | 2.35 | 1.38 | |
| Uric acid | 2.37 | 1.11 | |
| 2-Hydroxy-2-methylbutanedioic acid | 2.38 | 1.16 | |
| L-Serine | 2.4 | 1.41 | |
| Taurine | 2.44 | 1.68 | |
| Xanthosine | 2.51 | 1.3 | |
| gamma-Glutamylleucine | 2.56 | 1.41 | |
| Aniline | 2.59 | 2.06 | |
| trans-Aconitic acid | 2.76 | 1.1 | |
| gamma-Glutamyltyrosine | 2.82 | 1.43 | |
| N,N-Dimethylarginine | 2.85 | 1.41 | |
| Imidazoleacetic acid | 2.85 | 1.18 | |
| S-Adenosylhomocysteine | 2.99 | 1.42 | |
| Thiamine Pyrophosphate | 3.04 | 1.36 | |
| Pyrrole-2-carboxylic acid | 3.1 | 1.63 | |
| P-Aminobenzoate | 3.12 | 1.68 | |
| L-Dopa | 3.13 | 1.59 | |
| N1-Acetylspermine | 3.25 | 1.58 | |
| Adenosine | 3.25 | 1.48 | |
| Citrulline | 3.28 | 1.28 | |
| Proline-hydroxyproline | 3.42 | 1.61 | |
| Bilirubin | 3.58 | 1.38 | |
| Phenylpyruvic acid | 4.07 | 1.72 | |
| 6-Methylnicotinamide | 4.21 | 2.3 | |
| Lignoceric acid | 4.24 | 2.75 | |
| Ascorbic acid | 4.63 | 1.31 | |
| L-Kynurenine | 5.04 | 1.69 | |
| L-Ascorbate | 6.23 | 1.53 | |
| 3-Phenyllactic acid | 8.14 | 1.55 | |
| Glutaconic acid | 9.49 | 1.15 |
| Microbial-Related Metabolic Functions | Metabolites Enriched Pathways |
|---|---|
| ko00230 | Purine metabolism |
| ko00730 | Thiamine metabolism |
| ko00270 | Cysteine and methionine metabolism |
| ko00240 | Pyrimidine metabolism |
| ko00600 | Sphingolipid metabolism |
| ko00010 | Glycolysis/Gluconeogenesis |
| ko00970 | Aminoacyl-tRNA biosynthesis |
| ko00520 | Amino sugar and nucleotide sugar metabolism |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nie, S.; Wang, A.; Chen, X.; Gong, Y.; Yuan, Y. Microbial-Related Metabolites May Be Involved in Eight Major Biological Processes and Represent Potential Diagnostic Markers in Gastric Cancer. Cancers 2023, 15, 5271. https://doi.org/10.3390/cancers15215271
Nie S, Wang A, Chen X, Gong Y, Yuan Y. Microbial-Related Metabolites May Be Involved in Eight Major Biological Processes and Represent Potential Diagnostic Markers in Gastric Cancer. Cancers. 2023; 15(21):5271. https://doi.org/10.3390/cancers15215271
Chicago/Turabian StyleNie, Siru, Ang Wang, Xiaohui Chen, Yuehua Gong, and Yuan Yuan. 2023. "Microbial-Related Metabolites May Be Involved in Eight Major Biological Processes and Represent Potential Diagnostic Markers in Gastric Cancer" Cancers 15, no. 21: 5271. https://doi.org/10.3390/cancers15215271
APA StyleNie, S., Wang, A., Chen, X., Gong, Y., & Yuan, Y. (2023). Microbial-Related Metabolites May Be Involved in Eight Major Biological Processes and Represent Potential Diagnostic Markers in Gastric Cancer. Cancers, 15(21), 5271. https://doi.org/10.3390/cancers15215271