
Modulation of FDG Uptake by Cell Cycle Synchronization Using a T-Type Calcium Channel Inhibitor


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Mibefradil Treatment and Serum Starvation
2.3. Cell Cycle Analysis
2.4. Cellular Uptake
2.5. Animal Model
2.6. In Vivo PET Imaging
2.7. Statistical Analysis
3. Results
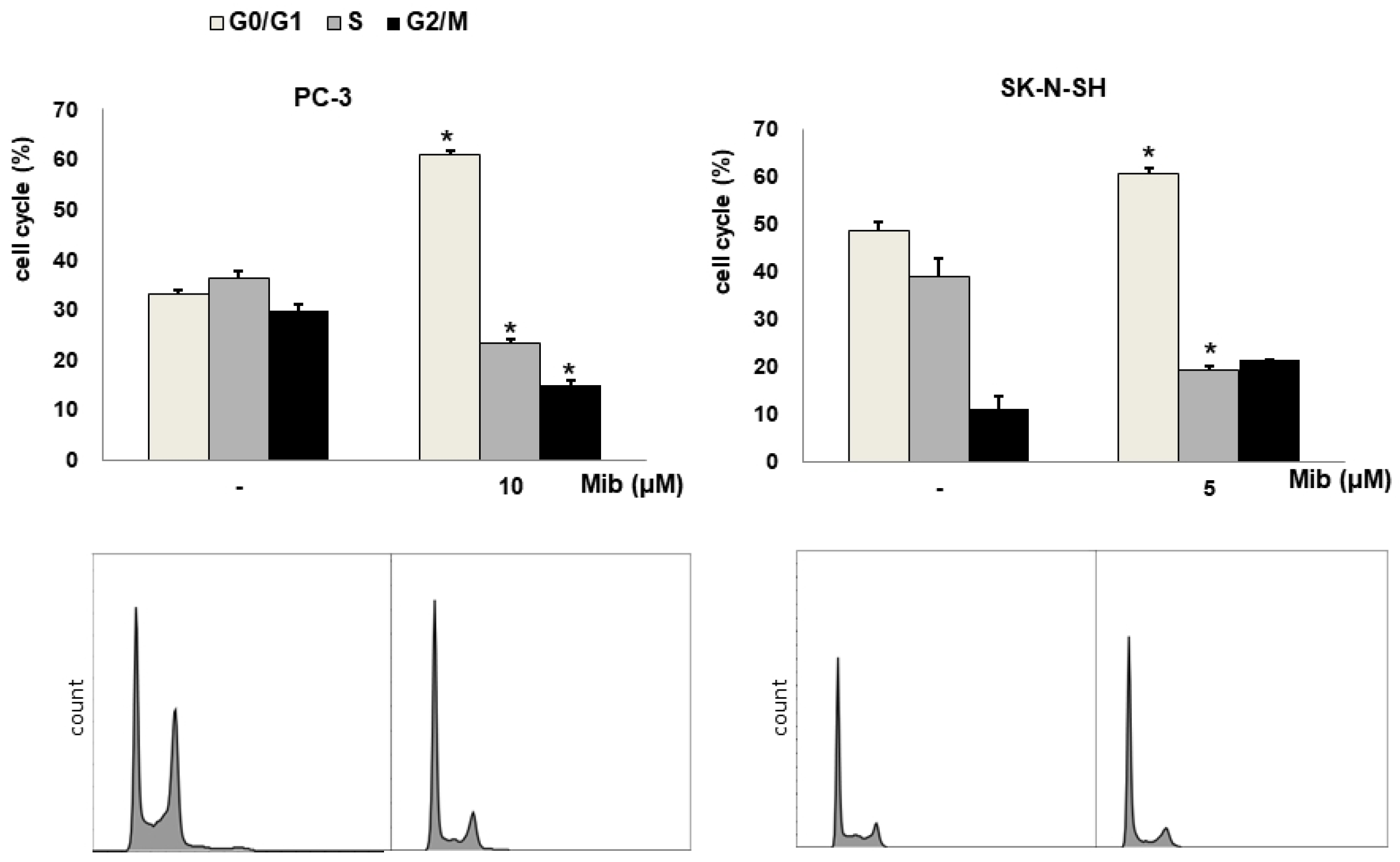
3.1. Cell Cycle Synchronization Induced by Mibefradil
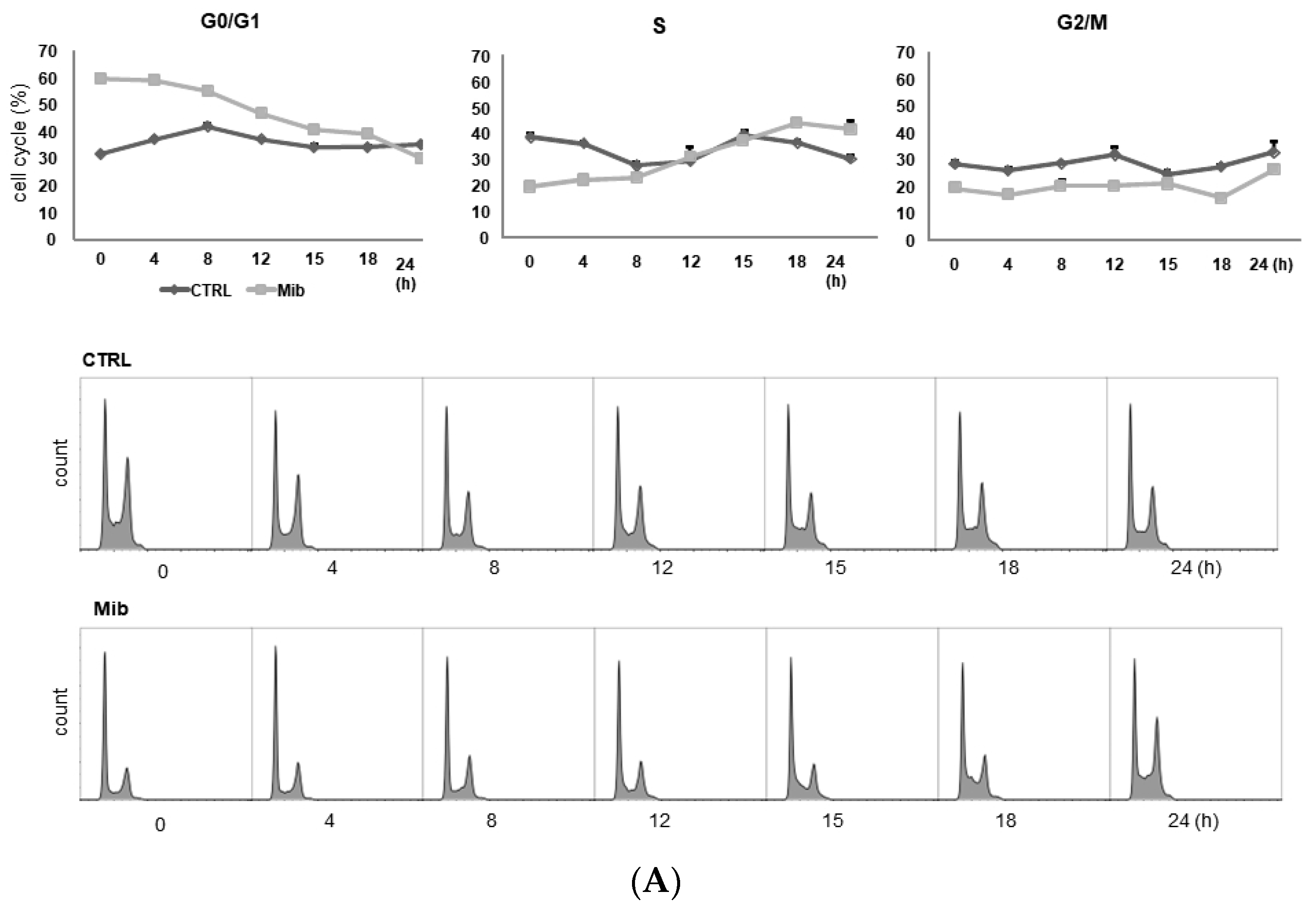
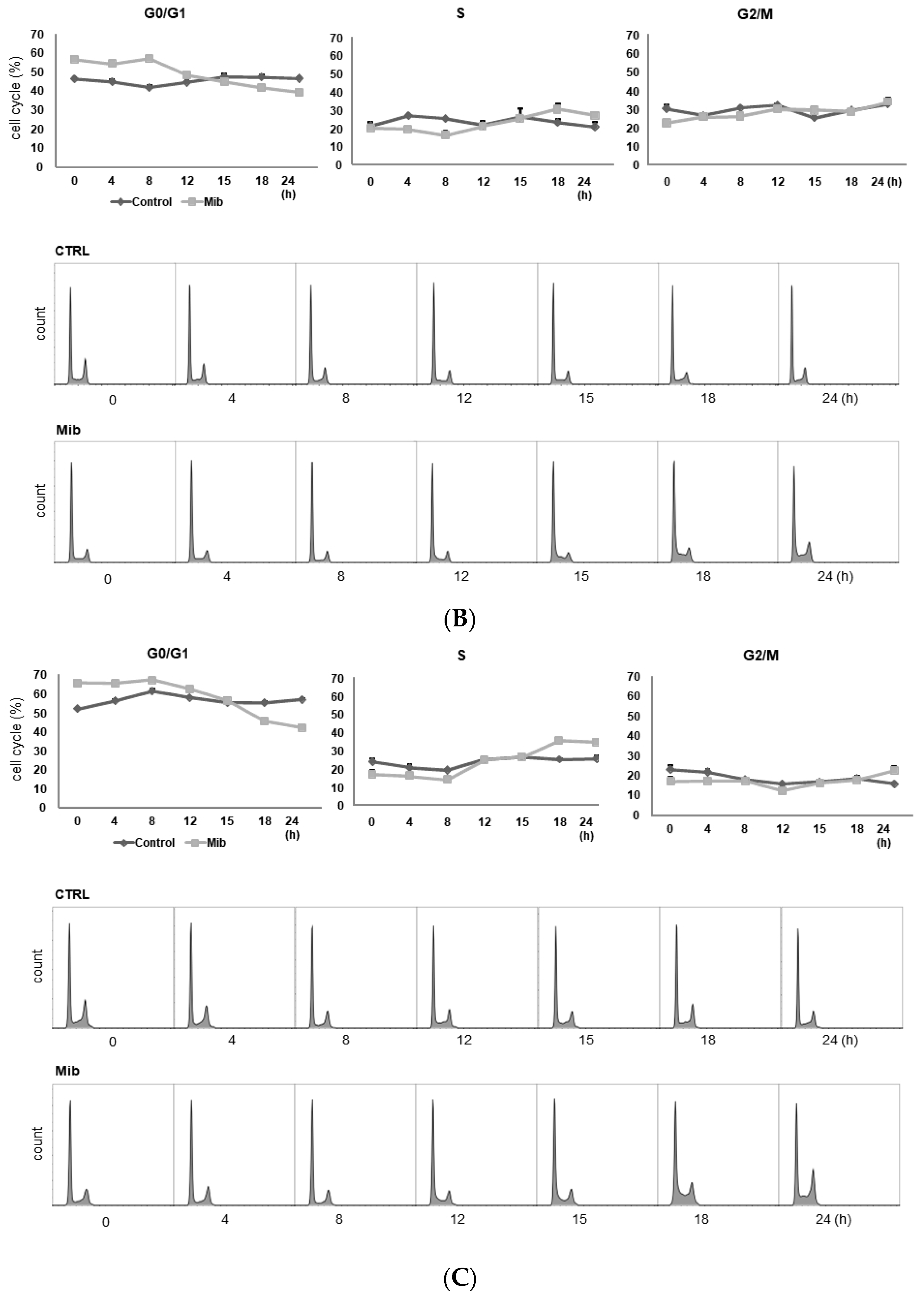
3.2. Comparison of the Cell Cycle Distribution between Serum Starvation and Mibefradil Treatment
3.3. Cell Cycle Distribution and Cellular [3H] DDG Uptake in PC-3 Cells after Mibefradil Treatment
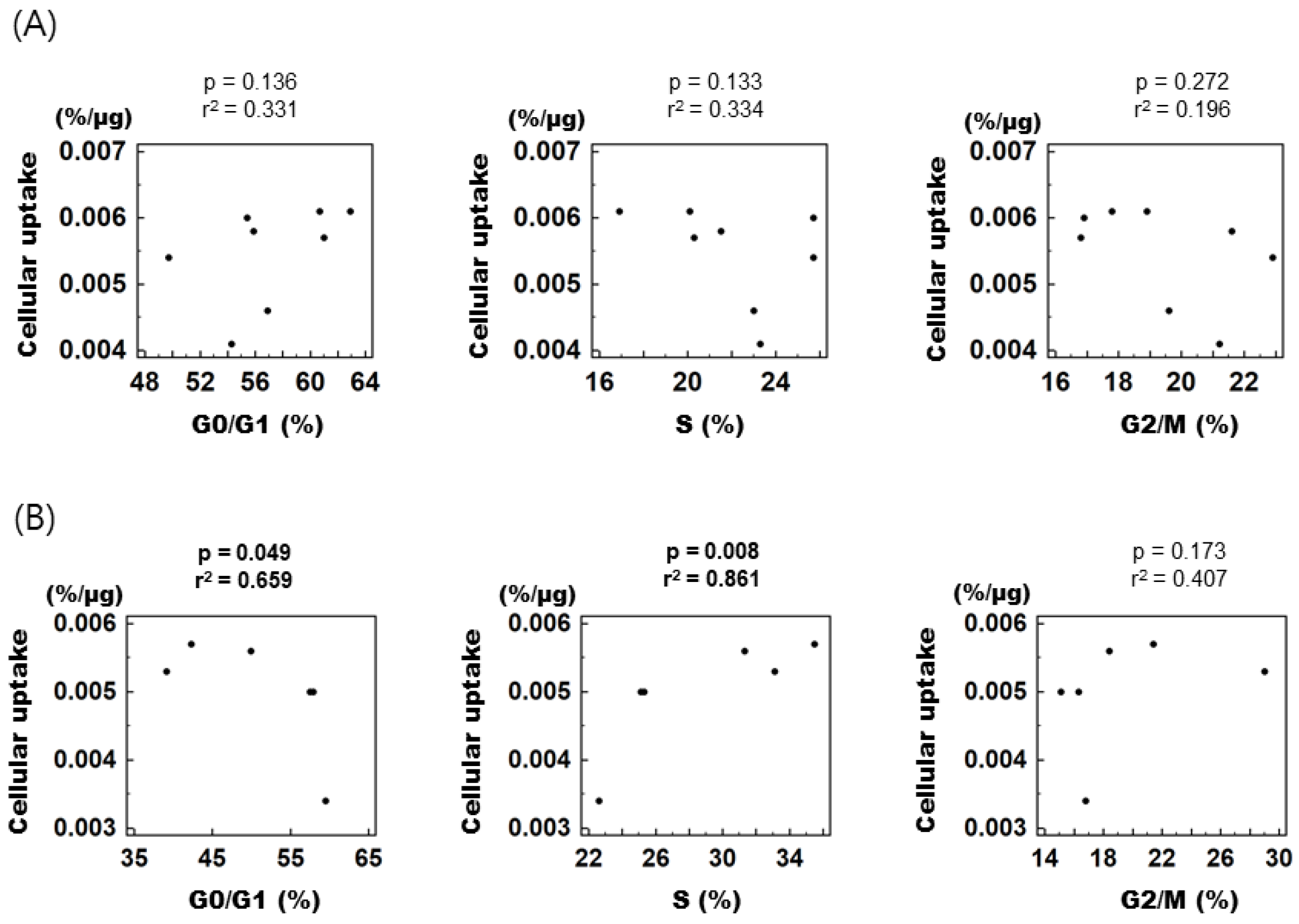
3.4. Correlation between Cell Cycle Distribution and Cellular [3H] DDG Uptake in PC-3 Cells after Mibefradil Treatment
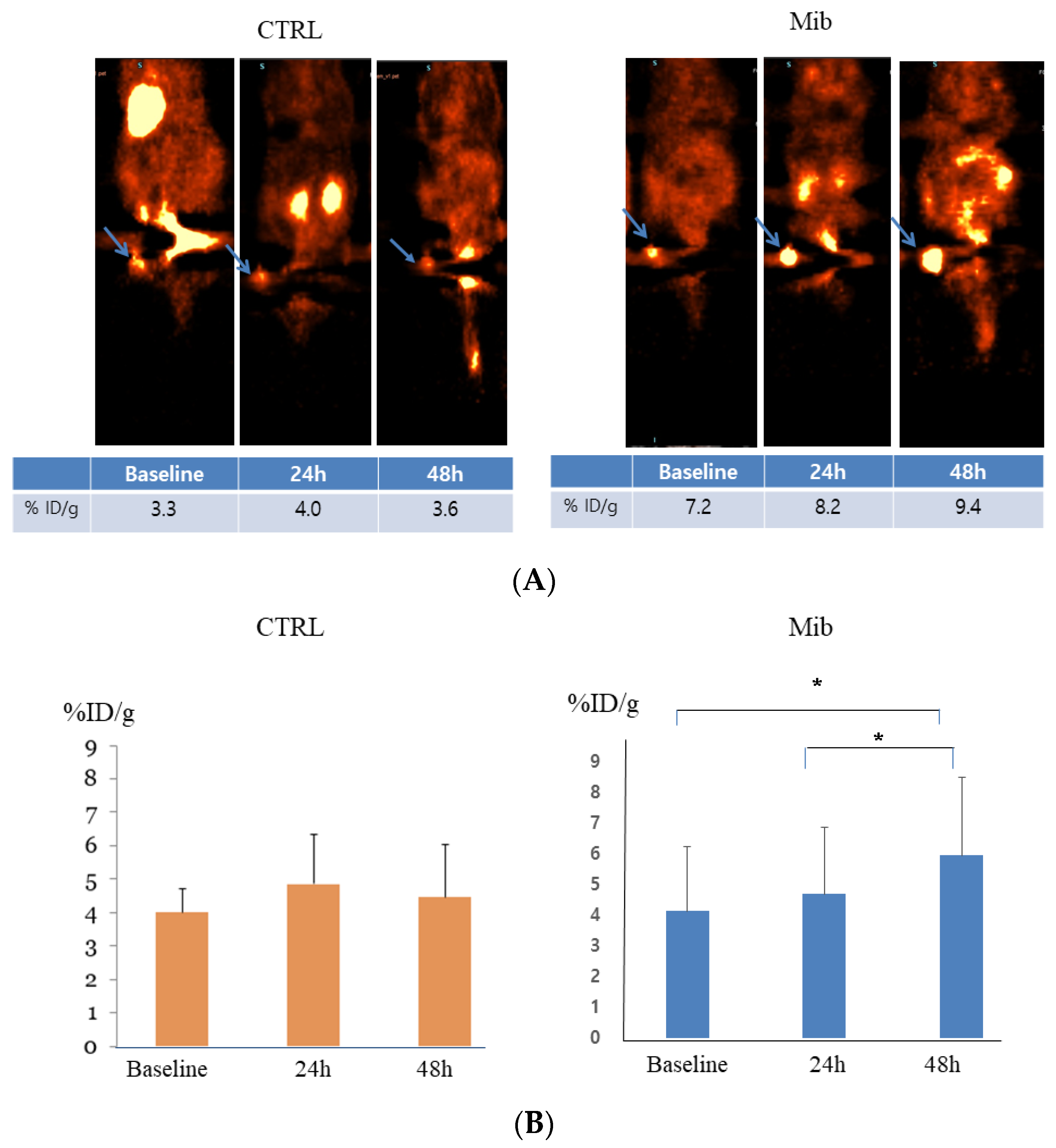
3.5. In Vivo PET Imaging after Mibefradil Treatment
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Schoder, H.; Larson, S.M.; Yeung, H.W. PET/CT in oncology: Integration into clinical management of lymphoma, melanoma, and gastrointestinal malignancies. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2004, 45 (Suppl. S1), 72S–81S. [Google Scholar]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- De Geus-Oei, L.F.; van Krieken, J.H.; Aliredjo, R.P.; Krabbe, P.F.; Frielink, C.; Verhagen, A.F.; Boerman, O.C.; Oyen, W.J. Biological correlates of FDG uptake in non-small cell lung cancer. Lung Cancer 2007, 55, 79–87. [Google Scholar] [CrossRef]
- Paudyal, B.; Oriuchi, N.; Paudyal, P.; Higuchi, T.; Nakajima, T.; Endo, K. Expression of glucose transporters and hexokinase II in cholangiocellular carcinoma compared using [18F]-2-fluro-2-deoxy-D-glucose positron emission tomography. Cancer Sci. 2008, 99, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Tomita, Y.; Qiu, Y.; Zhang, B.; Ueda, T.; Myoui, A.; Higuchi, I.; Yoshikawa, H.; Aozasa, K.; Hatazawa, J. 18F-FDG-PET of musculoskeletal tumors: A correlation with the expression of glucose transporter 1 and hexokinase II. Ann. Nucl. Med. 2008, 22, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Lawrentschuk, N.; Davis, I.D.; Bolton, D.M.; Scott, A.M. Positron emission tomography and molecular imaging of the prostate: An update. BJU Int. 2006, 97, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.N.; Fartoux, L.; Balogova, S.; Nataf, V.; Kerrou, K.; Gutman, F.; Huchet, V.; Ancel, D.; Grange, J.D.; Rosmorduc, O. Detection of hepatocellular carcinoma with PET/CT: A prospective comparison of 18F-fluorocholine and 18F-FDG in patients with cirrhosis or chronic liver disease. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2010, 51, 1699–1706. [Google Scholar] [CrossRef]
- Paudyal, B.; Paudyal, P.; Oriuchi, N.; Tsushima, Y.; Nakajima, T.; Endo, K. Clinical implication of glucose transport and metabolism evaluated by 18F-FDG PET in hepatocellular carcinoma. Int. J. Oncol. 2008, 33, 1047–1054. [Google Scholar]
- Torizuka, T.; Tamaki, N.; Inokuma, T.; Magata, Y.; Sasayama, S.; Yonekura, Y.; Tanaka, A.; Yamaoka, Y.; Yamamoto, K.; Konishi, J. In vivo assessment of glucose metabolism in hepatocellular carcinoma with FDG-PET. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1995, 36, 1811–1817. [Google Scholar]
- Choi, B.H.; Song, H.S.; An, Y.S.; Han, S.U.; Kim, J.H.; Yoon, J.K. Relation Between Fluorodeoxyglucose Uptake and Glucose Transporter-1 Expression in Gastric Signet Ring Cell Carcinoma. Nucl. Med. Mol. Imaging 2011, 45, 30–35. [Google Scholar] [CrossRef]
- Farolfi, A.; Calderoni, L.; Mattana, F.; Mei, R.; Telo, S.; Fanti, S.; Castellucci, P. Current and Emerging Clinical Applications of PSMA PET Diagnostic Imaging for Prostate Cancer. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2021, 62, 596–604. [Google Scholar] [CrossRef]
- Nyakale, N.; Filippi, L.; Aldous, C.; Sathekge, M. Update on PET Radiopharmaceuticals for Imaging Hepatocellular Carcinoma. Cancers 2023, 15, 1975. [Google Scholar] [CrossRef]
- Chen, H.; Pang, Y.; Li, J.; Kang, F.; Xu, W.; Meng, T.; Shang, Q.; Zhao, J.; Guan, Y.; Wu, H.; et al. Comparison of [(68)Ga]Ga-FAPI and [(18)F]FDG uptake in patients with gastric signet-ring-cell carcinoma: A multicenter retrospective study. Eur. Radiol. 2023, 33, 1329–1341. [Google Scholar] [CrossRef]
- Park, J.W.; Kim, J.H.; Kim, S.K.; Kang, K.W.; Park, K.W.; Choi, J.I.; Lee, W.J.; Kim, C.M.; Nam, B.H. A prospective evaluation of 18F-FDG and 11C-acetate PET/CT for detection of primary and metastatic hepatocellular carcinoma. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2008, 49, 1912–1921. [Google Scholar] [CrossRef]
- Deep, G.; Agarwal, R. New combination therapies with cell-cycle agents. Curr. Opin. Investig. Drugs 2008, 9, 591–604. [Google Scholar]
- Antal, L.; Martin-Caraballo, M. T-type Calcium Channels in Cancer. Cancers 2019, 11, 134. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Gray, L.S.; Dziegielewski, J. T-type calcium channels blockers as new tools in cancer therapies. Pflug. Arch. 2014, 466, 801–810. [Google Scholar] [CrossRef]
- Visa, A.; Alza, L.; Casas-Benito, A.; Herreros, J.; Canti, C. Targeting T-type channels in cancer: What is on and what is off? Drug. Discov. Today 2022, 27, 743–758. [Google Scholar] [CrossRef]
- Gray, L.S.; Macdonald, T.L. The pharmacology and regulation of T type calcium channels: New opportunities for unique therapeutics for cancer. Cell Calcium 2006, 40, 115–120. [Google Scholar] [CrossRef]
- Keir, S.T.; Reardon, D.A.; Saling, J.R.; Gray, L.S.; Bigner, D.D.; Friedman, H.S. Mibefradil, a novel therapy for glioblastoma multiforme: Interlaced therapy in a murine model [abstract]. Neuro-Oncology 2011, 13, iii108. [Google Scholar]
- Dziegielewska, B.; Casarez, E.V.; Yang, W.Z.; Gray, L.S.; Dziegielewski, J.; Slack-Davis, J.K. T-Type Ca2+ Channel Inhibition Sensitizes Ovarian Cancer to Carboplatin. Mol. Cancer 2016, 15, 460–470. [Google Scholar] [CrossRef]
- Panner, A.; Wurster, R.D. T-type calcium channels and tumor proliferation. Cell Calcium 2006, 40, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.T.; Zeng, X.B.; Pottle, J.E.; Lee, K.; Wang, A.R.; Yi, S.G.; Scruggs, J.A.; Sikka, S.S.; Li, M. Calcium signaling and T-type calcium channels in cancer cell cycling. World J. Gastroenterol. WJG 2008, 14, 4984–4991. [Google Scholar] [PubMed]
- Nakamura, H.; Hirata, T.; Kitamura, H.; Nishikawa, J. Correlation of the standardized uptake value in FDG-PET with the expression level of cell-cycle-related molecular biomarkers in resected non-small cell lung cancers. Ann. Thorac. Cardiovasc. Surg. Off. J. Assoc. Thorac. Cardiovasc. Surg. Asia 2009, 15, 304–310. [Google Scholar]
- Tchou, J.; Sonnad, S.S.; Bergey, M.R.; Basu, S.; Tomaszewski, J.; Alavi, A.; Schnall, M. Degree of tumor FDG uptake correlates with proliferation index in triple negative breast cancer. Mol. Imaging Biol. MIB Off. Publ. Acad. Mol. Imaging 2010, 12, 657–662. [Google Scholar] [CrossRef]
- Lapela, M.; Leskinen, S.; Minn, H.R.; Lindholm, P.; Klemi, P.J.; Soderstrom, K.O.; Bergman, J.; Haaparanta, M.; Ruotsalainen, U.; Solin, O.; et al. Increased glucose metabolism in untreated non-Hodgkin’s lymphoma: A study with positron emission tomography and fluorine-18-fluorodeoxyglucose. Blood 1995, 86, 3522–3527. [Google Scholar] [PubMed]
- Mullins, M.E.; Horowitz, B.Z.; Linden, D.H.; Smith, G.W.; Norton, R.L.; Stump, J. Life-threatening interaction of mibefradil and beta-blockers with dihydropyridine calcium channel blockers. JAMA J. Am. Med. Assoc. 1998, 280, 157–158. [Google Scholar]
- Bertolesi, G.E.; Shi, C.; Elbaum, L.; Jollimore, C.; Rozenberg, G.; Barnes, S.; Kelly, M.E. The Ca(2+) channel antagonists mibefradil and pimozide inhibit cell growth via different cytotoxic mechanisms. Mol. Pharmacol. 2002, 62, 210–219. [Google Scholar] [CrossRef]
- Direcks, W.G.; Berndsen, S.C.; Proost, N.; Peters, G.J.; Balzarini, J.; Spreeuwenberg, M.D.; Lammertsma, A.A.; Molthoff, C.F. [18F]FDG and [18F]FLT uptake in human breast cancer cells in relation to the effects of chemotherapy: An in vitro study. Br. J. Cancer 2008, 99, 481–487. [Google Scholar] [CrossRef]
- Morley, K.L.; Ferguson, P.J.; Koropatnick, J. Tangeretin and nobiletin induce G1 cell cycle arrest but not apoptosis in human breast and colon cancer cells. Cancer Lett. 2007, 251, 168–178. [Google Scholar] [CrossRef]
- Yoon, J.K.; Byeon, H.E.; Ko, S.A.; Park, B.N.; An, Y.S.; Lee, H.Y.; Lee, Y.W.; Lee, S.J. Cell cycle synchronisation using thiazolidinediones affects cellular glucose metabolism and enhances the therapeutic effect of 2-deoxyglucose in colon cancer. Sci. Rep. 2020, 10, 4713. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, S.; Salavati, A.; Segtnan, E.A.; Grupe, P.; Hoilund-Carlsen, P.F.; Alavi, A. Dual-time-point Imaging and Delayed-time-point Fluorodeoxyglucose-PET/Computed Tomography Imaging in Various Clinical Settings. PET Clin. 2016, 11, 65–84. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) † | [3H] DDG Uptake (%) * | Mean ± SE ‡ | ||
|---|---|---|---|---|
| Exp 1 (Passage 5) | Exp 2 (Passage 6) | Exp 4 (Passage 8) | ||
| 0 | 90.4% (0.569) | 71.0% (0.131) | 122.5% (0.132) | 91.3 ± 12.0% |
| 4 | 111.3% (0.469) | 63.6% (0.110) | 110.4% (0.539) | 95.1 ± 15.8% |
| 8 | 104.5% (0.769) | 106.1% (0.078) | 105.2% (0.407) | 105.2 ± 0.5% |
| 12 | 101.2% (0.902) | 100.6% (0.987) | 105.8% (0.247) | 102.5 ± 1.6% |
| 15 | 122.8% (0.052) | 141.2% (0.279) | 110.4% (0.209) | 124.8 ± 8.9% |
| 18 | 134.6% (0.012) | 131.8% (0.106) | 114.0% (0.048) | 126.8 ± 6.4% |
| 24 | 135.1% (0.001) | 147.2% (0.013) | 155.1% (0.047) | 145.8 ± 5.8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, J.-K.; Kang, W.J. Modulation of FDG Uptake by Cell Cycle Synchronization Using a T-Type Calcium Channel Inhibitor. Cancers 2023, 15, 5244. https://doi.org/10.3390/cancers15215244
Yoon J-K, Kang WJ. Modulation of FDG Uptake by Cell Cycle Synchronization Using a T-Type Calcium Channel Inhibitor. Cancers. 2023; 15(21):5244. https://doi.org/10.3390/cancers15215244
Chicago/Turabian StyleYoon, Joon-Kee, and Won Jun Kang. 2023. "Modulation of FDG Uptake by Cell Cycle Synchronization Using a T-Type Calcium Channel Inhibitor" Cancers 15, no. 21: 5244. https://doi.org/10.3390/cancers15215244
APA StyleYoon, J.-K., & Kang, W. J. (2023). Modulation of FDG Uptake by Cell Cycle Synchronization Using a T-Type Calcium Channel Inhibitor. Cancers, 15(21), 5244. https://doi.org/10.3390/cancers15215244