
Critical Roles of SRC-3 in the Development and Progression of Breast Cancer, Rendering It a Prospective Clinical Target


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. BCa and Estrogen Action
3. BCa Tumor Microenvironment
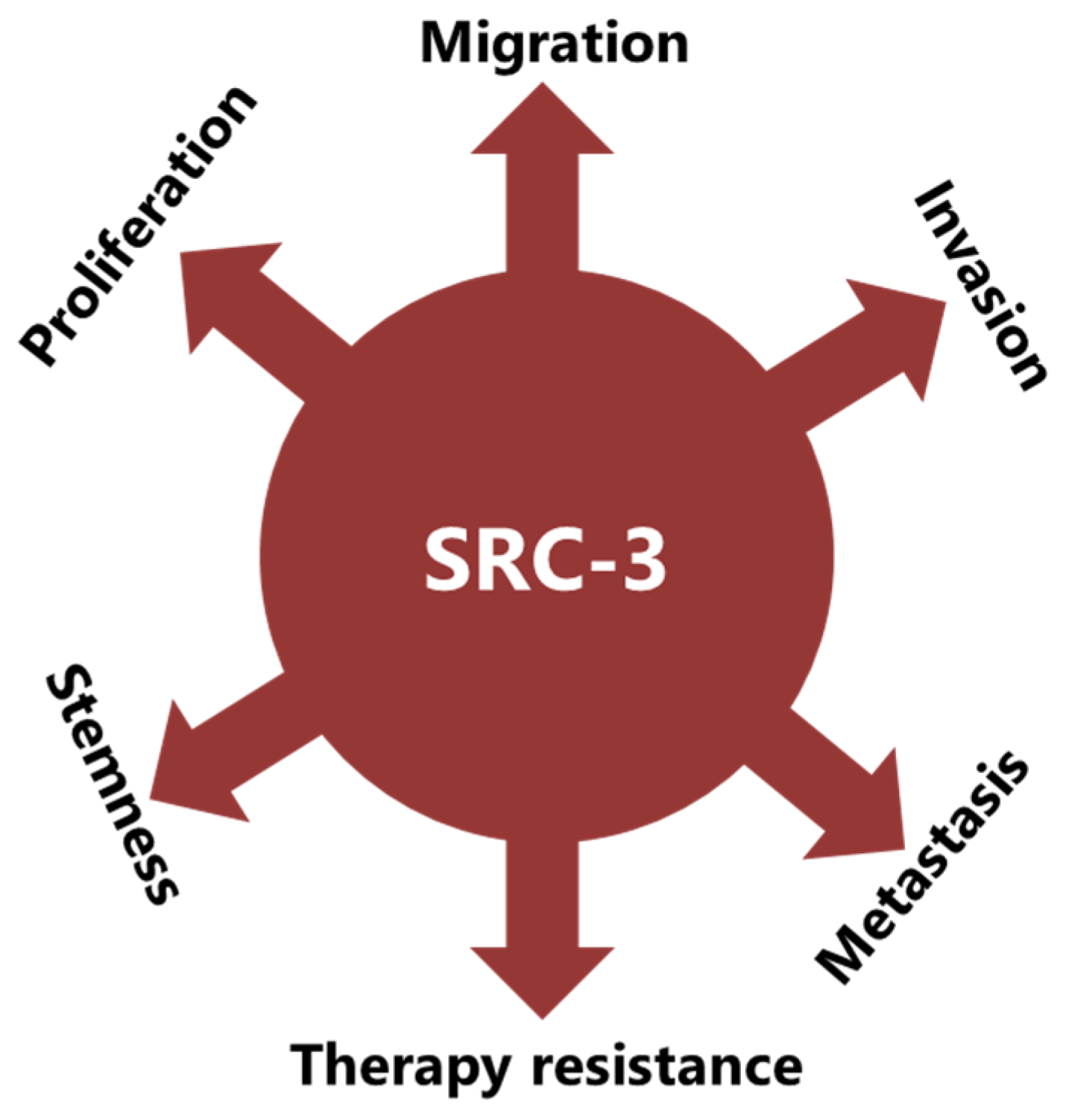
4. SRC-3 Has Multiple Roles in BCa Pathogenesis
4.1. SRC-3 Affects the Tumor Microenvironment
4.2. SRC-3 Promotes Stemness
4.3. SRC-3 Promotes Malignant Behaviors of Tumor Cells
5. SRC-3 May Contribute to Therapy Resistance in Multiple Ways in BCa
5.1. SRC-3 May Contribute to Hormone Therapy Resistance
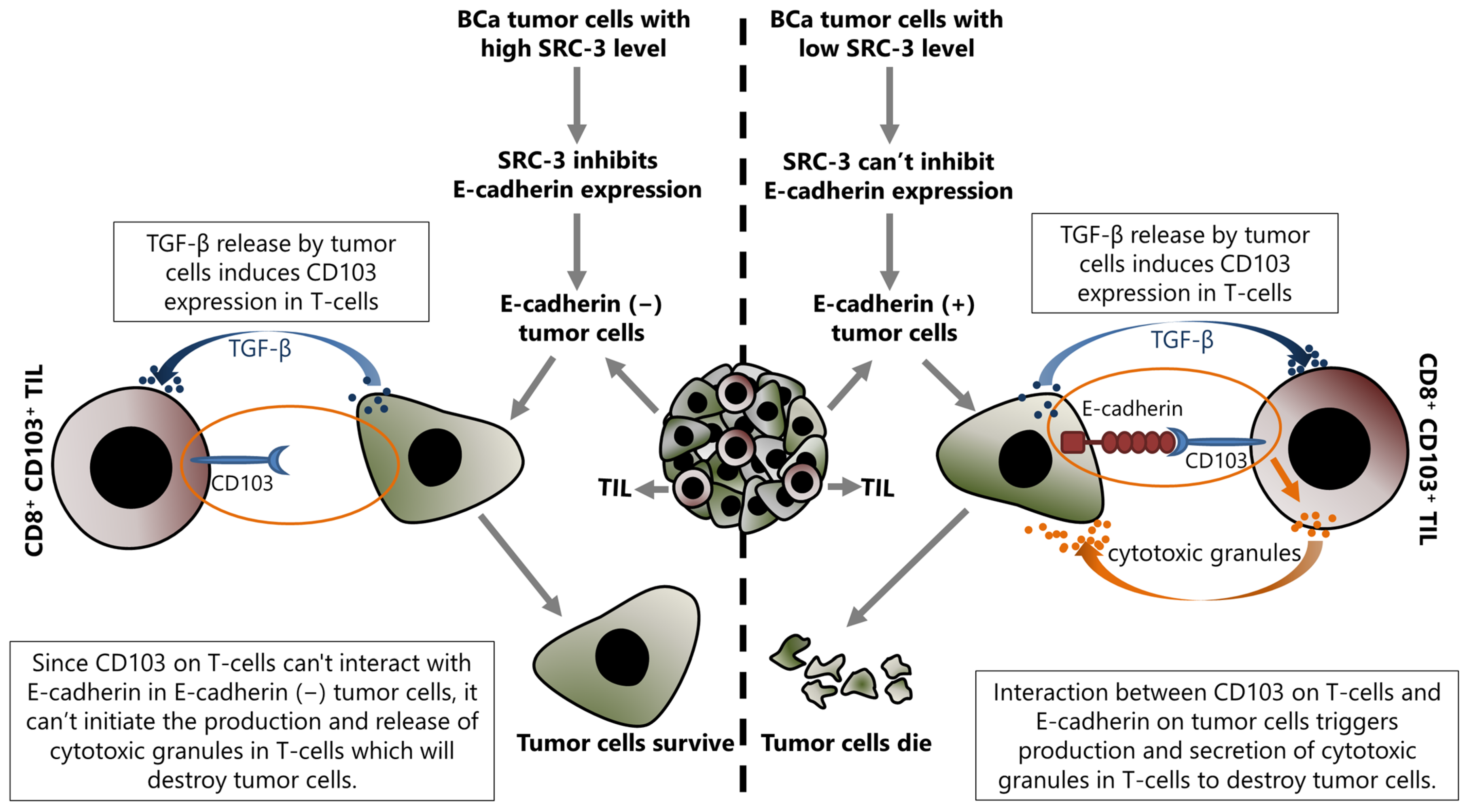
5.2. SRC-3 May Contribute to Immunotherapy Resistance
6. SRC-3 Is a Promising Target to Overcome Therapy Resistance in BCa
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Faltas, C.L.; LeBron, K.A.; Holz, M.K. Unconventional Estrogen Signaling in Health and Disease. Endocrinology 2020, 161, bqaa030. [Google Scholar] [CrossRef]
- Clusan, L.; Ferriere, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, R.C.; O’Malley, B.W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 2009, 9, 615–630. [Google Scholar] [CrossRef]
- Stashi, E.; York, B.; O’Malley, B.W. Steroid receptor coactivators: Servants and masters for control of systems metabolism. Trends Endocrinol. Metab. 2014, 25, 337–347. [Google Scholar] [CrossRef]
- Xu, J.; Liao, L.; Ning, G.; Yoshida-Komiya, H.; Deng, C.; O’Malley, B.W. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc. Natl. Acad. Sci. USA 2000, 97, 6379–6384. [Google Scholar] [CrossRef]
- Wu, R.C.; Qin, J.; Yi, P.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol. Cell 2004, 15, 937–949. [Google Scholar] [CrossRef]
- Zheng, F.F.; Wu, R.C.; Smith, C.L.; O’Malley, B.W. Rapid estrogen-induced phosphorylation of the SRC-3 coactivator occurs in an extranuclear complex containing estrogen receptor. Mol. Cell. Biol. 2005, 25, 8273–8284. [Google Scholar] [CrossRef]
- Anzick, S.L.; Kononen, J.; Walker, R.L.; Azorsa, D.O.; Tanner, M.M.; Guan, X.Y.; Sauter, G.; Kallioniemi, O.P.; Trent, J.M.; Meltzer, P.S. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 1997, 277, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.Y.; Xu, J.; Anzick, S.L.; Zhang, H.; Trent, J.M.; Meltzer, P.S. Hybrid selection of transcribed sequences from microdissected DNA: Isolation of genes within amplified region at 20q11-q13.2 in breast cancer. Cancer Res. 1996, 56, 3446–3450. [Google Scholar] [PubMed]
- Gojis, O.; Rudraraju, B.; Gudi, M.; Hogben, K.; Sousha, S.; Coombes, R.C.; Cleator, S.; Palmieri, C. The role of SRC-3 in human breast cancer. Nat. Rev. Clin. Oncol. 2010, 7, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Deng, C.X.; Chen, Q. SRC-3, a Steroid Receptor Coactivator: Implication in Cancer. Int. J. Mol. Sci. 2021, 22, 4760. [Google Scholar] [CrossRef]
- Nikolai, B.C.; Jain, P.; Cardenas, D.L.; York, B.; Feng, Q.; McKenna, N.J.; Dasgupta, S.; Lonard, D.M.; O’Malley, B.W. Steroid receptor coactivator 3 (SRC-3/AIB1) is enriched and functional in mouse and human Tregs. Sci. Rep. 2021, 11, 3441. [Google Scholar] [CrossRef]
- Rohira, A.D.; Yan, F.; Wang, L.; Wang, J.; Zhou, S.; Lu, A.; Yu, Y.; Xu, J.; Lonard, D.M.; O’Malley, B.W. Targeting SRC Coactivators Blocks the Tumor-Initiating Capacity of Cancer Stem-like Cells. Cancer Res. 2017, 77, 4293–4304. [Google Scholar] [CrossRef]
- Kiliti, A.J.; Sharif, G.M.; Martin, M.B.; Wellstein, A.; Riegel, A.T. AIB1/SRC-3/NCOA3 function in estrogen receptor alpha positive breast cancer. Front. Endocrinol. 2023, 14, 1250218. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Orrantia-Borunda, E.; Anchondo-Nunez, P.; Acuna-Aguilar, L.E.; Gomez-Valles, F.O.; Ramirez-Valdespino, C.A. Subtypes of Breast Cancer. In Breast Cancer, Mayrovitz, H.N., Ed.; Exon Publications: Brisbane, Australia, 2022. [Google Scholar]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Zattarin, E.; Leporati, R.; Ligorio, F.; Lobefaro, R.; Vingiani, A.; Pruneri, G.; Vernieri, C. Hormone Receptor Loss in Breast Cancer: Molecular Mechanisms, Clinical Settings, and Therapeutic Implications. Cells 2020, 9, 2644. [Google Scholar] [CrossRef]
- Kennecke, H.; McArthur, H.; Olivotto, I.A.; Speers, C.; Bajdik, C.; Chia, S.K.; Ellard, S.; Norris, B.; Hayes, M.; Barnett, J.; et al. Risk of early recurrence among postmenopausal women with estrogen receptor-positive early breast cancer treated with adjuvant tamoxifen. Cancer 2008, 112, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.F.; Olivotto, I.A.; Speers, C.; Norris, B.; Chia, S.K.; Bryce, C.; Gelmon, K.A. Late risk of relapse and mortality among postmenopausal women with estrogen responsive early breast cancer after 5 years of tamoxifen. Ann. Oncol. 2007, 18, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Verras, G.I.; Tchabashvili, L.; Mulita, F.; Grypari, I.M.; Sourouni, S.; Panagodimou, E.; Argentou, M.I. Micropapillary Breast Carcinoma: From Molecular Pathogenesis to Prognosis. Breast Cancer 2022, 14, 41–61. [Google Scholar] [CrossRef]
- Yang, Y.L.; Liu, B.B.; Zhang, X.; Fu, L. Invasive Micropapillary Carcinoma of the Breast: An Update. Arch. Pathol. Lab. Med. 2016, 140, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.Q.; Feng, J.H.; Zhao, Y.J. Clinicopathological features of invasive micropapillary carcinoma of the breast. Oncol. Lett. 2015, 9, 1163–1166. [Google Scholar] [CrossRef]
- Lewis, G.D.; Xing, Y.; Haque, W.; Patel, T.; Schwartz, M.R.; Chen, A.C.; Farach, A.; Hatch, S.S.; Butler, E.B.; Chang, J.C.; et al. The impact of molecular status on survival outcomes for invasive micropapillary carcinoma of the breast. Breast J. 2019, 25, 1171–1176. [Google Scholar] [CrossRef]
- Sinha, V.C.; Piwnica-Worms, H. Intratumoral Heterogeneity in Ductal Carcinoma In Situ: Chaos and Consequence. J. Mammary Gland. Biol. Neoplasia 2018, 23, 191–205. [Google Scholar] [CrossRef]
- Provenzano, E.; Hopper, J.L.; Giles, G.G.; Marr, G.; Venter, D.J.; Armes, J.E. Biological markers that predict clinical recurrence in ductal carcinoma in situ of the breast. Eur. J. Cancer 2003, 39, 622–630. [Google Scholar] [CrossRef]
- Roka, S.; Rudas, M.; Taucher, S.; Dubsky, P.; Bachleitner-Hofmann, T.; Kandioler, D.; Gnant, M.; Jakesz, R. High nuclear grade and negative estrogen receptor are significant risk factors for recurrence in DCIS. Eur. J. Surg. Oncol. 2004, 30, 243–247. [Google Scholar] [CrossRef]
- Zhou, W.; Jirstrom, K.; Amini, R.M.; Fjallskog, M.L.; Sollie, T.; Lindman, H.; Sorlie, T.; Blomqvist, C.; Warnberg, F. Molecular subtypes in ductal carcinoma in situ of the breast and their relation to prognosis: A population-based cohort study. BMC Cancer 2013, 13, 512. [Google Scholar] [CrossRef]
- Williams, K.E.; Barnes, N.L.P.; Cramer, A.; Johnson, R.; Cheema, K.; Morris, J.; Howe, M.; Bundred, N.J. Molecular phenotypes of DCIS predict overall and invasive recurrence. Ann. Oncol. 2015, 26, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Akrida, I.; Mulita, F. The clinical significance of HER2 expression in DCIS. Med. Oncol. 2022, 40, 16. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, T.J.; Blair, S.L.; Hosseini, A.; Harismendy, O.; Wallace, A.M. HER2-Overexpressing Ductal Carcinoma In Situ Associated with Increased Risk of Ipsilateral Invasive Recurrence, Receptor Discordance with Recurrence. Cancer Prev. Res. 2020, 13, 761–772. [Google Scholar] [CrossRef]
- Auchus, M.L.; Auchus, R.J. Human steroid biosynthesis for the oncologist. J. Investig. Med. 2012, 60, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.V.; Jacobson, H.I.; Walf, A.A.; Frye, C.A. Estrogen action: A historic perspective on the implications of considering alternative approaches. Physiol. Behav. 2010, 99, 151–162. [Google Scholar] [CrossRef]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Echeverria, P.C.; Picard, D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim. Biophys. Acta 2010, 1803, 641–649. [Google Scholar] [CrossRef]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genomics. 2006, 7, 497–508. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef]
- Li, C.; Briggs, M.R.; Ahlborn, T.E.; Kraemer, F.B.; Liu, J. Requirement of Sp1 and estrogen receptor alpha interaction in 17beta-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology 2001, 142, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Safe, S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam. Horm. 2001, 62, 231–252. [Google Scholar] [CrossRef] [PubMed]
- Stossi, F.; Likhite, V.S.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J. Biol. Chem. 2006, 281, 16272–16278. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [CrossRef] [PubMed]
- McKenna, N.J.; O’Malley, B.W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 108, 465–474. [Google Scholar] [CrossRef]
- Rae, J.M.; Johnson, M.D. What does an orphan G-protein-coupled receptor have to do with estrogen? Breast Cancer Res. 2005, 7, 243–244. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef]
- Filardo, E.J.; Thomas, P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: Its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology 2012, 153, 2953–2962. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERalpha) and beta (ERbeta): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J.A. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.T.; Jordan, V.C. The biological role of estrogen receptors alpha and beta in cancer. Crit. Rev. Oncol. Hematol. 2004, 50, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Bakas, P.; Liapis, A.; Vlahopoulos, S.; Giner, M.; Logotheti, S.; Creatsas, G.; Meligova, A.K.; Alexis, M.N.; Zoumpourlis, V. Estrogen receptor alpha and beta in uterine fibroids: A basis for altered estrogen responsiveness. Fertil. Steril. 2008, 90, 1878–1885. [Google Scholar] [CrossRef]
- Logotheti, S.; Papaevangeliou, D.; Michalopoulos, I.; Sideridou, M.; Tsimaratou, K.; Christodoulou, I.; Pyrillou, K.; Gorgoulis, V.; Vlahopoulos, S.; Zoumpourlis, V. Progression of mouse skin carcinogenesis is associated with increased ERalpha levels and is repressed by a dominant negative form of ERalpha. PLoS ONE 2012, 7, e41957. [Google Scholar] [CrossRef] [PubMed]
- Welboren, W.J.; Sweep, F.C.; Span, P.N.; Stunnenberg, H.G. Genomic actions of estrogen receptor alpha: What are the targets and how are they regulated? Endocr. Relat. Cancer 2009, 16, 1073–1089. [Google Scholar] [CrossRef] [PubMed]
- Dubik, D.; Dembinski, T.C.; Shiu, R.P. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res. 1987, 47, 6517–6521. [Google Scholar] [PubMed]
- Millour, J.; Constantinidou, D.; Stavropoulou, A.V.; Wilson, M.S.; Myatt, S.S.; Kwok, J.M.; Sivanandan, K.; Coombes, R.C.; Medema, R.H.; Hartman, J.; et al. FOXM1 is a transcriptional target of ERalpha and has a critical role in breast cancer endocrine sensitivity and resistance. Oncogene 2010, 29, 2983–2995. [Google Scholar] [CrossRef]
- Sabbah, M.; Courilleau, D.; Mester, J.; Redeuilh, G. Estrogen induction of the cyclin D1 promoter: Involvement of a cAMP response-like element. Proc. Natl. Acad. Sci. USA 1999, 96, 11217–11222. [Google Scholar] [CrossRef]
- Oesterreich, S.; Deng, W.; Jiang, S.; Cui, X.; Ivanova, M.; Schiff, R.; Kang, K.; Hadsell, D.L.; Behrens, J.; Lee, A.V. Estrogen-mediated down-regulation of E-cadherin in breast cancer cells. Cancer Res. 2003, 63, 5203–5208. [Google Scholar]
- Fujita, N.; Jaye, D.L.; Kajita, M.; Geigerman, C.; Moreno, C.S.; Wade, P.A. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003, 113, 207–219. [Google Scholar] [CrossRef]
- Vareslija, D.; Ward, E.; Purcell, S.P.; Cosgrove, N.S.; Cocchiglia, S.; O’Halloran, P.J.; Charmsaz, S.; Bane, F.T.; Brett, F.M.; Farrell, M.; et al. Comparative analysis of the AIB1 interactome in breast cancer reveals MTA2 as a repressive partner which silences E-Cadherin to promote EMT and associates with a pro-metastatic phenotype. Oncogene 2021, 40, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Lauritsen, K.J.; List, H.J.; Reiter, R.; Wellstein, A.; Riegel, A.T. A role for TGF-beta in estrogen and retinoid mediated regulation of the nuclear receptor coactivator AIB1 in MCF-7 breast cancer cells. Oncogene 2002, 21, 7147–7155. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Ben-Baruch, A. Host microenvironment in breast cancer development: Inflammatory cells, cytokines and chemokines in breast cancer progression: Reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2003, 5, 31–36. [Google Scholar] [CrossRef]
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. 2020, 2020, 6283796. [Google Scholar] [CrossRef]
- Li, J.J.; Tsang, J.Y.; Tse, G.M. Tumor Microenvironment in Breast Cancer-Updates on Therapeutic Implications and Pathologic Assessment. Cancers 2021, 13, 4233. [Google Scholar] [CrossRef]
- De Guillebon, E.; Dardenne, A.; Saldmann, A.; Seguier, S.; Tran, T.; Paolini, L.; Lebbe, C.; Tartour, E. Beyond the concept of cold and hot tumors for the development of novel predictive biomarkers and the rational design of immunotherapy combination. Int. J. Cancer 2020, 147, 1509–1518. [Google Scholar] [CrossRef]
- Farc, O.; Cristea, V. An overview of the tumor microenvironment, from cells to complex networks (Review). Exp. Ther. Med. 2021, 21, 96. [Google Scholar] [CrossRef]
- Dannenfelser, R.; Nome, M.; Tahiri, A.; Ursini-Siegel, J.; Vollan, H.K.M.; Haakensen, V.D.; Helland, A.; Naume, B.; Caldas, C.; Borresen-Dale, A.L.; et al. Data-driven analysis of immune infiltrate in a large cohort of breast cancer and its association with disease progression, ER activity, and genomic complexity. Oncotarget 2017, 8, 57121–57133. [Google Scholar] [CrossRef]
- Cunningham, M.; Gilkeson, G. Estrogen receptors in immunity and autoimmunity. Clin. Rev. Allergy Immunol. 2011, 40, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2015, 6, 635. [Google Scholar] [CrossRef] [PubMed]
- Okasha, S.A.; Ryu, S.; Do, Y.; McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Evidence for estradiol-induced apoptosis and dysregulated T cell maturation in the thymus. Toxicology 2001, 163, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Polanczyk, M.J.; Carson, B.D.; Subramanian, S.; Afentoulis, M.; Vandenbark, A.A.; Ziegler, S.F.; Offner, H. Cutting edge: Estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J. Immunol. 2004, 173, 2227–2230. [Google Scholar] [CrossRef] [PubMed]
- Staples, J.E.; Gasiewicz, T.A.; Fiore, N.C.; Lubahn, D.B.; Korach, K.S.; Silverstone, A.E. Estrogen receptor alpha is necessary in thymic development and estradiol-induced thymic alterations. J. Immunol. 1999, 163, 4168–4174. [Google Scholar] [CrossRef]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef]
- McMurray, R.W.; Ndebele, K.; Hardy, K.J.; Jenkins, J.K. 17-beta-estradiol suppresses IL-2 and IL-2 receptor. Cytokine 2001, 14, 324–333. [Google Scholar] [CrossRef]
- Somasundaram, A.; Rothenberger, N.J.; Stabile, L.P. The Impact of Estrogen in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 33–52. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Meng, Y.; Harlin, H. Immune suppression in the tumor microenvironment. J. Immunother. 2006, 29, 233–240. [Google Scholar] [CrossRef]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int. Immunol. 2007, 19, 337–343. [Google Scholar] [CrossRef]
- Prieto, G.A.; Rosenstein, Y. Oestradiol potentiates the suppressive function of human CD4 CD25 regulatory T cells by promoting their proliferation. Immunology 2006, 118, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Tai, P.; Wang, J.; Jin, H.; Song, X.; Yan, J.; Kang, Y.; Zhao, L.; An, X.; Du, X.; Chen, X.; et al. Induction of regulatory T cells by physiological level estrogen. J. Cell. Physiol. 2008, 214, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Askenasy, N.; Kaminitz, A.; Yarkoni, S. Mechanisms of T regulatory cell function. Autoimmun. Rev. 2008, 7, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.Y.; Wang, L.; Sun, C.; Li, D.J. Estrogen enhances the functions of CD4+CD25+Foxp3+ regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell. Mol. Immunol. 2011, 8, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Adurthi, S.; Kumar, M.M.; Vinodkumar, H.S.; Mukherjee, G.; Krishnamurthy, H.; Acharya, K.K.; Bafna, U.D.; Uma, D.K.; Abhishekh, B.; Krishna, S.; et al. Oestrogen Receptor-alpha binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci. Rep. 2017, 7, 17289. [Google Scholar] [CrossRef] [PubMed]
- Itahashi, K.; Irie, T.; Nishikawa, H. Regulatory T-cell development in the tumor microenvironment. Eur. J. Immunol. 2022, 52, 1216–1227. [Google Scholar] [CrossRef]
- Kang, M.J.; Kim, K.M.; Bae, J.S.; Park, H.S.; Lee, H.; Chung, M.J.; Moon, W.S.; Lee, D.G.; Jang, K.Y. Tumor-infiltrating PD1-Positive Lymphocytes and FoxP3-Positive Regulatory T Cells Predict Distant Metastatic Relapse and Survival of Clear Cell Renal Cell Carcinoma. Transl. Oncol. 2013, 6, 282–289. [Google Scholar] [CrossRef]
- Park, H.J.; Kusnadi, A.; Lee, E.J.; Kim, W.W.; Cho, B.C.; Lee, I.J.; Seong, J.; Ha, S.J. Tumor-infiltrating regulatory T cells delineated by upregulation of PD-1 and inhibitory receptors. Cell. Immunol. 2012, 278, 76–83. [Google Scholar] [CrossRef]
- Kim, H.R.; Park, H.J.; Son, J.; Lee, J.G.; Chung, K.Y.; Cho, N.H.; Shim, H.S.; Park, S.; Kim, G.; In Yoon, H.; et al. Tumor microenvironment dictates regulatory T cell phenotype: Upregulated immune checkpoints reinforce suppressive function. J. Immunother. Cancer 2019, 7, 339. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Cinier, J.; Hubert, M.; Besson, L.; Di Roio, A.; Rodriguez, C.; Lombardi, V.; Caux, C.; Menetrier-Caux, C. Recruitment and Expansion of Tregs Cells in the Tumor Environment-How to Target Them? Cancers 2021, 13, 1850. [Google Scholar] [CrossRef]
- Pedroza-Pacheco, I.; Madrigal, A.; Saudemont, A. Interaction between natural killer cells and regulatory T cells: Perspectives for immunotherapy. Cell. Mol. Immunol. 2013, 10, 222–229. [Google Scholar] [CrossRef]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-beta signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef]
- Stockis, J.; Roychoudhuri, R.; Halim, T.Y.F. Regulation of regulatory T cells in cancer. Immunology 2019, 157, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Dong, C. Cytokine Regulation and Function in T Cells. Annu. Rev. Immunol. 2021, 39, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F. Heterogeneity of Human CD4+ T Cells Against Microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic TH17 cells in the absence of TGF-beta signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef]
- Kono, M. New insights into the metabolism of Th17 cells. Immunol. Med. 2023, 46, 15–24. [Google Scholar] [CrossRef]
- Yang, J.; Sundrud, M.S.; Skepner, J.; Yamagata, T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol. Sci. 2014, 35, 493–500. [Google Scholar] [CrossRef]
- Bailey, S.R.; Nelson, M.H.; Himes, R.A.; Li, Z.; Mehrotra, S.; Paulos, C.M. Th17 cells in cancer: The ultimate identity crisis. Front. Immunol. 2014, 5, 276. [Google Scholar] [CrossRef]
- Agalioti, T.; Cortesi, F.; Gagliani, N. TH17 cell immune adaptation. Curr. Opin. Immunol. 2023, 83, 102333. [Google Scholar] [CrossRef]
- Wang, K.; Kim, M.K.; Di Caro, G.; Wong, J.; Shalapour, S.; Wan, J.; Zhang, W.; Zhong, Z.; Sanchez-Lopez, E.; Wu, L.W.; et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014, 41, 1052–1063. [Google Scholar] [CrossRef]
- Li, J.; Lau, G.K.; Chen, L.; Dong, S.S.; Lan, H.Y.; Huang, X.R.; Li, Y.; Luk, J.M.; Yuan, Y.F.; Guan, X.Y. Interleukin 17A promotes hepatocellular carcinoma metastasis via NF-kB induced matrix metalloproteinases 2 and 9 expression. PLoS ONE 2011, 6, e21816. [Google Scholar] [CrossRef]
- Gu, F.M.; Li, Q.L.; Gao, Q.; Jiang, J.H.; Zhu, K.; Huang, X.Y.; Pan, J.F.; Yan, J.; Hu, J.H.; Wang, Z.; et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol. Cancer 2011, 10, 150. [Google Scholar] [CrossRef]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene 2015, 34, 165–176. [Google Scholar] [CrossRef]
- Hu, X.; Liu, X.; Moisan, J.; Wang, Y.; Lesch, C.A.; Spooner, C.; Morgan, R.W.; Zawidzka, E.M.; Mertz, D.; Bousley, D.; et al. Synthetic RORgamma agonists regulate multiple pathways to enhance antitumor immunity. Oncoimmunology 2016, 5, e1254854. [Google Scholar] [CrossRef]
- Chen, J.G.; Xia, J.C.; Liang, X.T.; Pan, K.; Wang, W.; Lv, L.; Zhao, J.J.; Wang, Q.J.; Li, Y.Q.; Chen, S.P.; et al. Intratumoral expression of IL-17 and its prognostic role in gastric adenocarcinoma patients. Int. J. Biol. Sci. 2011, 7, 53–60. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Su, H.; Zhong, W.; Yuan, Y.; Yu, Z.; Fang, Y.; Zhou, H.; Li, C.; Huang, K. Interleukin-17 is a favorable prognostic marker for colorectal cancer. Clin. Transl. Oncol. 2015, 17, 50–56. [Google Scholar] [CrossRef]
- Majchrzak, K.; Nelson, M.H.; Bailey, S.R.; Bowers, J.S.; Yu, X.Z.; Rubinstein, M.P.; Himes, R.A.; Paulos, C.M. Exploiting IL-17-producing CD4+ and CD8+ T cells to improve cancer immunotherapy in the clinic. Cancer Immunol. Immunother. 2016, 65, 247–259. [Google Scholar] [CrossRef]
- Lu, L.; Pan, K.; Zheng, H.X.; Li, J.J.; Qiu, H.J.; Zhao, J.J.; Weng, D.S.; Pan, Q.Z.; Wang, D.D.; Jiang, S.S.; et al. IL-17A promotes immune cell recruitment in human esophageal cancers and the infiltrating dendritic cells represent a positive prognostic marker for patient survival. J. Immunother. 2013, 36, 451–458. [Google Scholar] [CrossRef]
- Pan, Y.; Yang, W.; Tang, B.; Wang, X.; Zhang, Q.; Li, W.; Li, L. The protective and pathogenic role of Th17 cell plasticity and function in the tumor microenvironment. Front. Immunol. 2023, 14, 1192303. [Google Scholar] [CrossRef]
- Cerboni, S.; Gehrmann, U.; Preite, S.; Mitra, S. Cytokine-regulated Th17 plasticity in human health and diseases. Immunology 2021, 163, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, D.; Foglierini, M.; Jarrossay, D.; Hu, D.; Weiner, H.L.; Kuchroo, V.K.; Lanzavecchia, A.; Notarbartolo, S.; Sallusto, F. An immunoregulatory and tissue-residency program modulated by c-MAF in human TH17 cells. Nat. Immunol. 2018, 19, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Fili, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Lai, Y.H.; Chen, H.Y.; Guo, H.R.; Su, I.J.; Chen, H.H. Interleukin-17-producing cell infiltration in the breast cancer tumour microenvironment is a poor prognostic factor. Histopathology 2013, 63, 225–233. [Google Scholar] [CrossRef]
- Wang, S.; Li, Z.; Hu, G. Prognostic role of intratumoral IL-17A expression by immunohistochemistry in solid tumors: A meta-analysis. Oncotarget 2017, 8, 66382–66391. [Google Scholar] [CrossRef]
- Du, J.W.; Xu, K.Y.; Fang, L.Y.; Qi, X.L. Interleukin-17, produced by lymphocytes, promotes tumor growth and angiogenesis in a mouse model of breast cancer. Mol. Med. Rep. 2012, 6, 1099–1102. [Google Scholar] [CrossRef]
- Avalos-Navarro, G.; Munoz-Valle, J.F.; Daneri-Navarro, A.; Quintero-Ramos, A.; Franco-Topete, R.A.; Moran-Mendoza, A.J.; Oceguera-Villanueva, A.; Bautista-Herrera, L.A.; Topete-Camacho, A.; Del Toro-Arreola, A. Circulating soluble levels of MIF in women with breast cancer in the molecular subtypes: Relationship with Th17 cytokine profile. Clin. Exp. Med. 2019, 19, 385–391. [Google Scholar] [CrossRef]
- Faucheux, L.; Grandclaudon, M.; Perrot-Dockes, M.; Sirven, P.; Berger, F.; Hamy, A.S.; Fourchotte, V.; Vincent-Salomon, A.; Mechta-Grigoriou, F.; Reyal, F.; et al. A multivariate Th17 metagene for prognostic stratification in T cell non-inflamed triple negative breast cancer. Oncoimmunology 2019, 8, e1624130. [Google Scholar] [CrossRef]
- Li, C.; Liang, Y.Y.; Feng, X.H.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Essential phosphatases and a phospho-degron are critical for regulation of SRC-3/AIB1 coactivator function and turnover. Mol. Cell 2008, 31, 835–849. [Google Scholar] [CrossRef]
- York, B.; Yu, C.; Sagen, J.V.; Liu, Z.; Nikolai, B.C.; Wu, R.C.; Finegold, M.; Xu, J.; O’Malley, B.W. Reprogramming the posttranslational code of SRC-3 confers a switch in mammalian systems biology. Proc. Natl. Acad. Sci. USA 2010, 107, 11122–11127. [Google Scholar] [CrossRef]
- Liao, L.; Kuang, S.Q.; Yuan, Y.; Gonzalez, S.M.; O’Malley, B.W.; Xu, J. Molecular structure and biological function of the cancer-amplified nuclear receptor coactivator SRC-3/AIB1. J. Steroid Biochem. Mol. Biol. 2002, 83, 3–14. [Google Scholar] [CrossRef]
- Wang, L.; Lonard, D.M.; O’Malley, B.W. The Role of Steroid Receptor Coactivators in Hormone Dependent Cancers and Their Potential as Therapeutic Targets. Horm. Cancer 2016, 7, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Tsai, S.Y.; Tsai, M.J. SRC-3/AIB1: Transcriptional coactivator in oncogenesis. Acta Pharmacol. Sin. 2006, 27, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Fu, J.; Xu, J.; O’Malley, B.W.; Wu, R.C. Steroid receptor coactivator 3 regulates autophagy in breast cancer cells through macrophage migration inhibitory factor. Cell Res. 2012, 22, 1003–1021. [Google Scholar] [CrossRef] [PubMed]
- Louie, M.C.; Zou, J.X.; Rabinovich, A.; Chen, H.W. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol. Cell. Biol. 2004, 24, 5157–5171. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liao, L.; Redmond, A.; Young, L.; Yuan, Y.; Chen, H.; O’Malley, B.W.; Xu, J. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol. Cell. Biol. 2008, 28, 5937–5950. [Google Scholar] [CrossRef]
- Goel, A.; Janknecht, R. Concerted activation of ETS protein ER81 by p160 coactivators, the acetyltransferase p300 and the receptor tyrosine kinase HER2/Neu. J. Biol. Chem. 2004, 279, 14909–14916. [Google Scholar] [CrossRef]
- Myers, E.; Hill, A.D.; Kelly, G.; McDermott, E.W.; O’Higgins, N.J.; Buggy, Y.; Young, L.S. Associations and interactions between Ets-1 and Ets-2 and coregulatory proteins, SRC-1, AIB1, and NCoR in breast cancer. Clin. Cancer Res. 2005, 11, 2111–2122. [Google Scholar] [CrossRef]
- He, C.; Shan, N.; Xu, P.; Ge, H.; Yuan, Y.; Liu, Y.; Zhang, P.; Wen, L.; Zhang, F.; Xiong, L.; et al. Hypoxia-induced Downregulation of SRC-3 Suppresses Trophoblastic Invasion and Migration Through Inhibition of the AKT/mTOR Pathway: Implications for the Pathogenesis of Preeclampsia. Sci. Rep. 2019, 9, 10349. [Google Scholar] [CrossRef]
- Long, W.; Foulds, C.E.; Qin, J.; Liu, J.; Ding, C.; Lonard, D.M.; Solis, L.M.; Wistuba, I.I.; Qin, J.; Tsai, S.Y.; et al. ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J. Clin. Invest. 2012, 122, 1869–1880. [Google Scholar] [CrossRef]
- Louet, J.F.; Coste, A.; Amazit, L.; Tannour-Louet, M.; Wu, R.C.; Tsai, S.Y.; Tsai, M.J.; Auwerx, J.; O’Malley, B.W. Oncogenic steroid receptor coactivator-3 is a key regulator of the white adipogenic program. Proc. Natl. Acad. Sci. USA 2006, 103, 17868–17873. [Google Scholar] [CrossRef]
- Coste, A.; Louet, J.F.; Lagouge, M.; Lerin, C.; Antal, M.C.; Meziane, H.; Schoonjans, K.; Puigserver, P.; O’Malley, B.W.; Auwerx, J. The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2008, 105, 17187–17192. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Hall, J.A.; Sun, C.M.; Cai, Q.; Ghyselinck, N.; Chambon, P.; Belkaid, Y.; Mathis, D.; Benoist, C. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity 2008, 29, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Jin, H.; Korn, T.; Liu, S.M.; Oukka, M.; Lim, B.; Kuchroo, V.K. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J. Immunol. 2008, 181, 2277–2284. [Google Scholar] [CrossRef]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, J.; Du, Q.; Xu, J.; Gwack, Y.; Sun, Z. SRC3 Is a Cofactor for RORgammat in Th17 Differentiation but Not Thymocyte Development. J. Immunol. 2019, 202, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Gilad, Y.; Lonard, D.M.; O’Malley, B.W. Steroid receptor coactivators—Their role in immunity. Front. Immunol. 2022, 13, 1079011. [Google Scholar] [CrossRef]
- Tanaka, K.; Martinez, G.J.; Yan, X.; Long, W.; Ichiyama, K.; Chi, X.; Kim, B.S.; Reynolds, J.M.; Chung, Y.; Tanaka, S.; et al. Regulation of Pathogenic T Helper 17 Cell Differentiation by Steroid Receptor Coactivator-3. Cell Rep. 2018, 23, 2318–2329. [Google Scholar] [CrossRef] [PubMed]
- Moraitis, A.N.; Giguere, V.; Thompson, C.C. Novel mechanism of nuclear receptor corepressor interaction dictated by activation function 2 helix determinants. Mol. Cell. Biol. 2002, 22, 6831–6841. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Peng, Z.; Guo, J.; Wang, Y.; Wang, S.; Jiang, H.; Wang, M.; Xie, Y.; Li, X.; Hu, M.; et al. CXCL10 Recruitment of gammadelta T Cells into the Hypoxic Bone Marrow Environment Leads to IL17 Expression and Multiple Myeloma Progression. Cancer Immunol. Res. 2023, 11, 1384–1399. [Google Scholar] [CrossRef]
- Liu, J.; Xie, Y.; Guo, J.; Li, X.; Wang, J.; Jiang, H.; Peng, Z.; Wang, J.; Wang, S.; Li, Q.; et al. Targeting NSD2-mediated SRC-3 liquid-liquid phase separation sensitizes bortezomib treatment in multiple myeloma. Nat. Commun. 2021, 12, 1022. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, C.V.; Rubio, M.F.; Fernandez Larrosa, P.N.; Panelo, L.C.; Azurmendi, P.J.; Ruiz Grecco, M.; Martinez-Noel, G.A.; Costas, M.A. The levels of RAC3 expression are up regulated by TNF in the inflammatory response. FEBS Open Bio 2014, 4, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Bleyer, Y.; Caron, R.; Seeley, J.J.; De Silva, N.S.; Schindler, C.W.; Hayden, M.S.; Klein, U.; Ghosh, S. The Alternative NF-kappaB Pathway in Regulatory T Cell Homeostasis and Suppressive Function. J. Immunol. 2018, 200, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Park, S.G.; Strickland, I.; Hayden, M.S.; Ghosh, S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009, 31, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Kameswaran, V.; Tone, Y.; Li, L.; Liou, H.C.; Greene, M.I.; Tone, M.; Chen, Y.H. Development of Foxp3+ regulatory t cells is driven by the c-Rel enhanceosome. Immunity 2009, 31, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhuo, M.; Lu, X.; Xia, X.; Zhao, Y.; Huang, Z.; Xu, J.; Li, W.; Yu, C. SRC-3 protects intestine from DSS-induced colitis by inhibiting inflammation and promoting goblet cell differentiation through enhancement of KLF4 expression. Int. J. Biol. Sci. 2018, 14, 2051–2064. [Google Scholar] [CrossRef] [PubMed]
- Mullany, L.K.; Rohira, A.D.; Leach, J.P.; Kim, J.H.; Monroe, T.O.; Ortiz, A.R.; Stork, B.; Gaber, M.W.; Sarkar, P.; Sikora, A.G.; et al. A steroid receptor coactivator stimulator (MCB-613) attenuates adverse remodeling after myocardial infarction. Proc. Natl. Acad. Sci. USA 2020, 117, 31353–31364. [Google Scholar] [CrossRef]
- Yu, C.; York, B.; Wang, S.; Feng, Q.; Xu, J.; O’Malley, B.W. An essential function of the SRC-3 coactivator in suppression of cytokine mRNA translation and inflammatory response. Mol. Cell 2007, 25, 765–778. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, T.; Xu, Y.; Zhu, J.; Jiang, Y.; Zhao, Y.; Xu, J.; Yu, C. Steroid receptor coactivator 3 is required for clearing bacteria and repressing inflammatory response in Escherichia coli-induced septic peritonitis. J. Immunol. 2010, 185, 5444–5452. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Biarnes, X.; Vidossich, P.; Rovira, C. The molecular mechanism of the catalase reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef]
- Colo, G.P.; Rosato, R.R.; Grant, S.; Costas, M.A. RAC3 down-regulation sensitizes human chronic myeloid leukemia cells to TRAIL-induced apoptosis. FEBS Lett. 2007, 581, 5075–5081. [Google Scholar] [CrossRef] [PubMed]
- Colo, G.P.; Rubio, M.F.; Nojek, I.M.; Werbajh, S.E.; Echeverria, P.C.; Alvarado, C.V.; Nahmod, V.E.; Galigniana, M.D.; Costas, M.A. The p160 nuclear receptor co-activator RAC3 exerts an anti-apoptotic role through a cytoplasmatic action. Oncogene 2008, 27, 2430–2444. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lu, X.; Chen, Y.; Li, M.; Mo, P.; Tong, Z.; Wang, W.; Wan, W.; Su, G.; Xu, J.; et al. Steroid Receptor Coactivator 3 Contributes to Host Defense against Enteric Bacteria by Recruiting Neutrophils via Upregulation of CXCL2 Expression. J. Immunol. 2017, 198, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Werbajh, S.; Nojek, I.; Lanz, R.; Costas, M.A. RAC-3 is a NF-kappa B coactivator. FEBS Lett. 2000, 485, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.C.; Qin, J.; Hashimoto, Y.; Wong, J.; Xu, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by I kappa B kinase. Mol. Cell. Biol. 2002, 22, 3549–3561. [Google Scholar] [CrossRef]
- Truong, T.H.; Hu, H.; Temiz, N.A.; Hagen, K.M.; Girard, B.J.; Brady, N.J.; Schwertfeger, K.L.; Lange, C.A.; Ostrander, J.H. Cancer Stem Cell Phenotypes in ER+ Breast Cancer Models Are Promoted by PELP1/AIB1 Complexes. Mol. Cancer Res. 2018, 16, 707–719. [Google Scholar] [CrossRef]
- Truong, T.H.; Benner, E.A.; Hagen, K.M.; Temiz, N.A.; Kerkvliet, C.P.; Wang, Y.; Cortes-Sanchez, E.; Yang, C.H.; Trousdell, M.C.; Pengo, T.; et al. PELP1/SRC-3-dependent regulation of metabolic PFKFB kinases drives therapy resistant ER+ breast cancer. Oncogene 2021, 40, 4384–4397. [Google Scholar] [CrossRef]
- Panelo, L.C.; Machado, M.S.; Rubio, M.F.; Jaworski, F.; Alvarado, C.V.; Paz, L.A.; Urtreger, A.J.; Vazquez, E.; Costas, M.A. High RAC3 expression levels are required for induction and maintaining of cancer cell stemness. Oncotarget 2018, 9, 5848–5860. [Google Scholar] [CrossRef]
- Dubrovska, A.; Hartung, A.; Bouchez, L.C.; Walker, J.R.; Reddy, V.A.; Cho, C.Y.; Schultz, P.G. CXCR4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through AhR signalling. Br. J. Cancer 2012, 107, 43–52. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Shien, T.; Ogiya, A.; Ishida, N.; Yamazaki, K.; Horii, R.; Horimoto, Y.; Masuda, N.; Yasojima, H.; Inao, T.; et al. Differences in expression of the cancer stem cell marker aldehyde dehydrogenase 1 among estrogen receptor-positive/human epidermal growth factor receptor type 2-negative breast cancer cases with early, late, and no recurrence. Breast Cancer Res. 2016, 18, 73. [Google Scholar] [CrossRef]
- Dancik, G.M.; Voutsas, I.F.; Vlahopoulos, S. Aldehyde Dehydrogenase Enzyme Functions in Acute Leukemia Stem Cells. Front. Biosci. (Schol. Ed.) 2022, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qiang, J.; Deng, Q.; Xia, J.; Deng, L.; Zhou, L.; Wang, D.; He, X.; Liu, Y.; Zhao, B.; et al. ALDH1A1 Activity in Tumor-Initiating Cells Remodels Myeloid-Derived Suppressor Cells to Promote Breast Cancer Progression. Cancer Res. 2021, 81, 5919–5934. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Duan, X.; Wang, Z.; Sun, Y.; Guan, Q.; Kang, L.; Zhang, Q.; Fang, L.; Li, J.; Wong, J. An acetylation-enhanced interaction between transcription factor Sox2 and the steroid receptor coactivators facilitates Sox2 transcriptional activity and function. J. Biol. Chem. 2021, 297, 101389. [Google Scholar] [CrossRef] [PubMed]
- Domenici, G.; Aurrekoetxea-Rodriguez, I.; Simoes, B.M.; Rabano, M.; Lee, S.Y.; Millan, J.S.; Comaills, V.; Oliemuller, E.; Lopez-Ruiz, J.A.; Zabalza, I.; et al. A Sox2-Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene 2019, 38, 3151–3169. [Google Scholar] [CrossRef]
- Leung, E.Y.; Askarian-Amiri, M.E.; Sarkar, D.; Ferraro-Peyret, C.; Joseph, W.R.; Finlay, G.J.; Baguley, B.C. Endocrine Therapy of Estrogen Receptor-Positive Breast Cancer Cells: Early Differential Effects on Stem Cell Markers. Front. Oncol. 2017, 7, 184. [Google Scholar] [CrossRef]
- Percharde, M.; Azuara, V. Essential roles for the nuclear receptor coactivator Ncoa3 in pluripotency. Cell Cycle 2013, 12, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Lavial, F.; Ng, J.H.; Kumar, V.; Tomaz, R.A.; Martin, N.; Yeo, J.C.; Gil, J.; Prabhakar, S.; Ng, H.H.; et al. Ncoa3 functions as an essential Esrrb coactivator to sustain embryonic stem cell self-renewal and reprogramming. Genes Dev. 2012, 26, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, M.; Liu, H.; Guo, H.; Wang, Y.; Cheng, H.; Chen, L. Role of nuclear receptor coactivator 3 (Ncoa3) in pluripotency maintenance. J. Biol. Chem. 2012, 287, 38295–38304. [Google Scholar] [CrossRef]
- Chitilian, J.M.; Thillainadesan, G.; Manias, J.L.; Chang, W.Y.; Walker, E.; Isovic, M.; Stanford, W.L.; Torchia, J. Critical components of the pluripotency network are targets for the p300/CBP interacting protein (p/CIP) in embryonic stem cells. Stem Cells 2014, 32, 204–215. [Google Scholar] [CrossRef]
- Hu, M.; Zeng, H.; Chen, S.; Xu, Y.; Wang, S.; Tang, Y.; Wang, X.; Du, C.; Shen, M.; Chen, F.; et al. SRC-3 is involved in maintaining hematopoietic stem cell quiescence by regulation of mitochondrial metabolism in mice. Blood 2018, 132, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Ren, Y.; Wang, K.; He, J. SRC-3 has a role in cancer other than as a nuclear receptor coactivator. Int. J. Biol. Sci. 2011, 7, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Torres-Arzayus, M.I.; Font de Mora, J.; Yuan, J.; Vazquez, F.; Bronson, R.; Rue, M.; Sellers, W.R.; Brown, M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 2004, 6, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.; Oh, A.S.; Wellstein, A.; Riegel, A.T. Impact of the nuclear receptor coactivator AIB1 isoform AIB1-Delta3 on estrogenic ligands with different intrinsic activity. Oncogene 2004, 23, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.; Wellstein, A.; Riegel, A.T. An isoform of the coactivator AIB1 that increases hormone and growth factor sensitivity is overexpressed in breast cancer. J. Biol. Chem. 2001, 276, 39736–39741. [Google Scholar] [CrossRef] [PubMed]
- Bautista, S.; Valles, H.; Walker, R.L.; Anzick, S.; Zeillinger, R.; Meltzer, P.; Theillet, C. In breast cancer, amplification of the steroid receptor coactivator gene AIB1 is correlated with estrogen and progesterone receptor positivity. Clin. Cancer Res. 1998, 4, 2925–2929. [Google Scholar]
- Hudelist, G.; Czerwenka, K.; Kubista, E.; Marton, E.; Pischinger, K.; Singer, C.F. Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast Cancer Res. Treat. 2003, 78, 193–204. [Google Scholar] [CrossRef]
- Lee, K.; Lee, A.; Song, B.J.; Kang, C.S. Expression of AIB1 protein as a prognostic factor in breast cancer. World J. Surg. Oncol. 2011, 9, 139. [Google Scholar] [CrossRef]
- Osborne, C.K.; Bardou, V.; Hopp, T.A.; Chamness, G.C.; Hilsenbeck, S.G.; Fuqua, S.A.; Wong, J.; Allred, D.C.; Clark, G.M.; Schiff, R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 2003, 95, 353–361. [Google Scholar] [CrossRef]
- Zhao, C.; Yasui, K.; Lee, C.J.; Kurioka, H.; Hosokawa, Y.; Oka, T.; Inazawa, J. Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer 2003, 98, 18–23. [Google Scholar] [CrossRef]
- Nakles, R.E.; Shiffert, M.T.; Diaz-Cruz, E.S.; Cabrera, M.C.; Alotaiby, M.; Miermont, A.M.; Riegel, A.T.; Furth, P.A. Altered AIB1 or AIB1Delta3 expression impacts ERalpha effects on mammary gland stromal and epithelial content. Mol. Endocrinol. 2011, 25, 549–563. [Google Scholar] [CrossRef]
- Tilli, M.T.; Reiter, R.; Oh, A.S.; Henke, R.T.; McDonnell, K.; Gallicano, G.I.; Furth, P.A.; Riegel, A.T. Overexpression of an N-terminally truncated isoform of the nuclear receptor coactivator amplified in breast cancer 1 leads to altered proliferation of mammary epithelial cells in transgenic mice. Mol. Endocrinol. 2005, 19, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Avivar, A.; Garcia-Macias, M.C.; Ascaso, E.; Herrera, G.; O’Connor, J.E.; Font de Mora, J. Moderate overexpression of AIB1 triggers pre-neoplastic changes in mammary epithelium. FEBS Lett. 2006, 580, 5222–5226. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.Q.; Liao, L.; Wang, S.; Medina, D.; O’Malley, B.W.; Xu, J. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res. 2005, 65, 7993–8002. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.Q.; Liao, L.; Zhang, H.; Lee, A.V.; O’Malley, B.W.; Xu, J. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004, 64, 1875–1885. [Google Scholar] [CrossRef]
- Planas-Silva, M.D.; Shang, Y.; Donaher, J.L.; Brown, M.; Weinberg, R.A. AIB1 enhances estrogen-dependent induction of cyclin D1 expression. Cancer Res. 2001, 61, 3858–3862. [Google Scholar]
- Suen, C.S.; Berrodin, T.J.; Mastroeni, R.; Cheskis, B.J.; Lyttle, C.R.; Frail, D.E. A transcriptional coactivator, steroid receptor coactivator-3, selectively augments steroid receptor transcriptional activity. J. Biol. Chem. 1998, 273, 27645–27653. [Google Scholar] [CrossRef]
- Tikkanen, M.K.; Carter, D.J.; Harris, A.M.; Le, H.M.; Azorsa, D.O.; Meltzer, P.S.; Murdoch, F.E. Endogenously expressed estrogen receptor and coactivator AIB1 interact in MCF-7 human breast cancer cells. Proc. Natl. Acad. Sci. USA 2000, 97, 12536–12540. [Google Scholar] [CrossRef]
- Yi, P.; Wang, Z.; Feng, Q.; Chou, C.K.; Pintilie, G.D.; Shen, H.; Foulds, C.E.; Fan, G.; Serysheva, I.; Ludtke, S.J.; et al. Structural and Functional Impacts of ER Coactivator Sequential Recruitment. Mol. Cell 2017, 67, 733–743.e4. [Google Scholar] [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5, e12792. [Google Scholar] [CrossRef]
- Zhao, Y.; Laws, M.J.; Guillen, V.S.; Ziegler, Y.; Min, J.; Sharma, A.; Kim, S.H.; Chu, D.; Park, B.H.; Oesterreich, S.; et al. Structurally Novel Antiestrogens Elicit Differential Responses from Constitutively Active Mutant Estrogen Receptors in Breast Cancer Cells and Tumors. Cancer Res. 2017, 77, 5602–5613. [Google Scholar] [CrossRef]
- Fereshteh, M.P.; Tilli, M.T.; Kim, S.E.; Xu, J.; O’Malley, B.W.; Wellstein, A.; Furth, P.A.; Riegel, A.T. The nuclear receptor coactivator amplified in breast cancer-1 is required for Neu (ErbB2/HER2) activation, signaling, and mammary tumorigenesis in mice. Cancer Res. 2008, 68, 3697–3706. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; McGlynn, L.M.; Campbell, F.M.; Muller, S.; Tovey, S.M.; Dunne, B.; Nielsen, K.V.; Cooke, T.G.; Bartlett, J.M. Amplified in breast cancer 1 in human epidermal growth factor receptor—Positive tumors of tamoxifen-treated breast cancer patients. Clin. Cancer Res. 2007, 13, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Ory, V.; Tassi, E.; Cavalli, L.R.; Sharif, G.M.; Saenz, F.; Baker, T.; Schmidt, M.O.; Mueller, S.C.; Furth, P.A.; Wellstein, A.; et al. The nuclear coactivator amplified in breast cancer 1 maintains tumor-initiating cells during development of ductal carcinoma in situ. Oncogene 2014, 33, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Torres-Arzayus, M.I.; Zhao, J.; Bronson, R.; Brown, M. Estrogen-dependent and estrogen-independent mechanisms contribute to AIB1-mediated tumor formation. Cancer Res. 2010, 70, 4102–4111. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Rose, D.W.; Hermanson, O.; Liu, F.; Herman, T.; Wu, W.; Szeto, D.; Gleiberman, A.; Krones, A.; Pratt, K.; et al. Regulation of somatic growth by the p160 coactivator p/CIP. Proc. Natl. Acad. Sci. USA 2000, 97, 13549–13554. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Yi, P.; Amazit, L.; LaMarca, H.L.; Ashcroft, F.; Kumar, R.; Mancini, M.A.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol. Cell 2010, 37, 321–332. [Google Scholar] [CrossRef]
- Yan, J.; Erdem, H.; Li, R.; Cai, Y.; Ayala, G.; Ittmann, M.; Yu-Lee, L.Y.; Tsai, S.Y.; Tsai, M.J. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 2008, 68, 5460–5468. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Pecina-Slaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef]
- Oda, H.; Takeichi, M. Evolution: Structural and functional diversity of cadherin at the adherens junction. J. Cell. Biol. 2011, 193, 1137–1146. [Google Scholar] [CrossRef]
- Kaszak, I.; Witkowska-Pilaszewicz, O.; Niewiadomska, Z.; Dworecka-Kaszak, B.; Ngosa Toka, F.; Jurka, P. Role of Cadherins in Cancer-A Review. Int. J. Mol. Sci. 2020, 21, 7624. [Google Scholar] [CrossRef] [PubMed]
- Varisli, L.; Tolan, V. Increased ROS alters E-/N-cadherin levels and promotes migration in prostate cancer cells. Bratisl. Lek. Listy 2022, 123, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Varisli, L.; Tolan, V.; Cen, J.H.; Vlahopoulos, S.; Cen, O. Dissecting the effects of androgen deprivation therapy on cadherin switching in advanced prostate cancer: A molecular perspective. Oncol. Res. 2022, 30, 137–155. [Google Scholar] [CrossRef]
- Ferreira Almeida, C.; Oliveira, A.; Joao Ramos, M.; Fernandes, P.A.; Teixeira, N.; Amaral, C. Estrogen receptor-positive (ER+) breast cancer treatment: Are multi-target compounds the next promising approach? Biochem. Pharmacol. 2020, 177, 113989. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative, G. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Heck, S.; Rom, J.; Thewes, V.; Becker, N.; Blume, B.; Sinn, H.P.; Deuschle, U.; Sohn, C.; Schneeweiss, A.; Lichter, P. Estrogen-related receptor alpha expression and function is associated with the transcriptional coregulator AIB1 in breast carcinoma. Cancer Res. 2009, 69, 5186–5193. [Google Scholar] [CrossRef]
- Lumachi, F.; Santeufemia, D.A.; Basso, S.M. Current medical treatment of estrogen receptor-positive breast cancer. World J. Biol. Chem. 2015, 6, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Ozyurt, R.; Ozpolat, B. Molecular Mechanisms of Anti-Estrogen Therapy Resistance and Novel Targeted Therapies. Cancers 2022, 14, 5206. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; O’Malley, B.W. Nuclear receptor modulation--role of coregulators in selective estrogen receptor modulator (SERM) actions. Steroids 2014, 90, 39–43. [Google Scholar] [CrossRef]
- Fawell, S.E.; White, R.; Hoare, S.; Sydenham, M.; Page, M.; Parker, M.G. Inhibition of estrogen receptor-DNA binding by the “pure” antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization. Proc. Natl. Acad. Sci. USA 1990, 87, 6883–6887. [Google Scholar] [CrossRef] [PubMed]
- Wijayaratne, A.L.; Nagel, S.C.; Paige, L.A.; Christensen, D.J.; Norris, J.D.; Fowlkes, D.M.; McDonnell, D.P. Comparative analyses of mechanistic differences among antiestrogens. Endocrinology 1999, 140, 5828–5840. [Google Scholar] [CrossRef]
- Yeh, W.L.; Shioda, K.; Coser, K.R.; Rivizzigno, D.; McSweeney, K.R.; Shioda, T. Fulvestrant-induced cell death and proteasomal degradation of estrogen receptor alpha protein in MCF-7 cells require the CSK c-Src tyrosine kinase. PLoS ONE 2013, 8, e60889. [Google Scholar] [CrossRef]
- Boszkiewicz, K.; Piwowar, A.; Petryszyn, P. Aromatase Inhibitors and Risk of Metabolic and Cardiovascular Adverse Effects in Breast Cancer Patients-A Systematic Review and Meta-Analysis. J. Clin. Med. 2022, 11, 3133. [Google Scholar] [CrossRef]
- Lonard, D.M.; Tsai, S.Y.; O’Malley, B.W. Selective estrogen receptor modulators 4-hydroxytamoxifen and raloxifene impact the stability and function of SRC-1 and SRC-3 coactivator proteins. Mol. Cell. Biol. 2004, 24, 14–24. [Google Scholar] [CrossRef]
- Su, Q.; Hu, S.; Gao, H.; Ma, R.; Yang, Q.; Pan, Z.; Wang, T.; Li, F. Role of AIB1 for tamoxifen resistance in estrogen receptor-positive breast cancer cells. Oncology 2008, 75, 159–168. [Google Scholar] [CrossRef]
- Hurtado, A.; Holmes, K.A.; Geistlinger, T.R.; Hutcheson, I.R.; Nicholson, R.I.; Brown, M.; Jiang, J.; Howat, W.J.; Ali, S.; Carroll, J.S. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature 2008, 456, 663–666. [Google Scholar] [CrossRef]
- Lebert, J.; Lilly, E.J. Developments in the Management of Metastatic HER2-Positive Breast Cancer: A Review. Curr. Oncol. 2022, 29, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Southey, M.C.; Venter, D.J. Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu. Cancer Res. 2001, 61, 903–907. [Google Scholar] [PubMed]
- Font de Mora, J.; Brown, M. AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol. Cell. Biol. 2000, 20, 5041–5047. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef]
- Lahusen, T.; Fereshteh, M.; Oh, A.; Wellstein, A.; Riegel, A.T. Epidermal growth factor receptor tyrosine phosphorylation and signaling controlled by a nuclear receptor coactivator, amplified in breast cancer 1. Cancer Res. 2007, 67, 7256–7265. [Google Scholar] [CrossRef]
- Han, S.J.; Jain, P.; Gilad, Y.; Xia, Y.; Sung, N.; Park, M.J.; Dean, A.M.; Lanz, R.B.; Xu, J.; Dacso, C.C.; et al. Steroid receptor coactivator 3 is a key modulator of regulatory T cell-mediated tumor evasion. Proc. Natl. Acad. Sci. USA 2023, 120, e2221707120. [Google Scholar] [CrossRef]
- Han, S.J.; Sung, N.; Wang, J.; O’Malley, B.W.; Lonard, D.M. Steroid receptor coactivator-3 inhibition generates breast cancer antitumor immune microenvironment. Breast Cancer Res. 2022, 24, 73. [Google Scholar] [CrossRef]
- Kunz, M.; Toksoy, A.; Goebeler, M.; Engelhardt, E.; Brocker, E.; Gillitzer, R. Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. J. Pathol. 1999, 189, 552–558. [Google Scholar] [CrossRef]
- Neo, S.Y.; Lundqvist, A. The Multifaceted Roles of CXCL9 Within the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1231, 45–51. [Google Scholar] [CrossRef]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef]
- Walser, T.C.; Ma, X.; Kundu, N.; Dorsey, R.; Goloubeva, O.; Fulton, A.M. Immune-mediated modulation of breast cancer growth and metastasis by the chemokine Mig (CXCL9) in a murine model. J. Immunother. 2007, 30, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Muller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Specht, K.; Harbeck, N.; Smida, J.; Annecke, K.; Reich, U.; Naehrig, J.; Langer, R.; Mages, J.; Busch, R.; Kruse, E.; et al. Expression profiling identifies genes that predict recurrence of breast cancer after adjuvant CMF-based chemotherapy. Breast Cancer Res. Treat. 2009, 118, 45–56. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hardenberg, J.B.; Braun, A.; Schon, M.P. A Yin and Yang in Epithelial Immunology: The Roles of the alphaE(CD103)beta7 Integrin in T Cells. J. Invest. Dermatol. 2018, 138, 23–31. [Google Scholar] [CrossRef]
- Hoffmann, J.C.; Schon, M.P. Integrin alphaE(CD103)beta7 in Epithelial Cancer. Cancers 2021, 13, 6211. [Google Scholar] [CrossRef]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Cepek, K.L.; Shaw, S.K.; Parker, C.M.; Russell, G.J.; Morrow, J.S.; Rimm, D.L.; Brenner, M.B. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature 1994, 372, 190–193. [Google Scholar] [CrossRef]
- Karecla, P.I.; Bowden, S.J.; Green, S.J.; Kilshaw, P.J. Recognition of E-cadherin on epithelial cells by the mucosal T cell integrin alpha M290 beta 7 (alpha E beta 7). Eur. J. Immunol. 1995, 25, 852–856. [Google Scholar] [CrossRef]
- Le Floc’h, A.; Jalil, A.; Vergnon, I.; Le Maux Chansac, B.; Lazar, V.; Bismuth, G.; Chouaib, S.; Mami-Chouaib, F. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J. Exp. Med. 2007, 204, 559–570. [Google Scholar] [CrossRef]
- Karecla, P.I.; Green, S.J.; Bowden, S.J.; Coadwell, J.; Kilshaw, P.J. Identification of a binding site for integrin alphaEbeta7 in the N-terminal domain of E-cadherin. J. Biol. Chem. 1996, 271, 30909–30915. [Google Scholar] [CrossRef]
- Shields, B.D.; Koss, B.; Taylor, E.M.; Storey, A.J.; West, K.L.; Byrum, S.D.; Mackintosh, S.G.; Edmondson, R.; Mahmoud, F.; Shalin, S.C.; et al. Loss of E-Cadherin Inhibits CD103 Antitumor Activity and Reduces Checkpoint Blockade Responsiveness in Melanoma. Cancer Res. 2019, 79, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, S.; Zeng, H.; Liu, Z.; Dong, W.; He, W.; Chen, X.; Dong, X.; Zheng, L.; Lin, T.; et al. CD103+ Tumor Infiltrating Lymphocytes Predict a Favorable Prognosis in Urothelial Cell Carcinoma of the Bladder. J. Urol. 2015, 194, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Austrup, F.; Rebstock, S.; Kilshaw, P.J.; Hamann, A. Transforming growth factor-beta 1-induced expression of the mucosa-related integrin alpha E on lymphocytes is not associated with mucosa-specific homing. Eur. J. Immunol. 1995, 25, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Pauls, K.; Schon, M.; Kubitza, R.C.; Homey, B.; Wiesenborn, A.; Lehmann, P.; Ruzicka, T.; Parker, C.M.; Schon, M.P. Role of integrin alphaE(CD103)beta7 for tissue-specific epidermal localization of CD8+ T lymphocytes. J. Investig. Dermatol. 2001, 117, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.W.; Green, S.J.; Carter, C.; Coadwell, J.; Kilshaw, P.J. Studies on transcriptional regulation of the mucosal T-cell integrin alphaEbeta7 (CD103). Immunology 2001, 103, 146–154. [Google Scholar] [CrossRef]
- Franciszkiewicz, K.; Le Floc’h, A.; Boutet, M.; Vergnon, I.; Schmitt, A.; Mami-Chouaib, F. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res. 2013, 73, 617–628. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef]
- Boutet, M.; Gauthier, L.; Leclerc, M.; Gros, G.; de Montpreville, V.; Theret, N.; Donnadieu, E.; Mami-Chouaib, F. TGFbeta Signaling Intersects with CD103 Integrin Signaling to Promote T-Lymphocyte Accumulation and Antitumor Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 1757–1769. [Google Scholar] [CrossRef]
- Gebhardt, T.; Mackay, L.K. Local immunity by tissue-resident CD8+ memory T cells. Front. Immunol. 2012, 3, 340. [Google Scholar] [CrossRef]
- Henson, S.M.; Akbar, A.N. KLRG1—more than a marker for T cell senescence. Age 2009, 31, 285–291. [Google Scholar] [CrossRef]
- Grundemann, C.; Bauer, M.; Schweier, O.; von Oppen, N.; Lassing, U.; Saudan, P.; Becker, K.F.; Karp, K.; Hanke, T.; Bachmann, M.F.; et al. Cutting edge: Identification of E-cadherin as a ligand for the murine killer cell lectin-like receptor G1. J. Immunol. 2006, 176, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Maruyama, T.; Saito, N.; Koganei, S.; Yamamoto, K.; Matsumoto, N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J. Exp. Med. 2006, 203, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wan, S.; Tao, K.; Wang, G.; Zhao, E. KLRG1 restricts memory T cell antitumor immunity. Oncotarget 2016, 7, 61670–61678. [Google Scholar] [CrossRef] [PubMed]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef]
- Meinicke, H.; Bremser, A.; Brack, M.; Schrenk, K.; Pircher, H.; Izcue, A. KLRG1 impairs regulatory T-cell competitive fitness in the gut. Immunology 2017, 152, 65–73. [Google Scholar] [CrossRef]
- Tessmer, M.S.; Fugere, C.; Stevenaert, F.; Naidenko, O.V.; Chong, H.J.; Leclercq, G.; Brossay, L. KLRG1 binds cadherins and preferentially associates with SHIP-1. Int. Immunol. 2007, 19, 391–400. [Google Scholar] [CrossRef]
- Schwartzkopff, S.; Woyciechowski, S.; Aichele, U.; Flecken, T.; Zhang, N.; Thimme, R.; Pircher, H. TGF-beta downregulates KLRG1 expression in mouse and human CD8+ T cells. Eur. J. Immunol. 2015, 45, 2212–2217. [Google Scholar] [CrossRef]
- Attig, S.; Hennenlotter, J.; Pawelec, G.; Klein, G.; Koch, S.D.; Pircher, H.; Feyerabend, S.; Wernet, D.; Stenzl, A.; Rammensee, H.G.; et al. Simultaneous infiltration of polyfunctional effector and suppressor T cells into renal cell carcinomas. Cancer Res. 2009, 69, 8412–8419. [Google Scholar] [CrossRef]
- Baitsch, L.; Legat, A.; Barba, L.; Fuertes Marraco, S.A.; Rivals, J.P.; Baumgaertner, P.; Christiansen-Jucht, C.; Bouzourene, H.; Rimoldi, D.; Pircher, H.; et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS ONE 2012, 7, e30852. [Google Scholar] [CrossRef]
- Sun, Y.; Jing, J.; Xu, H.; Xu, L.; Hu, H.; Tang, C.; Liu, S.; Wei, Q.; Duan, R.; Guo, J.; et al. N-cadherin inhibitor creates a microenvironment that protect TILs from immune checkpoints and Treg cells. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.L.; et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci. Transl. Med. 2011, 3, 111ra120. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef]
- Kolijn, K.; Verhoef, E.I.; Smid, M.; Bottcher, R.; Jenster, G.W.; Debets, R.; van Leenders, G. Epithelial-Mesenchymal Transition in Human Prostate Cancer Demonstrates Enhanced Immune Evasion Marked by IDO1 Expression. Cancer Res. 2018, 78, 4671–4679. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zhang, Z.; Ten Dijke, P. Harnessing epithelial-mesenchymal plasticity to boost cancer immunotherapy. Cell. Mol. Immunol. 2023, 20, 318–340. [Google Scholar] [CrossRef] [PubMed]
- Raj, G.V.; Sareddy, G.R.; Ma, S.; Lee, T.K.; Viswanadhapalli, S.; Li, R.; Liu, X.; Murakami, S.; Chen, C.C.; Lee, W.R.; et al. Estrogen receptor coregulator binding modulators (ERXs) effectively target estrogen receptor positive human breast cancers. eLife 2017, 6, e26857. [Google Scholar] [CrossRef]
- Song, X.; Chen, J.; Zhao, M.; Zhang, C.; Yu, Y.; Lonard, D.M.; Chow, D.C.; Palzkill, T.; Xu, J.; O’Malley, B.W.; et al. Development of potent small-molecule inhibitors to drug the undruggable steroid receptor coactivator-3. Proc. Natl. Acad. Sci. USA 2016, 113, 4970–4975. [Google Scholar] [CrossRef] [PubMed]
- Mc Ilroy, M.; Fleming, F.J.; Buggy, Y.; Hill, A.D.; Young, L.S. Tamoxifen-induced ER-alpha-SRC-3 interaction in HER2 positive human breast cancer; a possible mechanism for ER isoform specific recurrence. Endocr. Relat. Cancer 2006, 13, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Weiner, M.; Skoog, L.; Fornander, T.; Nordenskjold, B.; Sgroi, D.C.; Stal, O. Oestrogen receptor co-activator AIB1 is a marker of tamoxifen benefit in postmenopausal breast cancer. Ann. Oncol. 2013, 24, 1994–1999. [Google Scholar] [CrossRef]
- Lahusen, T.; Henke, R.T.; Kagan, B.L.; Wellstein, A.; Riegel, A.T. The role and regulation of the nuclear receptor co-activator AIB1 in breast cancer. Breast Cancer Res. Treat. 2009, 116, 225–237. [Google Scholar] [CrossRef]
- Sharif, G.M.; Campbell, M.J.; Nasir, A.; Sengupta, S.; Graham, G.T.; Kushner, M.H.; Kietzman, W.B.; Schmidt, M.O.; Pearson, G.W.; Loudig, O.; et al. An AIB1 Isoform Alters Enhancer Access and Enables Progression of Early-Stage Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 4230–4241. [Google Scholar] [CrossRef]
- Li, X.; Lonard, D.M.; Jung, S.Y.; Malovannaya, A.; Feng, Q.; Qin, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell 2006, 124, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.C.; Feng, Q.; Lonard, D.M.; O’Malley, B.W. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell 2007, 129, 1125–1140. [Google Scholar] [CrossRef] [PubMed]
- Ferry, C.; Gaouar, S.; Fischer, B.; Boeglin, M.; Paul, N.; Samarut, E.; Piskunov, A.; Pankotai-Bodo, G.; Brino, L.; Rochette-Egly, C. Cullin 3 mediates SRC-3 ubiquitination and degradation to control the retinoic acid response. Proc. Natl. Acad. Sci. USA 2011, 108, 20603–20608. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Su, L.; Zhang, F.; Zhu, X.; Zhu, Y.; Wei, L.; Jiao, X.; Hou, Y.; Chen, X.; Wang, W.; et al. Thevebioside, the active ingredient of traditional Chinese medicine, promotes ubiquitin-mediated SRC-3 degradation to induce NSCLC cells apoptosis. Cancer Lett. 2020, 493, 167–177. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, X.; Wen, L.; Yi, S.; Hu, J.; Ruan, J.; Zhao, F.; Cui, G.; Fang, J.; Chen, Y. Steroid receptor coactivator-3 is a pivotal target of gambogic acid in B-cell Non-Hodgkin lymphoma and an inducer of histone H3 deacetylation. Eur. J. Pharmacol. 2016, 789, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Yu, Y.; Chow, D.C.; Palzkill, T.; Madoux, F.; Hodder, P.; Chase, P.; Griffin, P.R.; O’Malley, B.W.; Lonard, D.M. Identification of verrucarin a as a potent and selective steroid receptor coactivator-3 small molecule inhibitor. PLoS ONE 2014, 9, e95243. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lonard, D.M.; Yu, Y.; Chow, D.C.; Palzkill, T.G.; O’Malley, B.W. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol. Endocrinol. 2011, 25, 2041–2053. [Google Scholar] [CrossRef]
- Wang, Y.; Lonard, D.M.; Yu, Y.; Chow, D.C.; Palzkill, T.G.; Wang, J.; Qi, R.; Matzuk, A.J.; Song, X.; Madoux, F.; et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res. 2014, 74, 1506–1517. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Cheng, B.; Ren, Y.; Cao, H.; Chen, J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur. J. Med. Chem. 2020, 199, 112377. [Google Scholar] [CrossRef]
- He, M.; Cao, C.; Ni, Z.; Liu, Y.; Song, P.; Hao, S.; He, Y.; Sun, X.; Rao, Y. PROTACs: Great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target Ther. 2022, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Cao, S.; Sun, Y.; Dong, Z.; Li, C.; Wang, H.; Yao, Y.; Yu, H.; Song, X.; et al. In vitro and in vivo degradation of programmed cell death ligand 1 (PD-L1) by a proteolysis targeting chimera (PROTAC). Bioorg. Chem. 2021, 111, 104833. [Google Scholar] [CrossRef]
- Qin, L.; Chen, J.; Lu, D.; Jain, P.; Yu, Y.; Cardenas, D.; Peng, X.; Yu, X.; Xu, J.; Wang, J.; et al. Development of improved SRC-3 inhibitors as breast cancer therapeutic agents. Endocr. Relat. Cancer 2021, 28, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Manmuan, S.; Sakunrangsit, N.; Ketchart, W. Salinomycin overcomes acquired tamoxifen resistance through AIB1 and inhibits cancer cell invasion in endocrine resistant breast cancer. Clin. Exp. Pharmacol. Physiol. 2017, 44, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Zhu, Y.; Wu, Z.; Cui, C.; Cai, F. Anticancer Mechanisms of Salinomycin in Breast Cancer and Its Clinical Applications. Front. Oncol. 2021, 11, 654428. [Google Scholar] [CrossRef] [PubMed]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef]
- Varisli, L.; Cen, O.; Vlahopoulos, S. Dissecting pharmacological effects of chloroquine in cancer treatment: Interference with inflammatory signaling pathways. Immunology 2020, 159, 257–278. [Google Scholar] [CrossRef]
- Stagni, V.; Kaminari, A.; Contadini, C.; Barila, D.; Sessa, R.L.; Sideratou, Z.; Vlahopoulos, S.A.; Tsiourvas, D. A Triphenylphosphonium-Functionalized Delivery System for an ATM Kinase Inhibitor That Ameliorates Doxorubicin Resistance in Breast Carcinoma Mammospheres. Cancers 2023, 15, 1474. [Google Scholar] [CrossRef]
- Stagni, V.; Kaminari, A.; Sideratou, Z.; Sakellis, E.; Vlahopoulos, S.A.; Tsiourvas, D. Targeting breast cancer stem-like cells using chloroquine encapsulated by a triphenylphosphonium-functionalized hyperbranched polymer. Int. J. Pharm. 2020, 585, 119465. [Google Scholar] [CrossRef]
- An, Y.; Wu, J.; Yang, B.; Zhu, Z.; Gao, M.; Yu, C.; Yang, C.J. Selection and Application of DNA Aptamer Against Oncogene Amplified in Breast Cancer 1. J. Mol. Evol. 2015, 81, 179–185. [Google Scholar] [CrossRef]
- Yusuf, A.N.M.; Amri, M.F.; Ugusman, A.; Hamid, A.A.; Wahab, N.A.; Mokhtar, M.H. Hyperandrogenism and Its Possible Effects on Endometrial Receptivity: A Review. Int. J. Mol. Sci. 2023, 24, 12026. [Google Scholar] [CrossRef] [PubMed]
- York, B.; Sagen, J.V.; Tsimelzon, A.; Louet, J.F.; Chopra, A.R.; Reineke, E.L.; Zhou, S.; Stevens, R.D.; Wenner, B.R.; Ilkayeva, O.; et al. Research resource: Tissue- and pathway-specific metabolomic profiles of the steroid receptor coactivator (SRC) family. Mol. Endocrinol. 2013, 27, 366–380. [Google Scholar] [CrossRef] [PubMed]
- McClendon, L.K.; Garcia, R.L.; Lazaro, T.; Robledo, A.; Vasandani, V.; Luna, Z.A.E.; Rao, A.S.; Srivatsan, A.; Lonard, D.M.; Dacso, C.C.; et al. A steroid receptor coactivator small molecule “stimulator” attenuates post-stroke ischemic brain injury. Front. Mol. Neurosci. 2022, 15, 1055295. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varisli, L.; Dancik, G.M.; Tolan, V.; Vlahopoulos, S. Critical Roles of SRC-3 in the Development and Progression of Breast Cancer, Rendering It a Prospective Clinical Target. Cancers 2023, 15, 5242. https://doi.org/10.3390/cancers15215242
Varisli L, Dancik GM, Tolan V, Vlahopoulos S. Critical Roles of SRC-3 in the Development and Progression of Breast Cancer, Rendering It a Prospective Clinical Target. Cancers. 2023; 15(21):5242. https://doi.org/10.3390/cancers15215242
Chicago/Turabian StyleVarisli, Lokman, Garrett M. Dancik, Veysel Tolan, and Spiros Vlahopoulos. 2023. "Critical Roles of SRC-3 in the Development and Progression of Breast Cancer, Rendering It a Prospective Clinical Target" Cancers 15, no. 21: 5242. https://doi.org/10.3390/cancers15215242
APA StyleVarisli, L., Dancik, G. M., Tolan, V., & Vlahopoulos, S. (2023). Critical Roles of SRC-3 in the Development and Progression of Breast Cancer, Rendering It a Prospective Clinical Target. Cancers, 15(21), 5242. https://doi.org/10.3390/cancers15215242