




Combining Mass Cytometry Data by CyTOFmerge Reveals Additional Cell Phenotypes in the Heterogeneous Ovarian Cancer Tumor Microenvironment: A Pilot Study
 , , , , and
, , , , and 
Abstract
:Simple Summary
Abstract
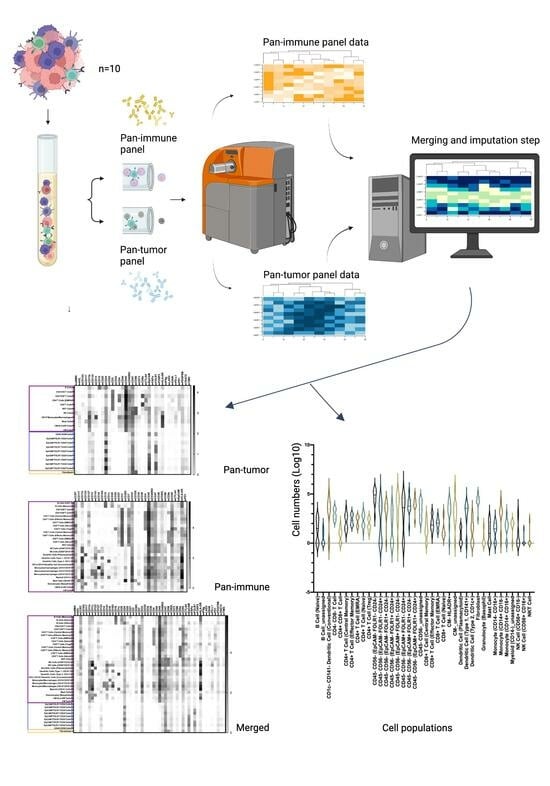
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Sample Processing
2.2.1. Tumor Tissues
2.2.2. Quality Control Samples
2.3. Ethical Statement
2.4. Antibodies for Mass Cytometry
2.4.1. The Pan-Tumor Panel
2.4.2. The Pan-Immune Panel
2.4.3. Antibody CONJUGATION and Validation
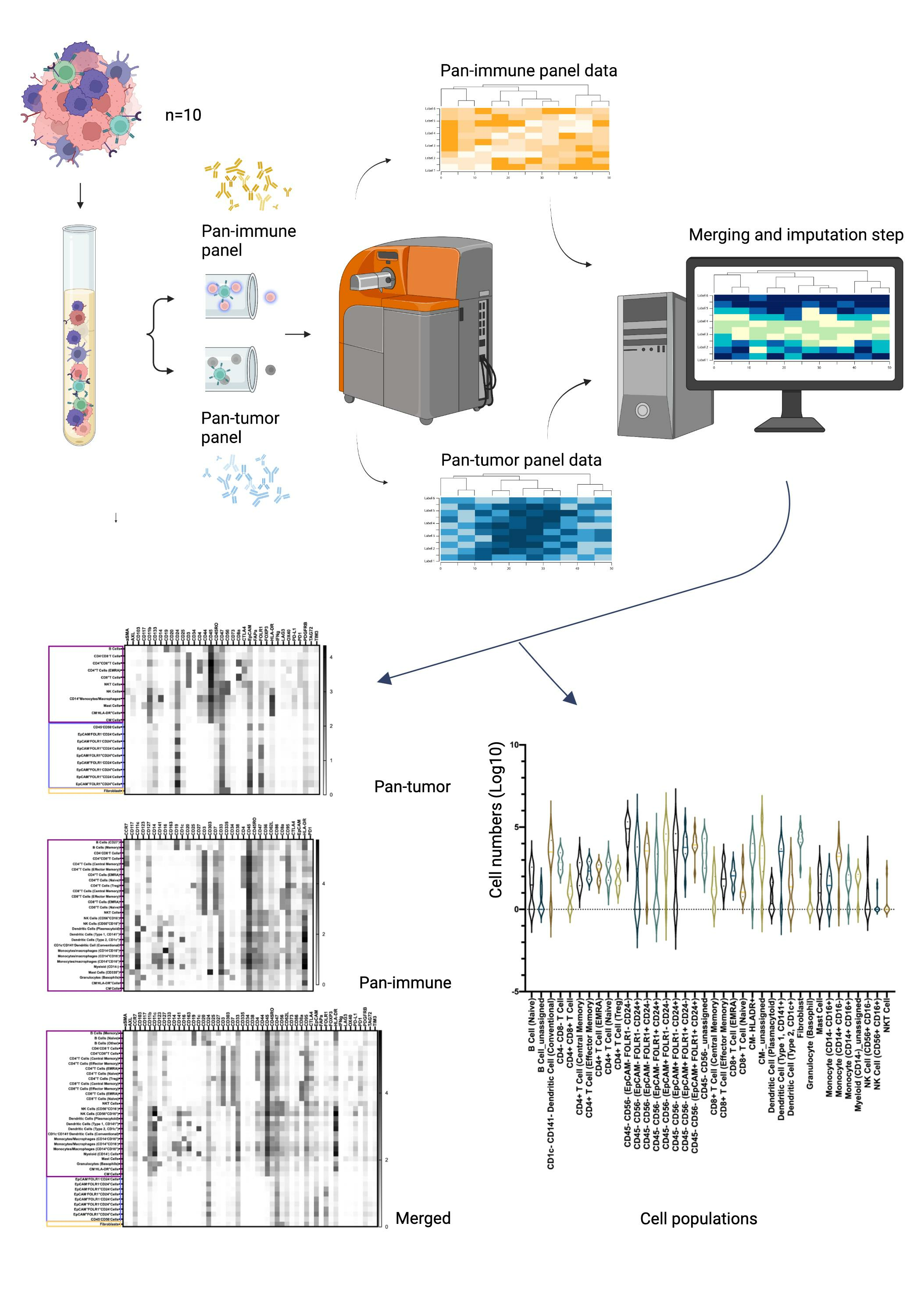
2.5. Mass Cytometry Analysis
2.6. Mass Cytometry Data Analysis
2.7. Statistical Analysis
3. Results
3.1. Patient Characteristics
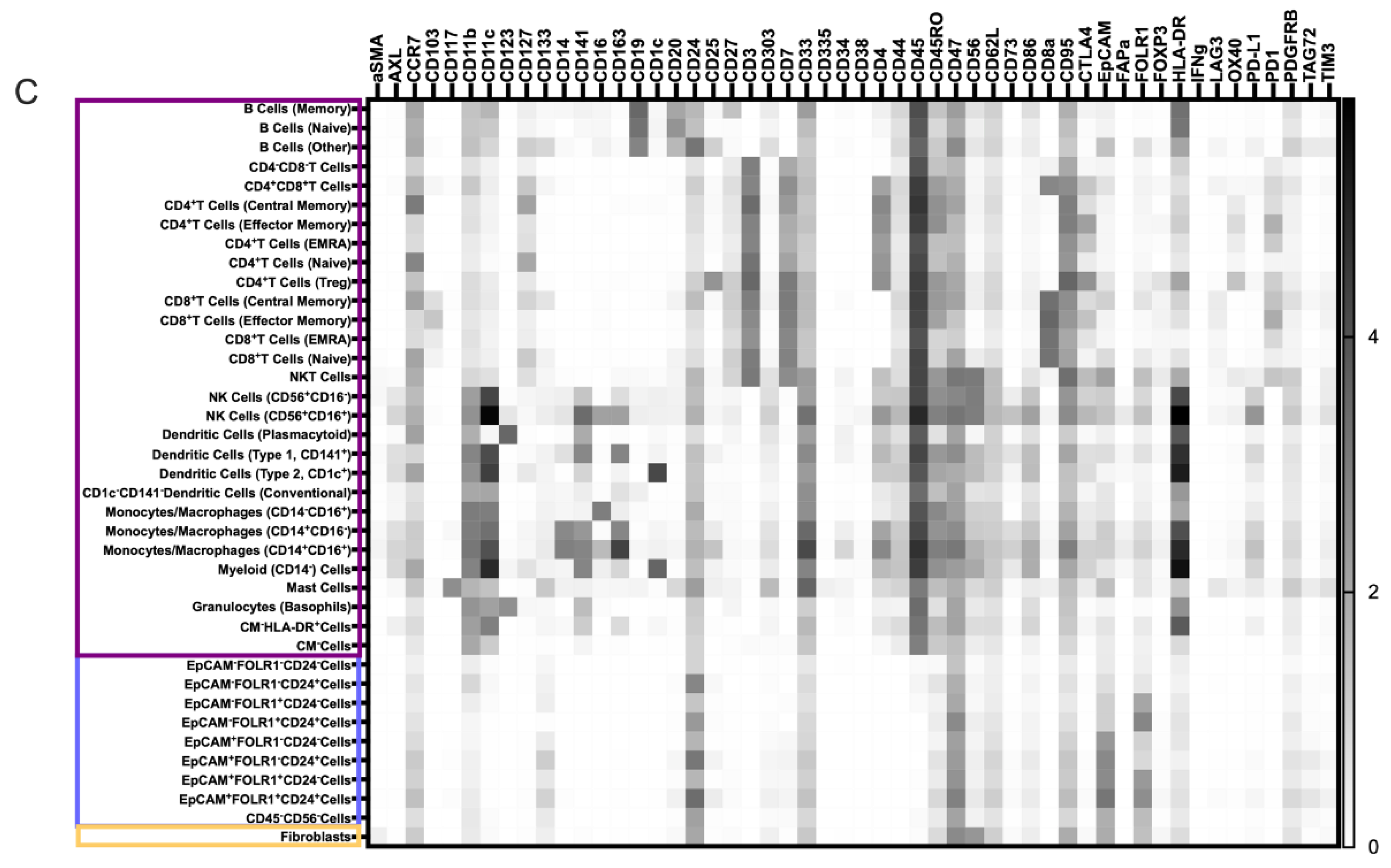
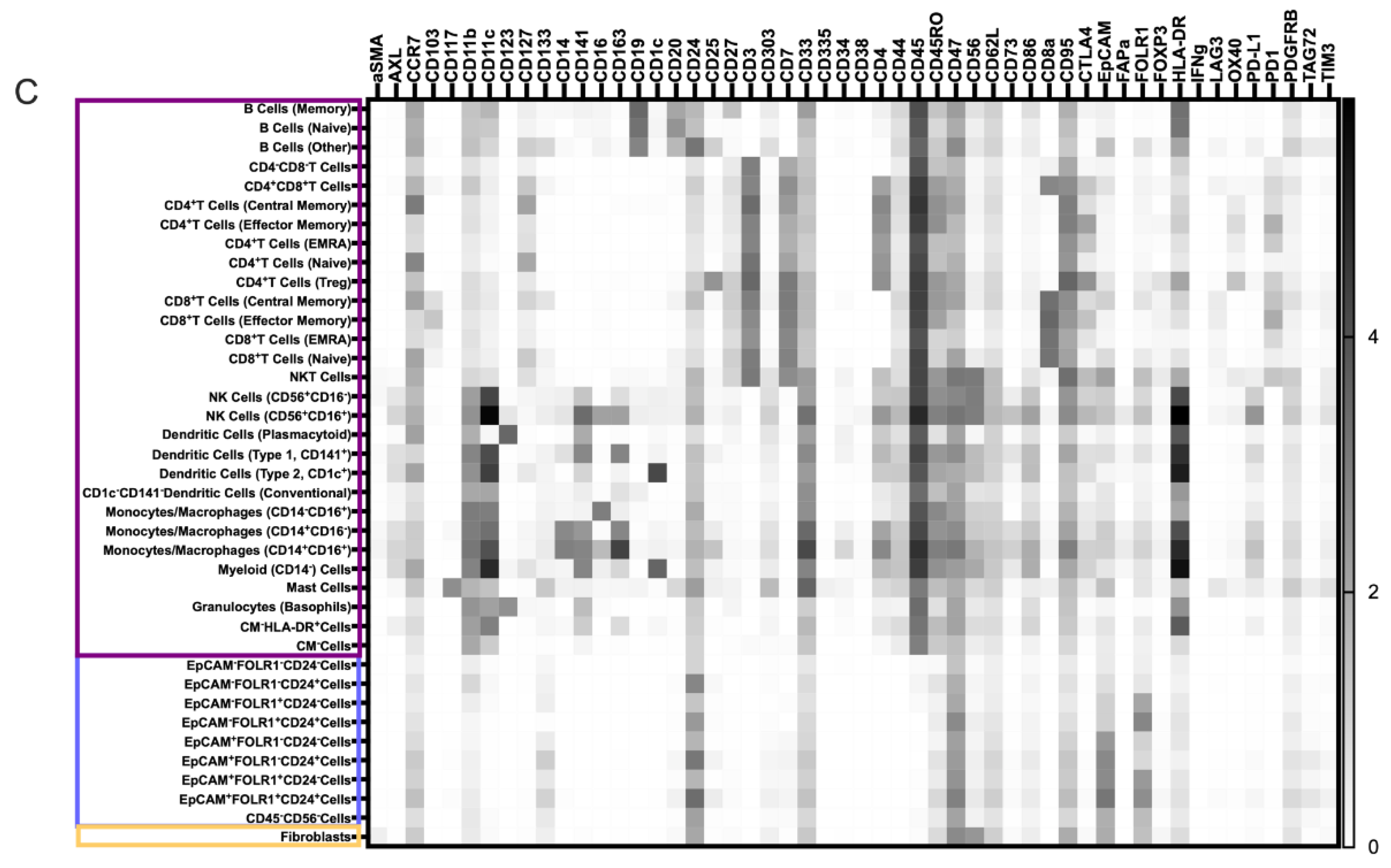
3.2. Characterization of the Ovarian TME with the Pan-Tumor Panel
3.3. Characterization of the Ovarian TiME with the Pan-Immune Panel
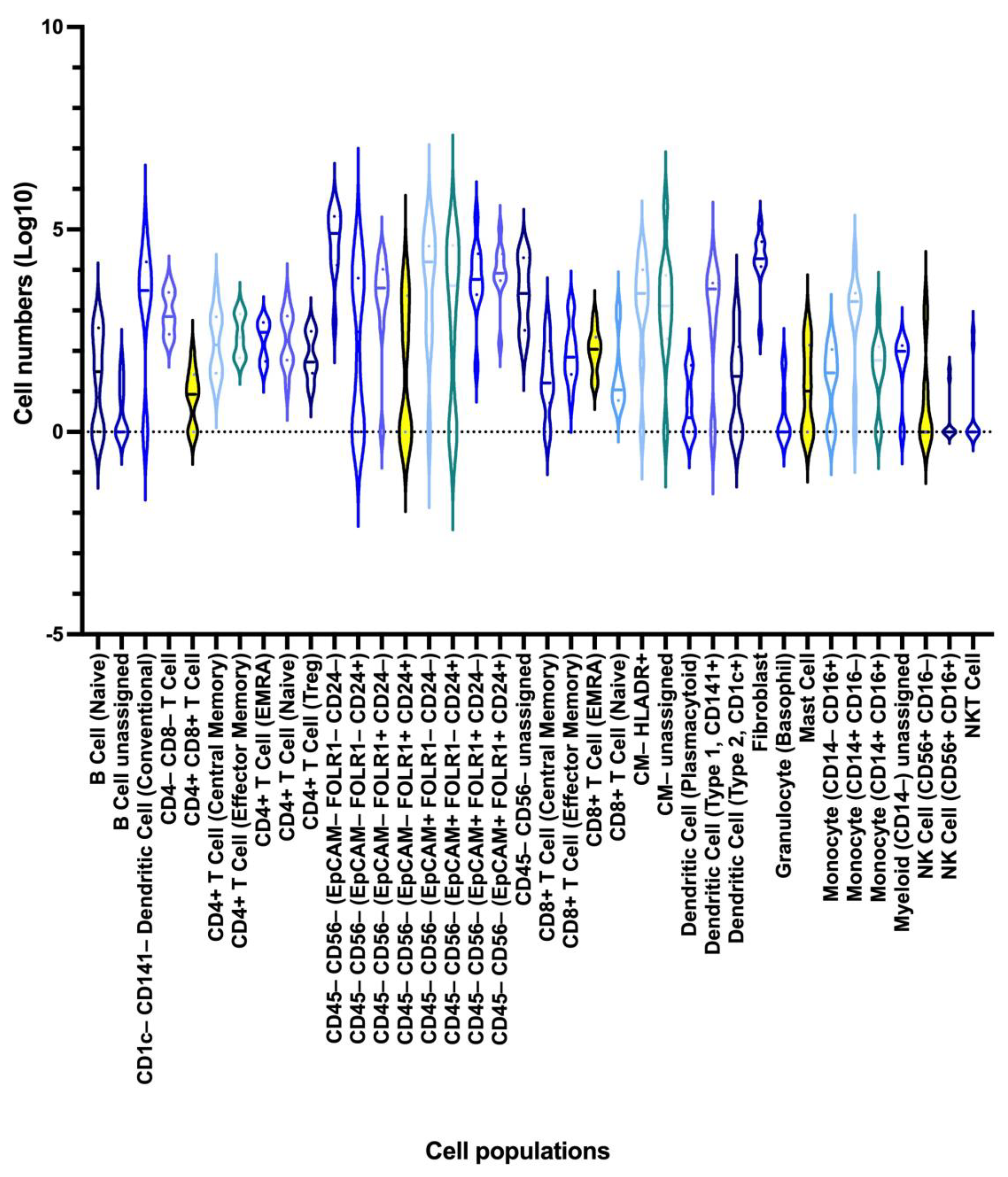
3.4. TME Information Generated by Merging the Pan-Tumor and Pan-Immune Data
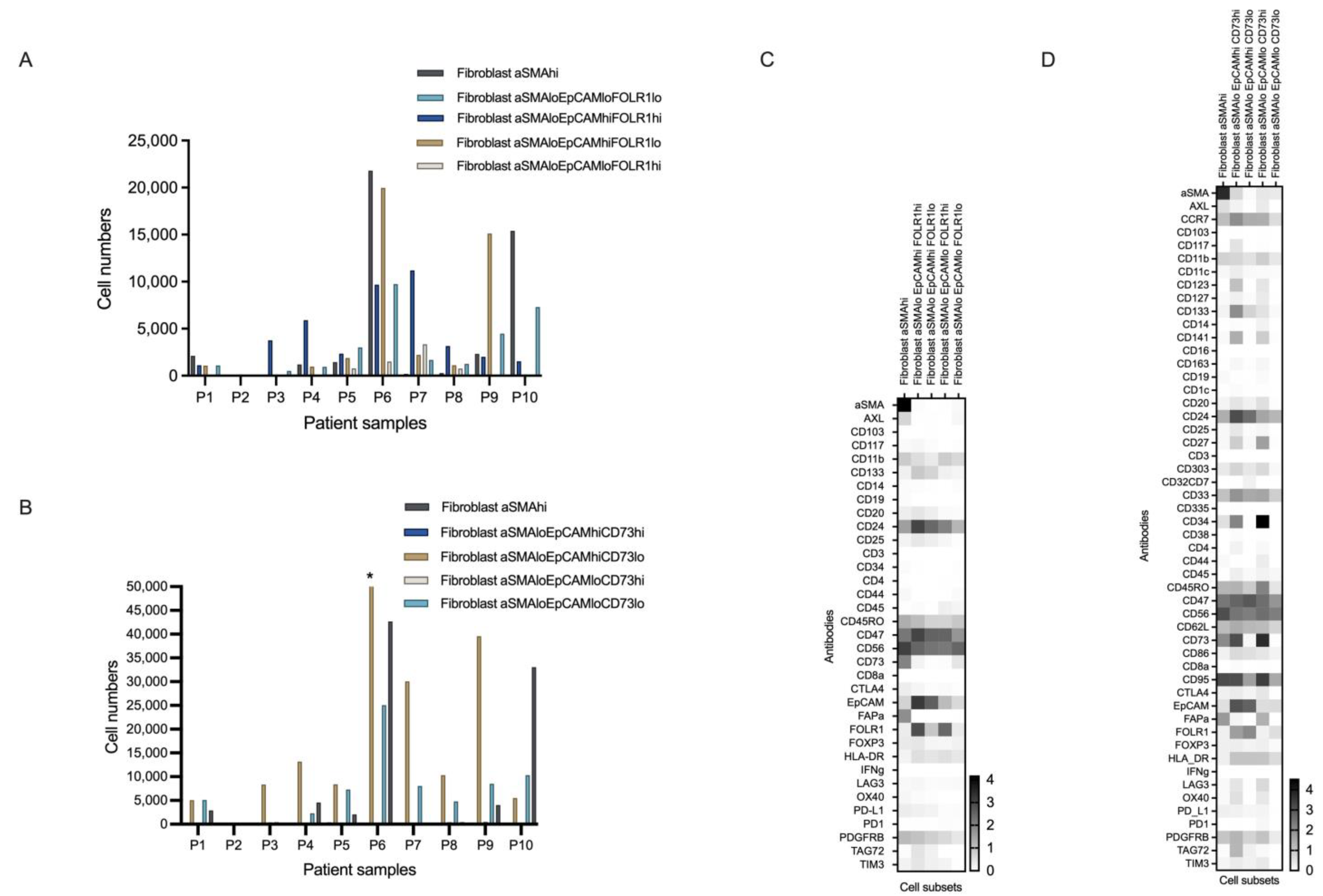
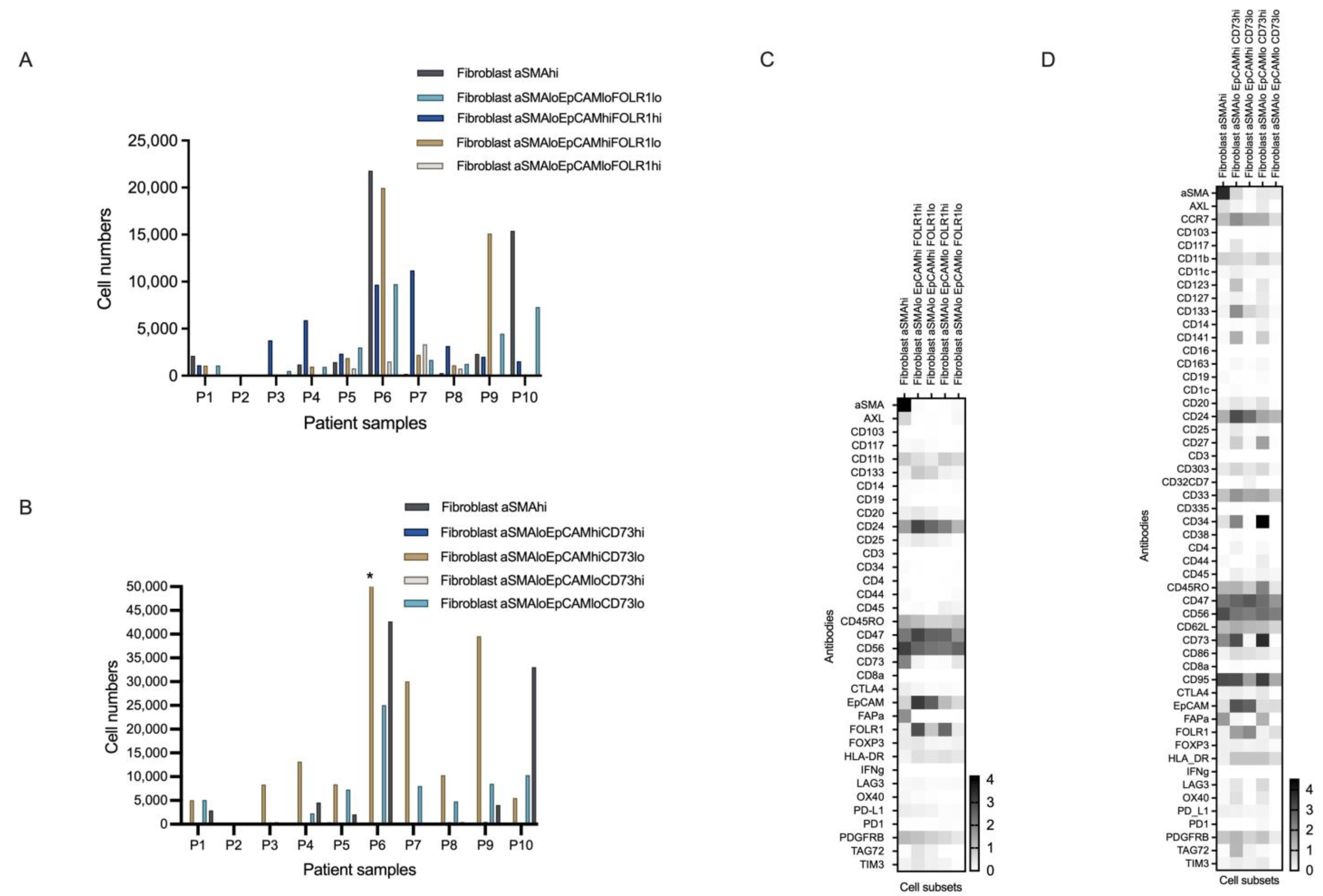
3.4.1. Novel Cell Subsets
3.4.2. Heterogeneity in the Cell Subset Composition of Tumor Samples
3.5. Association of the Detected Cell Subsets with Clinical Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Women’s Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Larsen, I.K. (Ed.) Cancer in Norway 2020—Cancer Incidence, Mortality, Survival and Prevalence in Norway; Cancer Registry of Norway: Oslo, Norway, 2021. [Google Scholar]
- Gourley, C.; Bookman, M.A. Evolving Concepts in the Management of Newly Diagnosed Epithelial Ovarian Cancer. J. Clin. Oncol. 2019, 37, 2386–2397. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Halbert, B.; Einstein, D.J. Hot or Not: Tumor Mutational Burden (TMB) as a Biomarker of Immunotherapy Response in Genitourinary Cancers. Urology 2021, 147, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Stack, M.S.; Nephew, K.P.; Burdette, J.E.; Mitra, A.K. The Tumor Microenvironment of High Grade Serous Ovarian Cancer. Cancers 2018, 11, 21. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Baci, D.; Bosi, A.; Gallazzi, M.; Rizzi, M.; Noonan, D.M.; Poggi, A.; Bruno, A.; Mortara, L. The Ovarian Cancer Tumor Immune Microenvironment (TIME) as Target for Therapy: A Focus on Innate Immunity Cells as Therapeutic Effectors. Int. J. Mol. Sci. 2020, 21, 3125. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Hornburg, M.; Desbois, M.; Lu, S.; Guan, Y.; Lo, A.A.; Kaufman, S.; Elrod, A.; Lotstein, A.; DesRochers, T.M.; Munoz-Rodriguez, J.L.; et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 2021, 39, 928–944. [Google Scholar] [CrossRef]
- Bou-Tayeh, B.; Miller, M.L. Ovarian tumors orchestrate distinct cellular compositions. Immunity 2021, 54, 1107–1109. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K. Immunotherapy in ovarian cancer. Ann. Oncol. 2017, 28 (Suppl. 8), viii1–viii7. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Herrera, F.G.; Ronet, C.; Ochoa de Olza, M.; Barras, D.; Crespo, I.; Andreatta, M.; Corria-Osorio, J.; Spill, A.; Benedetti, F.; Genolet, R.; et al. Low-Dose Radiotherapy Reverses Tumor Immune Desertification and Resistance to Immunotherapy. Cancer Discov. 2022, 12, 108–133. [Google Scholar] [CrossRef]
- Luo, X.; Xu, J.; Yu, J.; Yi, P. Shaping Immune Responses in the Tumor Microenvironment of Ovarian Cancer. Front. Immunol. 2021, 12, 692360. [Google Scholar] [CrossRef]
- Manica, M.; Kim, H.R.; Mathis, R.; Chouvarine, P.; Rutishauser, D.; Roditi, L.D.V.; Szalai, B.; Wagner, U.; Oehl, K.; Saba, K.; et al. Inferring clonal composition from multiple tumor biopsies. NPJ Syst. Biol. Appl. 2020, 6, 27. [Google Scholar] [CrossRef]
- Brady, L.; Kriner, M.; Coleman, I.; Morrissey, C.; Roudier, M.; True, L.D.; Gulati, R.; Plymate, S.R.; Zhou, Z.; Birditt, B.; et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat. Commun. 2021, 12, 1426. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef]
- Anandan, S.; Thomsen, L.C.V.; Gullaksen, S.-E.; Abdelaal, T.; Kleinmanns, K.; Skavland, J.; Bredholt, G.; Gjertsen, B.T.; McCormack, E.; Bjørge, L. Phenotypic Characterization by Mass Cytometry of the Microenvironment in Ovarian Cancer and Impact of Tumor Dissociation Methods. Cancers 2021, 13, 755. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, V.D.; Samusik, N.; Chen, T.J.; Savig, E.S.; Aghaeepour, N.; Quigley, D.A.; Huang, Y.-W.; Giangarrà, V.; Borowsky, A.D.; Hubbard, N.E.; et al. Commonly Occurring Cell Subsets in High-Grade Serous Ovarian Tumors Identified by Single-Cell Mass Cytometry. Cell Rep. 2018, 22, 1875–1888. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, V.D.; Huang, Y.-W.; Delgado-Gonzalez, A.; Chen, S.-Y.; Donoso, K.; Sachs, K.; Gentles, A.J.; Allard, G.M.; Kolahi, K.S.; Howitt, B.E.; et al. High-grade serous ovarian tumor cells modulate NK cell function to create an immune-tolerant microenvironment. Cell Rep. 2021, 36, 109632. [Google Scholar] [CrossRef] [PubMed]
- Casado, J.; Lehtonen, O.; Rantanen, V.; Kaipio, K.; Pasquini, L.; Häkkinen, A.; Petrucci, E.; Hynninen, J.; Hietanen, S.; Carpén, O.; et al. Agile workflow for interactive analysis of mass cytometry data. Bioinformatics 2021, 37, 1263–1268. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, A.; Cybulska, P.; Mager, K.L.; Koplev, S.; Cast, O.; Couturier, D.-L.; Memon, D.; Selenica, P.; Nikolovski, I.; Mazaheri, Y.; et al. Unraveling tumor–immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat. Genet. 2020, 52, 582–593. [Google Scholar] [CrossRef]
- Abdelaal, T.; Höllt, T.; van Unen, V.; Lelieveldt, B.P.F.; Koning, F.; Reinders, M.J.T.; Mahfouz, A. CyTOFmerge: Integrating mass cytometry data across multiple panels. Bioinformatics 2019, 35, 4063–4071. [Google Scholar] [CrossRef]
- Kleinmanns, K.; Gullaksen, S.-E.; Bredholt, G.; Davidson, B.; Torkildsen, C.F.; Grindheim, S.; Bjørge, L.; McCormack, E. Humanized Ovarian Cancer Patient-Derived Xenografts for Improved Preclinical Evaluation of Immunotherapies. Cancers 2022, 14, 3092. [Google Scholar] [CrossRef]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir, E.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef]
- Amir EA, D.; Lee, B.; Badoual, P.; Gordon, M.; Guo, X.V.; Merad, M.; Rahman, A.H. Development of a Comprehensive Antibody Staining Database Using a Standardized Analytics Pipeline. Front. Immunol. 2019, 10, 1315. [Google Scholar] [CrossRef]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef]
- Finak, G.; Langweiler, M.; Jaimes, M.; Malek, M.; Taghiyar, J.; Korin, Y.; Raddassi, K.; Devine, L.; Obermoser, G.; Pekalski, M.L.; et al. Standardizing Flow Cytometry Immunophenotyping Analysis from the Human ImmunoPhenotyping Consortium. Sci. Rep. 2016, 6, 20686. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 2015, 87, 636–645. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Liu, X.; Song, W.; Wong, B.Y.; Zhang, T.; Yu, S.; Lin, G.N.; Ding, X. A comparison framework and guideline of clustering methods for mass cytometry data. Genome Biol. 2019, 20, 297. [Google Scholar] [CrossRef] [PubMed]
- Toker, A.; Nguyen, L.T.; Stone, S.C.; Yang, S.Y.C.; Katz, S.R.; Shaw, P.A.; Clarke, B.A.; Ghazarian, D.; Al-Habeeb, A.; Easson, A.; et al. Regulatory T Cells in Ovarian Cancer Are Characterized by a Highly Activated Phenotype Distinct from that in Melanoma. Clin. Cancer Res. 2018, 24, 5685–5696. [Google Scholar] [CrossRef] [PubMed]
- Kverneland, A.H.; Pedersen, M.; Westergaard, M.C.W.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; van der Burg, S.H.; Milne, K.; et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef] [PubMed]
- Block, M.S.; Dietz, A.B.; Gustafson, M.P.; Kalli, K.R.; Erskine, C.L.; Youssef, B.; Vijay, G.V.; Allred, J.B.; Pavelko, K.D.; Strausbauch, M.A.; et al. Th17-inducing autologous dendritic cell vaccination promotes antigen-specific cellular and humoral immunity in ovarian cancer patients. Nat. Commun. 2020, 11, 5173. [Google Scholar] [CrossRef] [PubMed]
- Bobisse, S.; Genolet, R.; Roberti, A.; Tanyi, J.L.; Racle, J.; Stevenson, B.J.; Iseli, C.; Michel, A.; Le Bitoux, M.-A.; Guillaume, P.; et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8 + T cells in immunotherapy-naive ovarian cancer. Nat. Commun. 2018, 9, 1092. [Google Scholar] [CrossRef]
- Drakes, M.L.; Stiff, P.J. Regulation of Ovarian Cancer Prognosis by Immune Cells in the Tumor Microenvironment. Cancers 2018, 10, 302. [Google Scholar] [CrossRef]
- Schupp, J.; Krebs, F.K.; Zimmer, N.; Trzeciak, E.; Schuppan, D.; Tuettenberg, A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell. Immunol. 2019, 343, 103713. [Google Scholar] [CrossRef]
- Wu, L.; Deng, Z.; Peng, Y.; Han, L.; Liu, J.; Wang, L.; Li, B.; Zhao, J.; Jiao, S.; Wei, H. Ascites-derived IL-6 and IL-10 synergistically expand CD14(+)HLA-DR(-/low) myeloid-derived suppressor cells in ovarian cancer patients. Oncotarget 2017, 8, 76843–76856. [Google Scholar] [CrossRef]
- Duraiswamy, J.; Turrini, R.; Minasyan, A.; Barras, D.; Crespo, I.; Grimm, A.J.; Casado, J.; Genolet, R.; Benedetti, F.; Wicky, A.; et al. Myeloid antigen-presenting cell niches sustain antitumor T cells and license PD-1 blockade via CD28 costimulation. Cancer Cell 2021, 39, 1623–1642. [Google Scholar] [CrossRef] [PubMed]
- Farkkila, A.; Gulhan, D.C.; Casado, J.; Jacobson, C.A.; Nguyen, H.; Kochupurakkal, B.; Maliga, Z.; Yapp, C.; Chen, Y.-A.; Schapiro, D.; et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat. Commun. 2020, 11, 1459. [Google Scholar] [CrossRef]
- Yang, B.; Li, X.; Zhang, W.; Fan, J.; Zhou, Y.; Li, W.; Yin, J.; Yang, X.; Guo, E.; Li, X.; et al. Spatial heterogeneity of infiltrating T cells in high-grade serous ovarian cancer revealed by multi-omics analysis. Cell Rep. Med. 2022, 3, 100856. [Google Scholar] [CrossRef] [PubMed]
- The Impact of Real-Time Mass Spectrometry Data Analysis on the Field of Proteomics. 2022. Available online: https://hupo.org/News/12974741 (accessed on 6 July 2023).
- Kashima, Y.; Togashi, Y.; Fukuoka, S.; Kamada, T.; Irie, T.; Suzuki, A.; Nakamura, Y.; Shitara, K.; Minamide, T.; Yoshida, T.; et al. Potentiality of multiple modalities for single-cell analyses to evaluate the tumor microenvironment in clinical specimens. Sci. Rep. 2021, 11, 341. [Google Scholar] [CrossRef] [PubMed]
- Lyons, Y.A.; Wu, S.Y.; Overwijk, W.W.; Baggerly, K.A.; Sood, A.K. Immune cell profiling in cancer: Molecular approaches to cell-specific identification. NPJ Precis. Oncol. 2017, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ding, Y.; Wan, T.; Deng, T.; Huang, H.; Liu, J. Significance of CD47 and Its Association with Tumor Immune Microenvironment Heterogeneity in Ovarian Cancer. Front. Immunol. 2021, 12, 768115. [Google Scholar] [CrossRef]
- Kotnik, E.N.; Mullen, M.M.; Spies, N.C.; Li, T.; Inkman, M.; Zhang, J.; Martins-Rodrigues, F.; Hagemann, I.S.; McCourt, C.K.; Thaker, P.H.; et al. Genetic characterization of primary and metastatic high-grade serous ovarian cancer tumors reveals distinct features associated with survival. Commun. Biol. 2023, 6, 688. [Google Scholar] [CrossRef]
- Winterhoff, B.J.; Maile, M.; Mitra, A.K.; Sebe, A.; Bazzaro, M.; Geller, M.A.; Abrahante, J.E.; Klein, M.; Hellweg, R.; Mullany, S.A.; et al. Single cell sequencing reveals heterogeneity within ovarian cancer epithelium and cancer associated stromal cells. Gynecol Oncol. 2017, 144, 598–606. [Google Scholar] [CrossRef]
- Schreiber, J.; Boix, C.; Lee, J.W.; Li, H.; Guan, Y.; Chang, C.-C.; Chang, J.-C.; Hawkins-Hooker, A.; Schölkopf, B.; Schweikert, G.; et al. The ENCODE Imputation Challenge: A critical assessment of methods for cross-cell type imputation of epigenomic profiles. Genome Biol. 2023, 24, 79. [Google Scholar] [CrossRef]
- Pedersen, C.B.; Dam, S.H.; Barnkob, M.B.; Leipold, M.D.; Purroy, N.; Rassenti, L.Z.; Kipps, T.J.; Nguyen, J.; Lederer, J.A.; Gohil, S.H.; et al. cyCombine allows for robust integration of single-cell cytometry datasets within and across technologies. Nat. Commun. 2022, 13, 1698. [Google Scholar] [CrossRef]
- Pereira, A.L.; Lambotte, O.; Le Grand, R.; Cosma, A.; Tchitchek, N. CytoBackBone: An algorithm for merging of phenotypic information from different cytometric profiles. Bioinformatics 2019, 35, 4187–4189. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Tolstrup, D.; Dutertre, C.-A.; Morawski, P.A.; Campbell, D.J.; Ginhoux, F.; Newell, E.W.; Gottardo, R.; Headley, M.B. High-throughput single-cell quantification of hundreds of proteins using conventional flow cytometry and machine learning. Sci. Adv. 2021, 7, eabg0505. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Simonds, E.F.; Qiu, P.; Amir, A.D.; Krutzik, P.O.; Finck, R.; Bruggner, R.V.; Melamed, R.; Trejo, A.; Ornatsky, O.I.; et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011, 332, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Wang, C.; Roman, K.A.; Hoyt, C.C. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014, 70, 46–58. [Google Scholar] [CrossRef]
- Gerdes, M.J.; Sevinsky, C.J.; Sood, A.; Adak, S.; Bello, M.O.; Bordwell, A.; Can, A.; Corwin, A.; Dinn, S.; Filkins, R.J.; et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci. USA 2013, 110, 11982–11987. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Numbers | |
|---|---|---|
| Age | ||
| Median years (range) | 70 (54–78) | |
| Stage of cancer | ||
| I–II, III–IV | 4:6 | |
| Surgical outcomes | ||
| R < 1, R > 1 | 7:3 | |
| Clinical status at time of analysis | ||
| Disease-free, living with disease, dead | 6:2:2 |
| Dataset | Phenotype | Cell Subset | logFC | p Value | FDR |
|---|---|---|---|---|---|
| Pan-tumor | |||||
| Recurrence | B cells | −4.41 | 0.006 | 0.13 | |
| Pan-immune | |||||
| Clinical status | CD4+CD8+T cells | −5.94 | 0.0003 | 0.01 | |
| Plasmacytoid dendritic cells | −11.49 | 0.009 | 0.13 | ||
| Merged | |||||
| Recurrence | CD56+CD16−NK cells | −13.76 | 0.0018 | 0.07 | |
| NKT cells | −10.46 | 0.008 | 0.16 | ||
| Clinical status | EpCAM−FOLR1+CD24+cells | −14.64 | 0.0003 | 0.01 | |
| Plasmacytoid dendritic cells | −12.32 | 0.007 | 0.07 | ||
| CD56+CD16−NK cells | −13.50 | 0.007 | 0.07 | ||
| Unassigned cells | −10.18 | 0.008 | 0.07 | ||
| Unassigned HLA-DR+cells | −11.49 | 0.009 | 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomsen, L.C.V.; Kleinmanns, K.; Anandan, S.; Gullaksen, S.-E.; Abdelaal, T.; Iversen, G.A.; Akslen, L.A.; McCormack, E.; Bjørge, L. Combining Mass Cytometry Data by CyTOFmerge Reveals Additional Cell Phenotypes in the Heterogeneous Ovarian Cancer Tumor Microenvironment: A Pilot Study. Cancers 2023, 15, 5106. https://doi.org/10.3390/cancers15205106
Thomsen LCV, Kleinmanns K, Anandan S, Gullaksen S-E, Abdelaal T, Iversen GA, Akslen LA, McCormack E, Bjørge L. Combining Mass Cytometry Data by CyTOFmerge Reveals Additional Cell Phenotypes in the Heterogeneous Ovarian Cancer Tumor Microenvironment: A Pilot Study. Cancers. 2023; 15(20):5106. https://doi.org/10.3390/cancers15205106
Chicago/Turabian StyleThomsen, Liv Cecilie Vestrheim, Katrin Kleinmanns, Shamundeeswari Anandan, Stein-Erik Gullaksen, Tamim Abdelaal, Grete Alrek Iversen, Lars Andreas Akslen, Emmet McCormack, and Line Bjørge. 2023. "Combining Mass Cytometry Data by CyTOFmerge Reveals Additional Cell Phenotypes in the Heterogeneous Ovarian Cancer Tumor Microenvironment: A Pilot Study" Cancers 15, no. 20: 5106. https://doi.org/10.3390/cancers15205106
APA StyleThomsen, L. C. V., Kleinmanns, K., Anandan, S., Gullaksen, S.-E., Abdelaal, T., Iversen, G. A., Akslen, L. A., McCormack, E., & Bjørge, L. (2023). Combining Mass Cytometry Data by CyTOFmerge Reveals Additional Cell Phenotypes in the Heterogeneous Ovarian Cancer Tumor Microenvironment: A Pilot Study. Cancers, 15(20), 5106. https://doi.org/10.3390/cancers15205106