
Unraveling Resistance to Immunotherapy in MSI-High Colorectal Cancer
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Cell Types Involved in Tumor Immune Evasion in dMMR/MSI-H CRC
2.1. Regulatory T Cells
2.2. Myeloid-Derived Suppressor Cells
- Interaction with Tregs: there is evidence to suggest that MDSCs can stimulate the activation of Tregs through the release of the cytokine IL-10 [30], which leads to immunosuppression.
2.3. Tumor-Associated Macrophages
3. Mechanisms of Immune Evasion and Resistance to ICIs
3.1. Tumor Intrinsic Factors Related to Immune Evasion and ICI Resistance
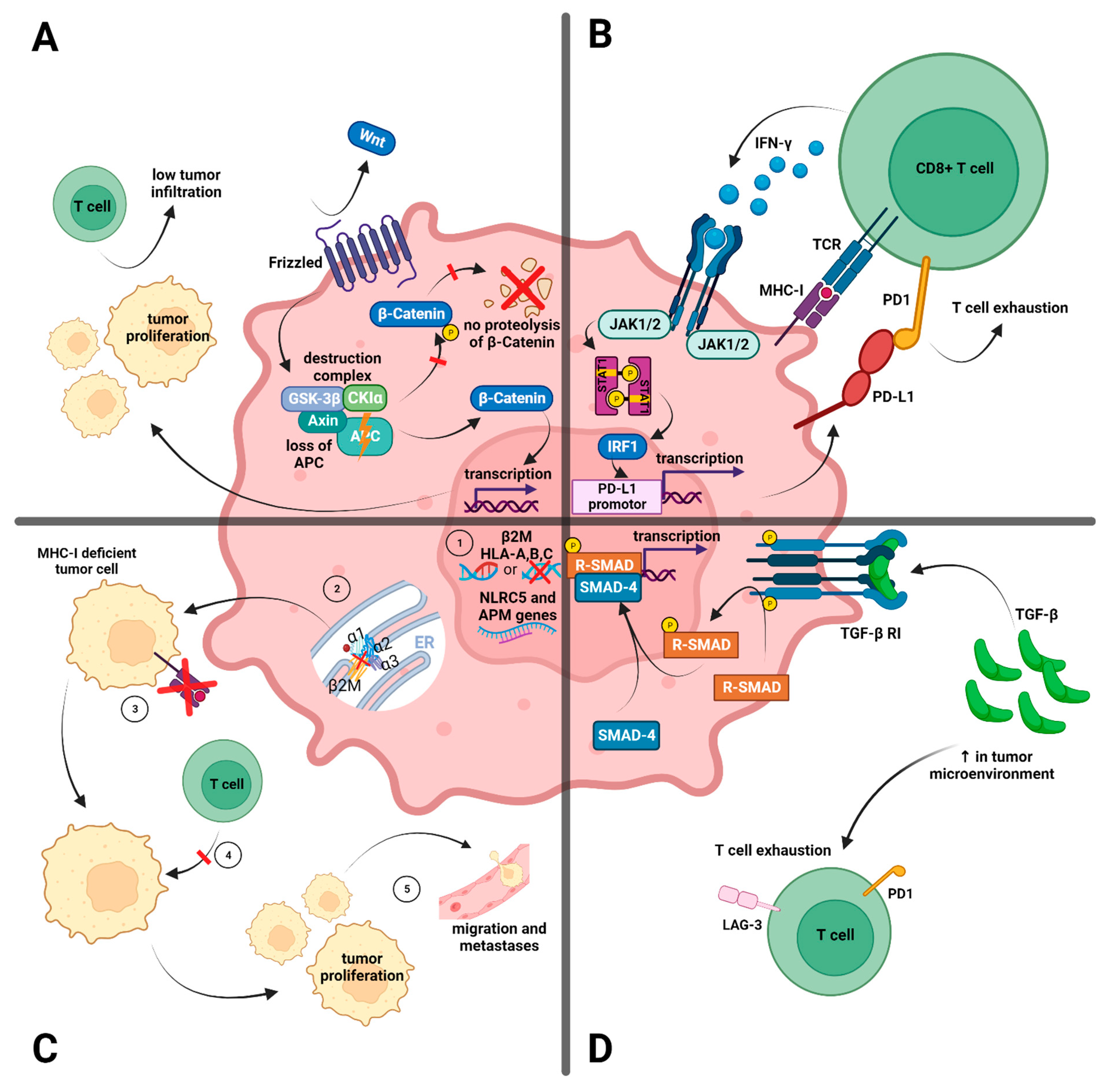
3.1.1. Antigen-Presenting Machinery
3.1.2. WNT/β-Catenin Signaling Pathway
3.1.3. Interferon-γ Signaling
3.1.4. The Transforming Growth Factor Beta (TGF-β)-Dependent Stromal Subset
3.1.5. Clinical, Histopathological, and Molecular Variations in dMMR/MSI-H CRCs
3.2. Extrinsic Factors Leading to Immune Evasion and ICI Resistance
3.2.1. Variable Expression of Immune Checkpoints
3.2.2. Gut Microbiota
3.2.3. Immunoscore
4. Uncertainties in dMMR/MSI-H Diagnosis
4.1. Discordance between Diagnostic Methods
4.2. Intratumoral and Intertumoral Heterogeneity May Contribute to Therapy Resistance
4.3. Lynch Syndrome versus Sporadic MSI-H
5. Conclusions and Prospect
Author Contributions
Funding
Conflicts of Interest
Lexical of Abbreviations
| APC | adenomatous polyposis coli |
| CD | cluster of differentiation |
| CMS | consensus molecular subtypes |
| CRC | colorectal cancer |
| CTLA-4 | cytotoxic T lymphocyte antigen 4 |
| CXCL3 | chemokine (C-X-C motif) ligand 3 |
| dMMR | deficient mismatch repair |
| FOXP3 | Forkhead-Box-Protein P3 |
| HLA | human leukocyte antigen |
| HNPCC | hereditary non-polyposis colorectal cancer |
| IDO | indoleamine 2,3-dioxygenase |
| ICI | immune checkpoint inhibitor |
| IFN-γ | interferon-γ |
| IL | interleukin |
| JAK | Janus Kinase |
| LAG-3 | lymphocyte activation gene 3 |
| MAPK | Raf/MEK/ERK |
| MDSC | myeloid-derived suppressor cell |
| MHC | main histocompatibility complex |
| MLH1 | MutL homolog 1 |
| MMR | mismatch repair |
| MSH2 | MutS homolog 2 |
| MSH6 | MutS homolog 6 |
| MSI-H | microsatellite instability-high |
| MSS | microsatellite stability |
| NGS | next-generation sequencing |
| ORR | overall response rate |
| pMMR | proficient mismatch repair |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death ligand |
| PMS2 | PMS1 homolog 2 |
| STING | stimulator of interferon genes |
| TAM | tumor-associated macrophage |
| TGF-β | transforming growth factor β |
| TIL | tumor-infiltrating lymphocyte |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TMB | tumor mutational burden |
| Tregs | regulatory T cells |
| β2M | beta2-microglobulin |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.; Desantis, C.; Jemal, A. Colorectal cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 104–117. [Google Scholar] [CrossRef]
- World Health Organization. Cancer Today 2020. Available online: https://gco.iarc.fr/today/online-analysis-pie?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=total&sex=0&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=7&group_cancer=1&include_nmsc=0&include_nmsc_other=1&half_pie=0&donut=0 (accessed on 12 February 2022).
- Cunningham, D.; Atkin, W.; Lenz, H.J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef] [PubMed]
- European Cancer Information System. Estimates of Cancer Incidence and Mortality in 2020. Available online: https://ecis.jrc.ec.europa.eu/explorer.php?$0-0$1-All$2-All$4-1,2$3-12$6-0,85$5-2020,2020$7-7$CEstByCountry$X0_8-3$X0_19-AE27$X0_20-No$CEstBySexByCountry$X1_8-3$X1_19-AE27$X1_-1-1$CEstByIndiByCountry$X2_8-3$X2_19-AE27$X2_20-No$CEstRelative$X3_8-3$X3_9-AE27$X3_19-AE27$CEstByCountryTable$X4_19-AE27 (accessed on 12 February 2022).
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Venderbosch, S.; Nagtegaal, I.D.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Herman, J.G.; Umar, A.; Polyak, K.; Graff, J.R.; Ahuja, N.; Issa, J.-P.J.; Markowitz, S.; Willson, J.K.; Hamilton, S.R.; Kinzler, K.W. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 6870–6875. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Sargent, D.J. Molecular Pathways: Microsatellite Instability in Colorectal Cancer: Prognostic, Predictive, and Therapeutic Implications. Clin. Cancer Res. 2012, 18, 1506. [Google Scholar]
- Andre, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.M.; Garcia-Carbonero, R.; Alcaide, J.; Gibbs, P.; et al. Final overall survival for the phase III KN177 study: Pembrolizumab versus chemotherapy in microsatellite instability-high/mismatch repair deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2021, 39, 3500. [Google Scholar] [CrossRef]
- Lenz, H.J.; Van Cutsem, E.; Luisa Limon, M.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; García-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef]
- Arai, Y.; Saito, H.; Ikeguchi, M. Upregulation of TIM-3 and PD-1 on CD4+ and CD8+ T Cells Associated with Dysfunction of Cell-Mediated Immunity after Colorectal Cancer Operation. Yonago Acta Med. 2012, 55, 1–9. [Google Scholar] [PubMed]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally arising Foxp3-expressing CD25+ CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 2005, 6, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Vlad, C.; Kubelac, P.; Fetica, B.; Vlad, D.; Irimie, A.; Achimas-Cadariu, P. The prognostic value of FOXP3+ T regulatory cells in colorectal cancer. J. Buon. 2015, 20, 114–119. [Google Scholar]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Waniczek, D.; Lorenc, Z.; Śnietura, M.; Wesecki, M.; Kopec, A.; Muc-Wierzgoń, M. Tumor-Associated Macrophages and Regulatory T Cells Infiltration and the Clinical Outcome in Colorectal Cancer. Arch. Immunol. Ther. Exp. 2017, 65, 445–454. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Mahalingam, J.; Chiang, J.-M.; Su, P.-J.; Chu, Y.-Y.; Lai, H.-Y.; Fang, J.-H.; Huang, C.-T.; Chiu, C.-T.; Lin, C.-Y. Activated but not resting regulatory T cells accumulated in tumor microenvironment and correlated with tumor progression in patients with colorectal cancer. Int. J. Cancer. 2013, 132, 1341–1350. [Google Scholar] [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E. Two FOXP3+ CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.M.; Jiang, X.; Seo, Y.D.; Kenerson, H.L.; Yan, X.; Lausted, C.; Meng, C.; Jabbari, N.; Labadie, K.P.; Daniel, S.K. IL-10 blockade reactivates antitumor immunity in human colorectal cancer liver metastases. Cancer Res. 2019, 79, 4489. [Google Scholar] [CrossRef]
- Michel, S.; Benner, A.; Tariverdian, M.; Wentzensen, N.; Hoefler, P.; Pommerencke, T.; Grabe, N.; von Knebel Doeberitz, M.; Kloor, M. High density of FOXP3-positive T cells infiltrating colorectal cancers with microsatellite instability. Br. J. Cancer 2008, 99, 1867–1873. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory CheckpointsImmune Checkpoints in Human Colorectal Cancer. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.; Colombo, M.; Frey, A.; Greten, T.; Mandruzzato, S.; Murray, P.; Ochoa, A.; Ostrand-Rosenberg, S. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+ CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015, 16, 64. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Forssell, J.; Oberg, A.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef]
- Narayanan, S.; Kawaguchi, T.; Peng, X.; Qi, Q.; Liu, S.; Yan, L.; Takabe, K. Tumor infiltrating lymphocytes and macrophages improve survival in microsatellite unstable colorectal cancer. Sci. Rep. 2019, 9, 13455. [Google Scholar] [CrossRef]
- Erreni, M.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) and inflammation in colorectal cancer. Cancer Microenviron. 2011, 4, 141–154. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Korehisa, S.; Oki, E.; Iimori, M.; Nakaji, Y.; Shimokawa, M.; Saeki, H.; Okano, S.; Oda, Y.; Maehara, Y. Clinical significance of programmed cell death-ligand 1 expression and the immune microenvironment at the invasive front of colorectal cancers with high microsatellite instability. Int. J. Cancer 2018, 142, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Georgoudaki, A.-M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Bicknell, D.C.; Rowan, A.; Bodmer, W.F. Beta 2-microglobulin gene mutations: A study of established colorectal cell lines and fresh tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 4751–4755. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung CancerAntigen-Processing Defects and Resistance to PD-1 Blockade. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [PubMed]
- Gurjao, C.; Liu, D.; Hofree, M.; AlDubayan, S.H.; Wakiro, I.; Su, M.-J.; Felt, K.; Gjini, E.; Brais, L.K.; Rotem, A. Intrinsic Resistance to Immune Checkpoint Blockade in a Mismatch Repair–Deficient Colorectal CancerResistance to Immune Checkpoint Blockade in a MSI-H CRC. Cancer Immunol. Res. 2019, 7, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Middha, S.; Yaeger, R.; Shia, J.; Stadler, Z.K.; King, S.; Guercio, S.; Paroder, V.; Bates, D.D.; Rana, S.; Diaz, L.A., Jr. Majority of B2M-mutant and-deficient colorectal carcinomas achieve clinical benefit from immune checkpoint inhibitor therapy and are microsatellite instability-high. JCO Precis. Oncol. 2019, 3, PO.18.00321. [Google Scholar] [CrossRef] [PubMed]
- Chun, E.; Lavoie, S.; Michaud, M.; Gallini, C.A.; Kim, J.; Soucy, G.; Odze, R.; Glickman, J.N.; Garrett, W.S. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. 2015, 12, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Clendenning, M.; Huang, A.; Jayasekara, H.; Lorans, M.; Preston, S.; O’Callaghan, N.; Pope, B.J.; Macrae, F.A.; Winship, I.M.; Milne, R.L. Somatic mutations of the coding microsatellites within the beta-2-microglobulin gene in mismatch repair-deficient colorectal cancers and adenomas. Fam. Cancer 2018, 17, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Michel, S.; Buckowitz, B.; Rüschoff, J.; Büttner, R.; Holinski-Feder, E.; Dippold, W.; Wagner, R.; Tariverdian, M.; Benner, A. Beta2-microglobulin mutations in microsatellite unstable colorectal tumors. Int. J. Cancer 2007, 121, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Dierssen, J.W.F.; de Miranda, N.F.; Ferrone, S.; van Puijenbroek, M.; Cornelisse, C.J.; Fleuren, G.J.; van Wezel, T.; Morreau, H. HNPCC versus sporadic microsatellite-unstable colon cancers follow different routes toward loss of HLA class I expression. BMC Cancer 2007, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K. Genetic Mechanisms of Immune Evasion in Colorectal CancerGenetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, M.; Janikovits, J.; von Knebel Doeberitz, M.; Kloor, M. Complex pattern of immune evasion in MSI colorectal cancer. Oncoimmunology 2018, 7, e1445453. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Andre, T.; El-Refai, S.M.; Kopetz, S.; Tabernero, J.; Sinicrope, F.A.; Tie, J.; George, T.J.; VanCutsem, E.; Mauer, E. Impact of RAS mutations on immunologic characteristics of the tumor microenvironment (TME) in patients with microsatellite instability-high (MSI-H) or mismatch-repair–deficient (dMMR) colorectal cancer (CRC). Am. Soc. Clin. Oncol. 2022, 40, 3067. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [PubMed]
- Sers, C.; Kuner, R.; Falk, C.S.; Lund, P.; Sueltmann, H.; Braun, M.; Buness, A.; Ruschhaupt, M.; Conrad, J.; Mang-Fatehi, S. Down-regulation of HLA Class I and NKG2D ligands through a concerted action of MAPK and DNA methyltransferases in colorectal cancer cells. Int. J. Cancer 2009, 125, 1626–1639. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; de Reyniès, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautès-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.-J.; Lonardi, S.; Zagonel, V.; Van Cutsem, E.; Limon, M.L.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; Garcia-Alfonso, P.; Neyns, B. Subgroup analyses of patients (pts) with microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC) treated with nivolumab (NIVO) plus low-dose ipilimumab (IPI) as first-line (1L) therapy: Two-year clinical update. Am. Soc. Clin. Oncol. 2021, 39, 58. [Google Scholar]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef] [PubMed]
- Goldsberry, W.N.; Meza-Perez, S.; Londoño, A.I.; Katre, A.A.; Mott, B.T.; Roane, B.M.; Goel, N.; Wall, J.A.; Cooper, S.J.; Norian, L.A. Inhibiting WNT ligand production for improved immune recognition in the ovarian tumor microenvironment. Cancers 2020, 12, 766. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Tinsley, H.N.; Keeton, A.; Qu, Z.; Piazza, G.A.; Li, Y. Suppression of Wnt/β-catenin signaling inhibits prostate cancer cell proliferation. Eur. J. Pharmacol. 2009, 602, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Wherry, E.J. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 2016, 165, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Albacker, L.A.; Wu, J.; Smith, P.; Warmuth, M.; Stephens, P.J.; Zhu, P.; Yu, L.; Chmielecki, J. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS ONE 2017, 12, e0176181. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.A.; Zibelman, M.; Lindsay, T.; Feldman, R.A.; Saul, M.; Gatalica, Z.; Korn, W.M.; Heath, E.I. An emerging landscape for canonical and actionable molecular alterations in primary and metastatic prostate cancer. Mol. Cancer Ther. 2020, 19, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef] [PubMed]
- Sucker, A.; Zhao, F.; Pieper, N.; Heeke, C.; Maltaner, R.; Stadtler, N.; Real, B.; Bielefeld, N.; Howe, S.; Weide, B.; et al. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat. Commun. 2017, 8, 15440. [Google Scholar] [CrossRef] [PubMed]
- Prud’homme, G.J. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab. Investig. J. Tech. Methods Pathol. 2007, 87, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Endo, E.; Okayama, H.; Saito, K.; Nakajima, S.; Yamada, L.; Ujiie, D.; Kase, K.; Fujita, S.; Endo, H.; Sakamoto, W.; et al. A TGFβ-Dependent Stromal Subset Underlies Immune Checkpoint Inhibitor Efficacy in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Colorectal Cancer. Mol. Cancer Res. MCR 2020, 18, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Noonan, K.A.; Pham, V.; Bedi, R.; Zhavoronkov, A.; Ozerov, I.V.; Makarev, E.; Artemov, A.V.; Wysocki, P.T.; Mehra, R.; et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat. Commun. 2018, 9, 741. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.D.; Delorenzi, M.; Tejpar, S.; Yan, P.; Klingbiel, D.; Fiocca, R.; d’Ario, G.; Cisar, L.; Labianca, R.; Cunningham, D.; et al. Integrated analysis of molecular and clinical prognostic factors in stage II/III colon cancer. J. Natl. Cancer Inst. 2012, 104, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Voorneveld, P.W.; Jacobs, R.J.; Kodach, L.L.; Hardwick, J.C. A Meta-Analysis of SMAD4 Immunohistochemistry as a Prognostic Marker in Colorectal Cancer. Transl. Oncol. 2015, 8, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Klingbiel, D.; Saridaki, Z.; Ceppa, P.; Curto, M.; McKee, T.A.; Roth, A.; Tejpar, S.; Delorenzi, M.; Bosman, F.T.; et al. Reduced Expression of SMAD4 Is Associated with Poor Survival in Colon Cancer. Clin. Cancer Res. 2016, 22, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Cloyd, J.M.; Vicente, D.; Omichi, K.; Chun, Y.S.; Kopetz, S.E.; Maru, D.; Conrad, C.; Tzeng, C.D.; Wei, S.H.; et al. SMAD4 gene mutation predicts poor prognosis in patients undergoing resection for colorectal liver metastases. Eur. J. Surg. Oncol. 2018, 44, 684–692. [Google Scholar]
- Yoo, S.-Y.; Lee, J.-A.; Shin, Y.; Cho, N.-Y.; Bae, J.M.; Kang, G.H. Clinicopathological Characterization and Prognostic Implication of SMAD4 Expression in Colorectal Carcinoma. J. Pathol. Transl. Med. 2019, 53, 289–297. [Google Scholar] [CrossRef]
- Isaksson-Mettävainio, M.; Palmqvist, R.; Dahlin, A.M.; Van Guelpen, B.; Rutegård, J.; Öberg, Å.; Henriksson, M.L. High SMAD4 levels appear in microsatellite instability and hypermethylated colon cancers, and indicate a better prognosis. Int. J. Cancer. 2012, 131, 779–788. [Google Scholar] [CrossRef]
- Goldstein, J.; Tran, B.; Ensor, J.; Gibbs, P.; Wong, H.L.; Wong, S.F.; Vilar, E.; Tie, J.; Broaddus, R.; Kopetz, S.; et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann. Oncol. 2014, 25, 1032–1038. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef]
- Temraz, S.; Nassar, F.; Nasr, R.; Charafeddine, M.; Mukherji, D.; Shamseddine, A. Gut microbiome: A promising biomarker for immunotherapy in colorectal cancer. Int. J. Mol. Sci. 2019, 20, 4155. [Google Scholar] [CrossRef]
- Hamada, T.; Zhang, X.; Mima, K.; Bullman, S.; Sukawa, Y.; Nowak, J.A.; Kosumi, K.; Masugi, Y.; Twombly, T.S.; Cao, Y. Fusobacterium nucleatum in Colorectal Cancer Relates to Immune Response Differentially by Tumor Microsatellite Instability StatusFusobacterium, MSI, and Immunity in Colorectal Cancer. Cancer Immunol. Res. 2018, 6, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Derosa, L.; Hellmann, M.D.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.J.; Arbour, K.C.; Chaft, J.E.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Huemer, F.; Rinnerthaler, G.; Westphal, T.; Hackl, H.; Hutarew, G.; Gampenrieder, S.P.; Weiss, L.; Greil, R. Impact of antibiotic treatment on immune-checkpoint blockade efficacy in advanced non-squamous non-small cell lung cancer. Oncotarget 2018, 9, 16512–16520. [Google Scholar] [CrossRef] [PubMed]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Galon, J.; Fuchs, C.S.; Dranoff, G. Cancer immunology--analysis of host and tumor factors for personalized medicine. Nat. Rev. Clin. Oncol. 2011, 8, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Louafi, S.; Bardier, A.; Charlotte, F.; Vaillant, J.C.; Ménégaux, F.; Rosenzwajg, M.; Lemoine, F.; Klatzmann, D.; Taieb, J. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut 2009, 58, 520. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.M.; Banerjea, A.; Feakins, R.; Li, S.R.; Bustin, S.A.; Dorudi, S. Tumour-infiltrating lymphocytes in colorectal cancer with microsatellite instability are activated and cytotoxic. Br. J. Surg. 2004, 91, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Pagès, F.; Marincola, F.M.; Angell, H.K.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.; Botti, G.; et al. Cancer classification using the Immunoscore: A worldwide task force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Wirta, E.-V.; Seppälä, T.; Friman, M.; Väyrynen, J.; Ahtiainen, M.; Kautiainen, H.; Kuopio, T.; Kellokumpu, I.; Mecklin, J.-P.; Böhm, J. Immunoscore in mismatch repair-proficient and -deficient colon cancer. J. Pathol. Clin. Res. 2017, 3, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Huebner, L.J.; Finnes, H.D.; Muranyi, A.; Clements, J.; Singh, S.; Hubbard, J.M.; McWilliams, R.R.; Shanmugam, K.; Sinicrope, F.A. Intratumoral CD3+ and CD8+ T-Cell Densities in Patients with DNA Mismatch Repair–Deficient Metastatic Colorectal Cancer Receiving Programmed Cell Death-1 Blockade. JCO Precis. Oncol. 2019, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Yu, X.; Xue, L.; Ge, X.; Zhao, W.; Peng, W. Intrinsic β-catenin signaling suppresses CD8(+) T-cell infiltration in colorectal cancer. Biomed. Pharmacother. 2019, 115, 108921. [Google Scholar] [CrossRef] [PubMed]
- Engel, K.B.; Moore, H.M. Effects of preanalytical variables on the detection of proteins by immunohistochemistry in formalin-fixed, paraffin-embedded tissue. Arch. Pathol. Lab. Med. 2011, 135, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Shia, J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J. Mol. Diagn. 2008, 10, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Snowsill, T.; Coelho, H.; Huxley, N.; Jones-Hughes, T.; Briscoe, S.; Frayling, I.M.; Hyde, C. Molecular testing for Lynch syndrome in people with colorectal cancer: Systematic reviews and economic evaluation. Health Technol. Assess. 2017, 21, 1–238. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Nagasaka, T.; Hamelin, R.; Boland, C.R. An optimized pentaplex PCR for detecting DNA mismatch repair-deficient colorectal cancers. PLoS ONE 2010, 5, e9393. [Google Scholar] [CrossRef]
- Xicola, R.M.; Llor, X.; Pons, E.; Castells, A.; Alenda, C.; Piñol, V.; Andreu, M.; Castellví-Bel, S.; Payá, A.; Jover, R.; et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J. Natl. Cancer Inst. 2007, 99, 244–252. [Google Scholar] [CrossRef]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Hain, E.; Buhard, O.; Guilloux, A.; Bardier, A.; Kaci, R.; Bertheau, P.; Renaud, F.; Bibeau, F.; Fléjou, J.F.; et al. 537P-Assessment of local clinical practice for testing of mismatch repair deficiency in metastatic colorectal cancer: The need for new diagnostic guidelines prior to immunotherapy. Ann. Oncol. 2018, 29, viii179–viii180. [Google Scholar] [CrossRef]
- Yuan, L.; Chi, Y.; Chen, W.; Chen, X.; Wei, P.; Sheng, W.; Zhou, X.; Shi, D. Immunohistochemistry and microsatellite instability analysis in molecular subtyping of colorectal carcinoma based on mismatch repair competency. Int. J. Clin. Exp. Med. 2015, 8, 20988–21000. [Google Scholar]
- Chen, M.L.; Chen, J.Y.; Hu, J.; Chen, Q.; Yu, L.X.; Liu, B.R.; Qian, X.P.; Yang, M. Comparison of microsatellite status detection methods in colorectal carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 1431–1438. [Google Scholar] [PubMed]
- Wang, Y.; Shi, C.; Eisenberg, R.; Vnencak-Jones, C.L. Differences in Microsatellite Instability Profiles between Endometrioid and Colorectal Cancers: A Potential Cause for False-Negative Results? J. Mol. Diagn. 2017, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, L.I.; Ligtenberg, M.J.; Willems, R.W.; Hermens, R.P.; Blokx, W.A.; Dubois, S.V.; van der Linden, H.; Meijer, J.W.; Mlynek-Kersjes, M.L.; Hoogerbrugge, N.; et al. Interpretation of immunohistochemistry for mismatch repair proteins is only reliable in a specialized setting. Am. J. Surg. Pathol. 2008, 32, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Verma, L.; Kane, M.F.; Brassett, C.; Schmeits, J.; Evans, D.G.; Kolodner, R.D.; Maher, E.R. Mononucleotide microsatellite instability and germline MSH6 mutation analysis in early onset colorectal cancer. J. Med. Genet. 1999, 36, 678–682. [Google Scholar]
- Pyatt, R.; Chadwick, R.B.; Johnson, C.K.; Adebamowo, C.; de la Chapelle, A.; Prior, T.W. Polymorphic variation at the BAT-25 and BAT-26 loci in individuals of African origin. Implications for microsatellite instability testing. Am. J. Pathol. 1999, 155, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [PubMed]
- He, W.-Z.; Hu, W.-M.; Wang, F.; Rong, Y.-M.; Yang, L.; Xie, Q.-K.; Yang, Y.-Z.; Jiang, C.; Qiu, H.-J.; Lu, J.-B. Comparison of mismatch repair status between primary and matched metastatic sites in patients with colorectal cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 1174–1183. [Google Scholar] [CrossRef]
- Moreira, L.; Balaguer, F.; Lindor, N.; de la Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA 2012, 308, 1555–1565. [Google Scholar] [CrossRef]
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.; Caron, O.; Colas, C.; Entz-Werle, N.; Gerdes, A.M.; Goldberg, Y.; Ilencikova, D.; Muleris, M.; et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J. Med. Genet. 2014, 51, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Roca, A.; Giner-Calabuig, M.; Murcia, O.; Castillejo, A.; Soto, J.L.; García-Heredia, A.; Jover, R. Lynch-like Syndrome: Potential Mechanisms and Management. Cancers 2022, 14, 1115. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Chalabi, M.; Verschoor, Y.; van den Berg, J.; Sikorska, K.; Beets, G.; Lent, A.; Grootscholten, M.; Aalbers, A.; Buller, N.; Marsman, H. LBA7 Neoadjuvant immune checkpoint inhibition in locally advanced MMR-deficient colon cancer: The NICHE-2 study. Ann. Oncol. 2022, 33, S1389. [Google Scholar] [CrossRef]
- Colle, R.; Lonardi, S.; Cachanado, M.; Overman, M.J.; Elez, E.; Fakih, M.; Corti, F.; Jayachandran, P.; Svrcek, M.; Dardenne, A.; et al. Impact of Lynch syndrome, BRAFV600E, and RAS mutations on outcomes in MSI/dMMR metastatic colorectal cancer (mCRC) treated with immune checkpoint inhibitors (ICI): Analysis of combined international cohorts. J. Clin. Oncol. 2023, 41, 171. [Google Scholar] [CrossRef]
- Nakayama, Y.; Iijima, T.; Inokuchi, T.; Kojika, E.; Takao, M.; Takao, A.; Koizumi, K.; Horiguchi, S.-I.; Hishima, T.; Yamaguchi, T. Clinicopathological features of sporadic MSI colorectal cancer and Lynch syndrome: A single-center retrospective cohort study. Int. J. Clin. Oncol. 2021, 26, 1881–1889. [Google Scholar] [CrossRef]
- Liu, G.C.; Liu, R.Y.; Yan, J.P.; An, X.; Jiang, W.; Ling, Y.H.; Chen, J.W.; Bei, J.X.; Zuo, X.Y.; Cai, M.Y.; et al. The Heterogeneity Between Lynch-Associated and Sporadic MMR Deficiency in Colorectal Cancers. J. Natl. Cancer Inst. 2018, 110, 975–984. [Google Scholar]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra52. [Google Scholar] [CrossRef]
- Chon, H.J.; Kim, H.; Noh, J.H.; Yang, H.; Lee, W.S.; Kong, S.J.; Lee, S.J.; Lee, Y.S.; Kim, W.R.; Kim, J.H. STING signaling is a potential immunotherapeutic target in colorectal cancer. J. Cancer 2019, 10, 4932. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Liu, C.; Luo, S.; Cao, T.; Lin, B.; Zhou, M.; Zhang, X.; Wang, S.; Zheng, T.; Li, X. STING agonist and IDO inhibitor combination therapy inhibits tumor progression in murine models of colorectal cancer. Cell. Immunol. 2021, 366, 104384. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, W.S.; Kong, S.J.; Kim, C.G.; Kim, J.H.; Chang, S.K.; Kim, S.; Kim, G.; Chon, H.J.; Kim, C. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Investig. 2019, 129, 4350–4364. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Genetic Defects (“Hard” Lesions) | Non-Genetic Defects (“Soft-Lesions”) | |
|---|---|---|
| Mutations | LOH at | Transcriptional downregulation: HLA-I genes |
| HLA-I heavy chain | chr 6 | |
| B2M | ||
| IFN pathway | chr 15 | β2M genes IFN pathway APM genes |
| APM genes | ||
| Mutation Type | Lynch Syndrome | CMMRD | Lynch-like Syndrome |
|---|---|---|---|
| Germline | One allele of MMR gene | Both alleles of MMR gene | None |
| Somatic | Second allele of MMR gene | None | Both alleles of MMR gene |
| Target | Regimen | Phase | Setting | Identifier |
|---|---|---|---|---|
| ICI combinations or refractory to first ICI | Nivolumab/Ipilimumab | II | ICI resistant | NCT05310643 |
| Nivolumab/Ipilimumab | III | ICI naive | NCT04008030 | |
| Nivolumab/Ipilimumab | II | ICI naive | NCT04730544 | |
| IBI310 (anti-CTLA-4)/Sintilimab | II | ICI naive | NCT04258111 | |
| Pembrolizumab +/− Quavonlimab +/− Favezelimab | II | n.a. | NCT04895722 | |
| +/− Vibostolimab +/− MK-4830 (anti-ILT4) | ||||
| Cadonilimab | I/II | ICI resistant | NCT05426005 | |
| ICIs plus novel agents | Pembrolizumab/Encorafenib | |||
| M7824 (anti-PD-L1/TGF-β trap fusion protein) | II | ICI naive | NCT05217446 | |
| I/II | n.a. | NCT03436563 | ||
| Pembrolizumab + NC410 (LAIR-2 Fc protein) | ||||
| I/II | n.a. | NCT05572684 | ||
| Pembrolizumab + ATRC-101 (anti-RNP) | ||||
| N-803 (IL-15 superagonist) +/− pembrolizumab | I | n.a. | NCT04244552 | |
| +/− nivolumab +/− atezolizumab +/− durvalumab | II | ICI resistant | NCT03228667 | |
| +/− avelumab, respectively | ||||
| Tiselizumab + KFA115 (immunomodulatory agent) | I | ICI naive | NCT05544929 | |
| ICIs plus cytotoxic and anti VEGF agents | Pembrolizumab + bevacizumab + FOLFIRI | II | ICI naive | NCT05035381 |
| Atezolizumab + bevacizumab + FOLFOX | III | ICI naive | NCT02997228 | |
| Toripalimab + bevacizumab + irinotecan | I/II | ICI naive | NCT04988191 | |
| Toripalimab + oxaliplatin + capecitabine | II | ICI naive | NCT04301557 | |
| Camrelizumab + apatinib | II | ICI naive | NCT04715633 | |
| ICIs plus radiotherapy | Sintilimab + RT | I/II | ICI naive | NCT04636008 |
| Nivolumab + ipilimumab + RT | II | ICI naive | NCT03104439 | |
| ICI plus COX inhibitor | Toripalimab + celecoxib | I/II | ICI naive | NCT03926338 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heregger, R.; Huemer, F.; Steiner, M.; Gonzalez-Martinez, A.; Greil, R.; Weiss, L. Unraveling Resistance to Immunotherapy in MSI-High Colorectal Cancer. Cancers 2023, 15, 5090. https://doi.org/10.3390/cancers15205090
Heregger R, Huemer F, Steiner M, Gonzalez-Martinez A, Greil R, Weiss L. Unraveling Resistance to Immunotherapy in MSI-High Colorectal Cancer. Cancers. 2023; 15(20):5090. https://doi.org/10.3390/cancers15205090
Chicago/Turabian StyleHeregger, Ronald, Florian Huemer, Markus Steiner, Alejandra Gonzalez-Martinez, Richard Greil, and Lukas Weiss. 2023. "Unraveling Resistance to Immunotherapy in MSI-High Colorectal Cancer" Cancers 15, no. 20: 5090. https://doi.org/10.3390/cancers15205090
APA StyleHeregger, R., Huemer, F., Steiner, M., Gonzalez-Martinez, A., Greil, R., & Weiss, L. (2023). Unraveling Resistance to Immunotherapy in MSI-High Colorectal Cancer. Cancers, 15(20), 5090. https://doi.org/10.3390/cancers15205090