
A Combination of Alectinib and DNA-Demethylating Agents Synergistically Inhibits Anaplastic-Lymphoma-Kinase-Positive Anaplastic Large-Cell Lymphoma Cell Proliferation
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Proliferation Assay
2.4. LINE1 Methylation Assay by Bisulfite Pyrosequencing
2.5. Determining the Synergistic Anti-Tumor Effect of the Combination Treatment
2.6. Apoptosis Assays
2.7. Cell Cycle Analysis
2.8. Western Blotting
2.9. RNA Sequencing (Seq) and Analysis
2.10. Xenograft Mouse Model
2.11. Statistics
3. Results
3.1. Efficacy of DNA-Demethylating-Agent Monotherapy at Inhibiting ALCL Cell Growth In Vitro
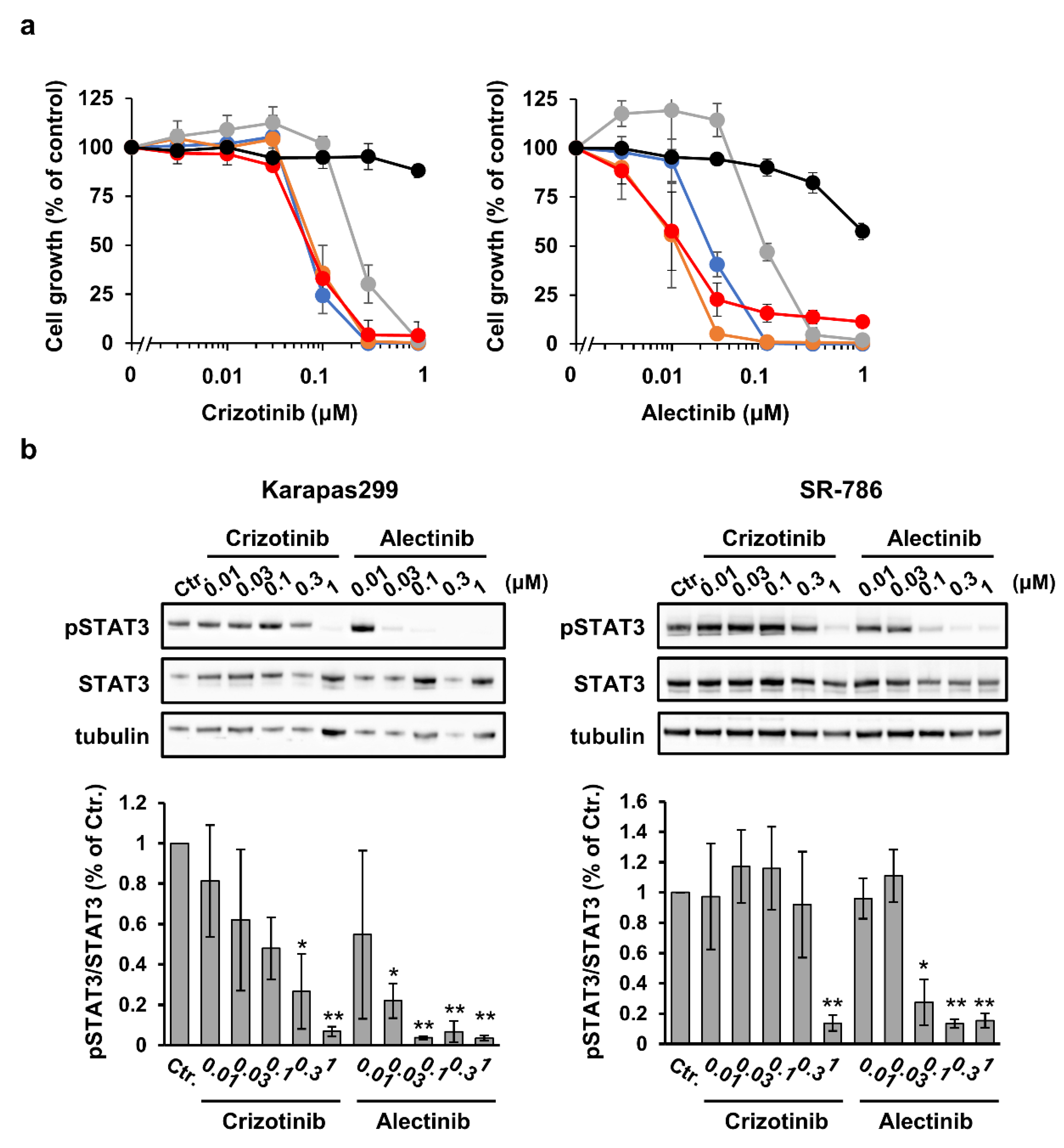
3.2. Efficacy of ALK Tyrosine Kinase Inhibitor Monotherapy at Inhibiting ALCL Cell Growth In Vitro
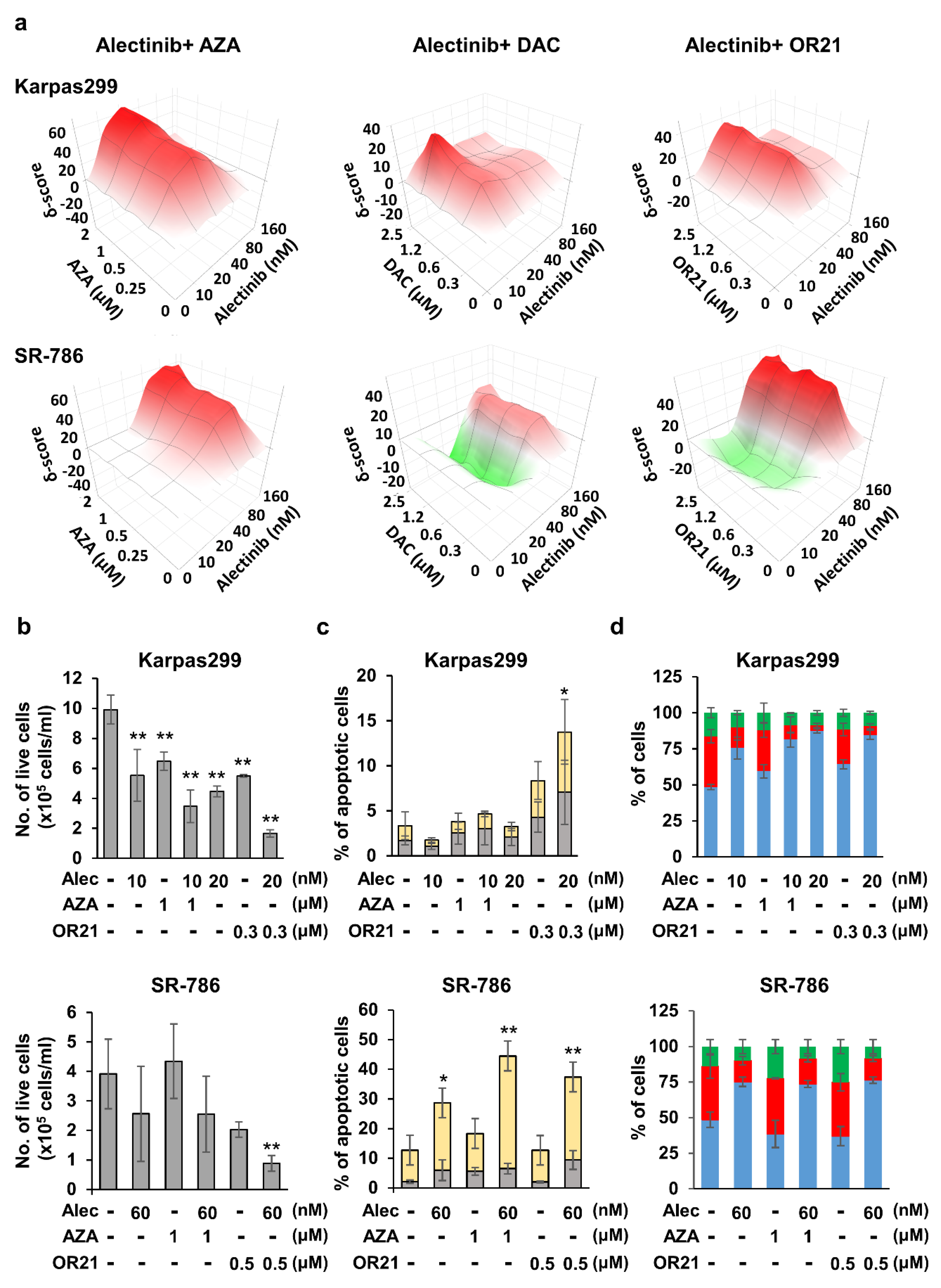
3.3. DNA-Demethylating Agents Synergize with Alectinib to Inhibit the Growth of ALCL Cells
3.4. The DNA-Demethylating Agent and Alectinib Combination Induces Gene Expression Reprogramming in Karpas299 Cells
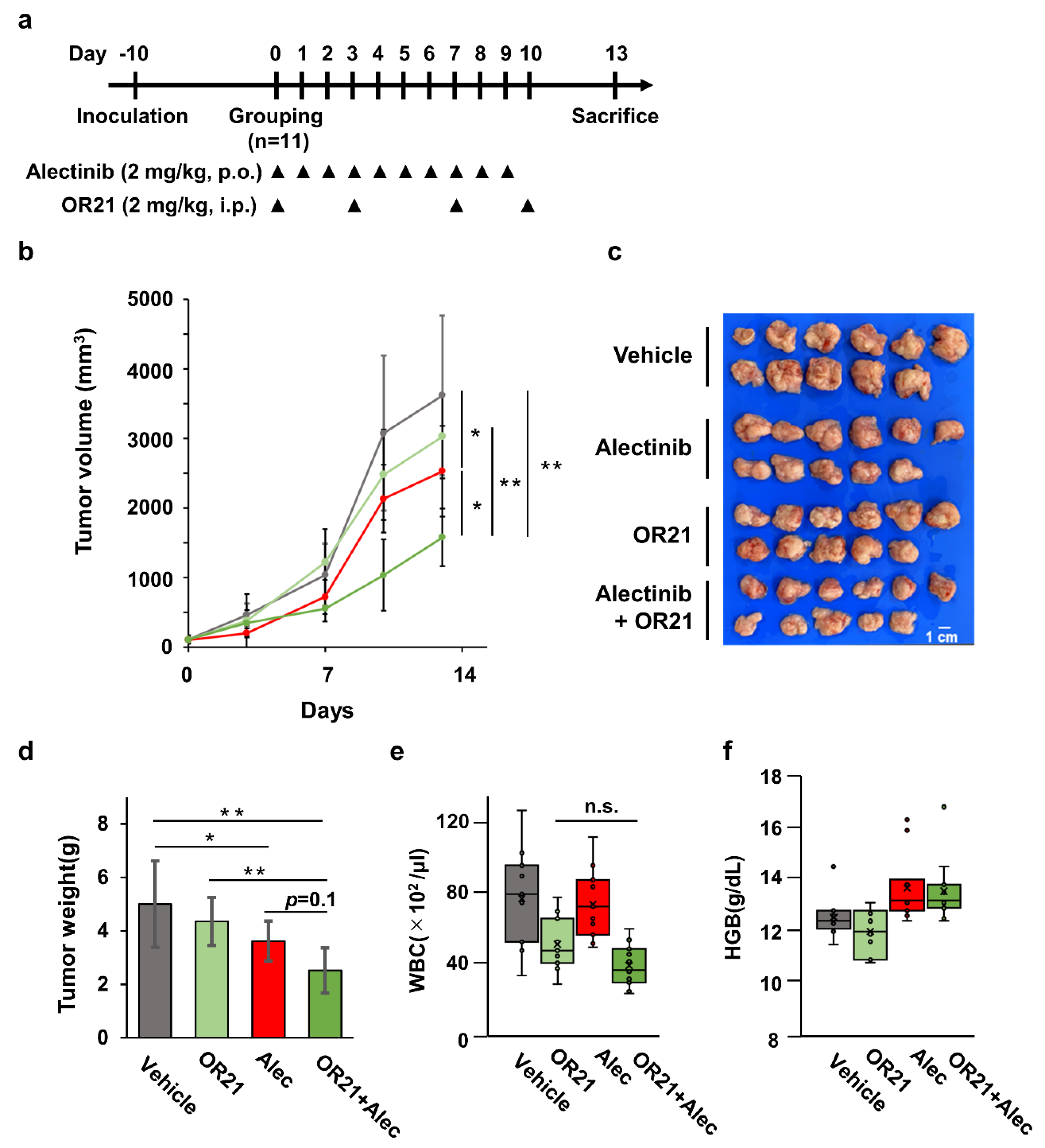
3.5. Combination Treatment with OR21 and Alectinib Suppresses Tumor Cell Growth in a Xenograft Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.R.; Chien, P.N.; Nam, S.Y.; Heo, C.Y. Anaplastic Large Cell Lymphoma: Molecular Pathogenesis and Treatment. Cancers 2022, 14, 1650. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Lamant, L.; Kenner, L.; Brugieres, L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br. J. Haematol. 2016, 173, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 2007, 6 Pt 1, 3314–3322. [Google Scholar] [CrossRef]
- Davies, K.D.; Le, A.T.; Theodoro, M.F.; Skokan, M.C.; Aisner, D.L.; Berge, E.M.; Terracciano, L.M.; Cappuzzo, F.; Incarbone, M.; Roncalli, M.; et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin. Cancer Res.Off. J. Am. Assoc. Cancer Res. 2012, 18, 4570–4579. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Orlov, S.; Zhang, L.; Braiteh, F.; Huang, H.; Esaki, T.; Horibe, K.; Ahn, J.S.; Beck, J.T.; Edenfield, W.J.; et al. Long-term effects of crizotinib in ALK-positive tumors (excluding NSCLC): A phase 1b open-label study. Am. J. Hematol. 2018, 93, 607–614. [Google Scholar] [CrossRef]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Fukano, R.; Mori, T.; Sekimizu, M.; Choi, I.; Kada, A.; Saito, A.M.; Asada, R.; Takeuchi, K.; Terauchi, T.; Tateishi, U.; et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: An open-label phase II trial. Cancer Sci. 2020, 111, 4540–4547. [Google Scholar] [CrossRef]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, H.Y.; Woetmann, A.; Raghunath, P.N.; Odum, N.; Wasik, M.A. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood 2006, 108, 1058–1064. [Google Scholar] [CrossRef]
- Hassler, M.R.; Klisaroska, A.; Kollmann, K.; Steiner, I.; Bilban, M.; Schiefer, A.I.; Sexl, V.; Egger, G. Antineoplastic activity of the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine in anaplastic large cell lymphoma. Biochimie 2012, 94, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Redl, E.; Sheibani-Tezerji, R.; Cardona, C.J.; Hamminger, P.; Timelthaler, G.; Hassler, M.R.; Zrimsek, M.; Lagger, S.; Dillinger, T.; Hofbauer, L.; et al. Requirement of DNMT1 to orchestrate epigenomic reprogramming for NPM-ALK-driven lymphomagenesis. Life Sci. Alliance 2021, 4, e202000794. [Google Scholar] [CrossRef]
- Hassler, M.R.; Pulverer, W.; Lakshminarasimhan, R.; Redl, E.; Hacker, J.; Garland, G.D.; Merkel, O.; Schiefer, A.I.; Simonitsch-Klupp, I.; Kenner, L.; et al. Insights into the Pathogenesis of Anaplastic Large-Cell Lymphoma through Genome-wide DNA Methylation Profiling. Cell Rep. 2016, 17, 596–608. [Google Scholar] [CrossRef]
- Rassidakis, G.Z.; Lai, R.; McDonnell, T.J.; Cabanillas, F.; Sarris, A.H.; Medeiros, L.J. Overexpression of Mcl-1 in anaplastic large cell lymphoma cell lines and tumors. Am. J. Pathol. 2002, 160, 2309–2310. [Google Scholar] [CrossRef]
- Rust, R.; Harms, G.; Blokzijl, T.; Boot, M.; Diepstra, A.; Kluiver, J.; Visser, L.; Peh, S.C.; Lim, M.; Kamps, W.A.; et al. High expression of Mcl-1 in ALK positive and negative anaplastic large cell lymphoma. J. Clin. Pathol. 2005, 58, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Desjobert, C.; Renalier, M.H.; Bergalet, J.; Dejean, E.; Joseph, N.; Kruczynski, A.; Soulier, J.; Espinos, E.; Meggetto, F.; Cavaille, J.; et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood 2011, 117, 6627–6637. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Amin, H.M.; Frantz, C.; Franko, B.; Lee, J.; Lin, Q.; Lai, R. Restoration of shp1 expression by 5-AZA-2′-deoxycytidine is associated with downregulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell lymphoma. Leukemia 2006, 20, 1602–1609. [Google Scholar] [CrossRef]
- Hoareau-Aveilla, C.; Valentin, T.; Daugrois, C.; Quelen, C.; Mitou, G.; Quentin, S.; Jia, J.; Spicuglia, S.; Ferrier, P.; Ceccon, M.; et al. Reversal of microRNA-150 silencing disadvantages crizotinib-resistant NPM-ALK(+) cell growth. J. Clin. Investig. 2015, 125, 3505–3518. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Sako, M.; Kimura, K.; Iida, N.; Takeshima, H.; Nakata, Y.; Kono, Y.; Ushijima, T. Novel prodrugs of decitabine with greater metabolic stability and less toxicity. Clin. Epigenetics 2019, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Yamashita, S.; Ureshino, H.; Kamachi, K.; Kurahashi, Y.; Fukuda-Kurahashi, Y.; Yoshida, N.; Hattori, N.; Nakamura, H.; Sato, A.; et al. Targeting aberrant DNA hypermethylation as a driver of ATL leukemogenesis by using the new oral demethylating agent OR-2100. Blood 2020, 136, 871–884. [Google Scholar] [CrossRef]
- Kamachi, K.; Ureshino, H.; Watanabe, T.; Yoshida, N.; Yamamoto, Y.; Kurahashi, Y.; Fukuda-Kurahashi, Y.; Hayashi, Y.; Hirai, H.; Yamashita, S.; et al. Targeting DNMT1 by demethylating agent OR-2100 increases tyrosine kinase inhibitors-sensitivity and depletes leukemic stem cells in chronic myeloid leukemia. Cancer Lett. 2022, 526, 273–283. [Google Scholar] [CrossRef]
- Ureshino, H.; Kurahashi, Y.; Watanabe, T.; Yamashita, S.; Kamachi, K.; Yamamoto, Y.; Fukuda-Kurahashi, Y.; Yoshida-Sakai, N.; Hattori, N.; Hayashi, Y.; et al. Silylation of deoxynucleotide analog yields an orally available drug with anti-leukemia effects. Mol. Cancer Ther. 2021, 20, 1412–1421. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose-Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef]
- Kamachi, K.; Ureshino, H.; Watanabe, T.; Yoshida-Sakai, N.; Fukuda-Kurahashi, Y.; Kawasoe, K.; Hoshiko, T.; Yamamoto, Y.; Kurahashi, Y.; Kimura, S. Combination of a New Oral Demethylating Agent, OR2100, and Venetoclax for Treatment of Acute Myeloid Leukemia. Cancer Res. Commun. 2023, 3, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, S.; Omar, W.A.; Tomaszewski, B.; De Prins, S.; Jacobs, G.; Koppen, G.; Mathers, J.C.; Langie, S.A. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS ONE 2013, 8, e79044. [Google Scholar] [CrossRef]
- Wang, Y.; He, J.; Xu, M.; Xue, Q.; Zhu, C.; Liu, J.; Zhang, Y.; Shi, W. Holistic View of ALK TKI Resistance in ALK-Positive Anaplastic Large Cell Lymphoma. Front. Oncol. 2022, 12, 815654. [Google Scholar] [CrossRef]
- Surana, R.; Sikka, S.; Cai, W.; Shin, E.M.; Warrier, S.R.; Tan, H.J.; Arfuso, F.; Fox, S.A.; Dharmarajan, A.M.; Kumar, A.P. Secreted frizzled related proteins: Implications in cancers. Biochim. Biophys. Acta 2014, 1845, 53–65. [Google Scholar] [CrossRef]
- Yu, J.; Xie, Y.; Li, M.; Zhou, F.; Zhong, Z.; Liu, Y.; Wang, F.; Qi, J. Association between SFRP promoter hypermethylation and different types of cancer: A systematic review and meta-analysis. Oncol. Lett. 2019, 18, 3481–3492. [Google Scholar] [CrossRef]
- Anand, M.; Lai, R.; Gelebart, P. beta-catenin is constitutively active and increases STAT3 expression/activation in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica 2011, 96, 253–261. [Google Scholar] [CrossRef]
- Richardson, A.I.; Yin, C.C.; Cui, W.; Li, N.; Medeiros, L.J.; Li, L.; Zhang, D. p53 and beta-Catenin Expression Predict Poorer Prognosis in Patients With Anaplastic Large-Cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2019, 19, e385–e392. [Google Scholar] [CrossRef]
- Savona, M.R.; Odenike, O.; Amrein, P.C.; Steensma, D.P.; DeZern, A.E.; Michaelis, L.C.; Faderl, S.; Harb, W.; Kantarjian, H.; Lowder, J.; et al. An oral fixed-dose combination of decitabine and cedazuridine in myelodysplastic syndromes: A multicentre, open-label, dose-escalation, phase 1 study. Lancet Haematol. 2019, 6, e194–e203. [Google Scholar] [CrossRef]
- Li, L.H.; Olin, E.J.; Buskirk, H.H.; Reineke, L.M. Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res. 1970, 30, 2760–2769. [Google Scholar] [PubMed]
- Arosio, G.; Sharma, G.G.; Villa, M.; Mauri, M.; Crespiatico, I.; Fontana, D.; Manfroni, C.; Mastini, C.; Zappa, M.; Magistroni, V.; et al. Synergistic Drug Combinations Prevent Resistance in ALK+ Anaplastic Large Cell Lymphoma. Cancers 2021, 13, 4422. [Google Scholar] [CrossRef] [PubMed]
- Mitou, G.; Frentzel, J.; Desquesnes, A.; Le Gonidec, S.; AlSaati, T.; Beau, I.; Lamant, L.; Meggetto, F.; Espinos, E.; Codogno, P.; et al. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget 2015, 6, 30149–30164. [Google Scholar] [CrossRef]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Aryal, D.K.; Wardman, J.H.; Gupta, A.; Gagnidze, K.; Rodriguiz, R.M.; Kumar, S.; Wetsel, W.C.; Pintar, J.E.; Fricker, L.D.; et al. GPR171 is a hypothalamic G protein-coupled receptor for BigLEN, a neuropeptide involved in feeding. Proc. Natl. Acad. Sci. USA 2013, 110, 16211–16216. [Google Scholar] [CrossRef]
- Bobeck, E.N.; Gomes, I.; Pena, D.; Cummings, K.A.; Clem, R.L.; Mezei, M.; Devi, L.A. The BigLEN-GPR171 Peptide Receptor System Within the Basolateral Amygdala Regulates Anxiety-Like Behavior and Contextual Fear Conditioning. Neuropsychopharmacology 2017, 42, 2527–2536. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Torphy, R.J.; Sun, Y.; Miller, E.N.; Ho, F.; Borcherding, N.; Wu, T.; Torres, R.M.; Zhang, W.; Schulick, R.D.; et al. The GPR171 pathway suppresses T cell activation and limits antitumor immunity. Nat. Commun. 2021, 12, 5857. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawasoe, K.; Watanabe, T.; Yoshida-Sakai, N.; Yamamoto, Y.; Kurahashi, Y.; Kidoguchi, K.; Ureshino, H.; Kamachi, K.; Fukuda-Kurahashi, Y.; Kimura, S. A Combination of Alectinib and DNA-Demethylating Agents Synergistically Inhibits Anaplastic-Lymphoma-Kinase-Positive Anaplastic Large-Cell Lymphoma Cell Proliferation. Cancers 2023, 15, 5089. https://doi.org/10.3390/cancers15205089
Kawasoe K, Watanabe T, Yoshida-Sakai N, Yamamoto Y, Kurahashi Y, Kidoguchi K, Ureshino H, Kamachi K, Fukuda-Kurahashi Y, Kimura S. A Combination of Alectinib and DNA-Demethylating Agents Synergistically Inhibits Anaplastic-Lymphoma-Kinase-Positive Anaplastic Large-Cell Lymphoma Cell Proliferation. Cancers. 2023; 15(20):5089. https://doi.org/10.3390/cancers15205089
Chicago/Turabian StyleKawasoe, Kazunori, Tatsuro Watanabe, Nao Yoshida-Sakai, Yuta Yamamoto, Yuki Kurahashi, Keisuke Kidoguchi, Hiroshi Ureshino, Kazuharu Kamachi, Yuki Fukuda-Kurahashi, and Shinya Kimura. 2023. "A Combination of Alectinib and DNA-Demethylating Agents Synergistically Inhibits Anaplastic-Lymphoma-Kinase-Positive Anaplastic Large-Cell Lymphoma Cell Proliferation" Cancers 15, no. 20: 5089. https://doi.org/10.3390/cancers15205089
APA StyleKawasoe, K., Watanabe, T., Yoshida-Sakai, N., Yamamoto, Y., Kurahashi, Y., Kidoguchi, K., Ureshino, H., Kamachi, K., Fukuda-Kurahashi, Y., & Kimura, S. (2023). A Combination of Alectinib and DNA-Demethylating Agents Synergistically Inhibits Anaplastic-Lymphoma-Kinase-Positive Anaplastic Large-Cell Lymphoma Cell Proliferation. Cancers, 15(20), 5089. https://doi.org/10.3390/cancers15205089