
Histone Deacetylase Inhibitors for Peripheral T-Cell Lymphomas


Abstract
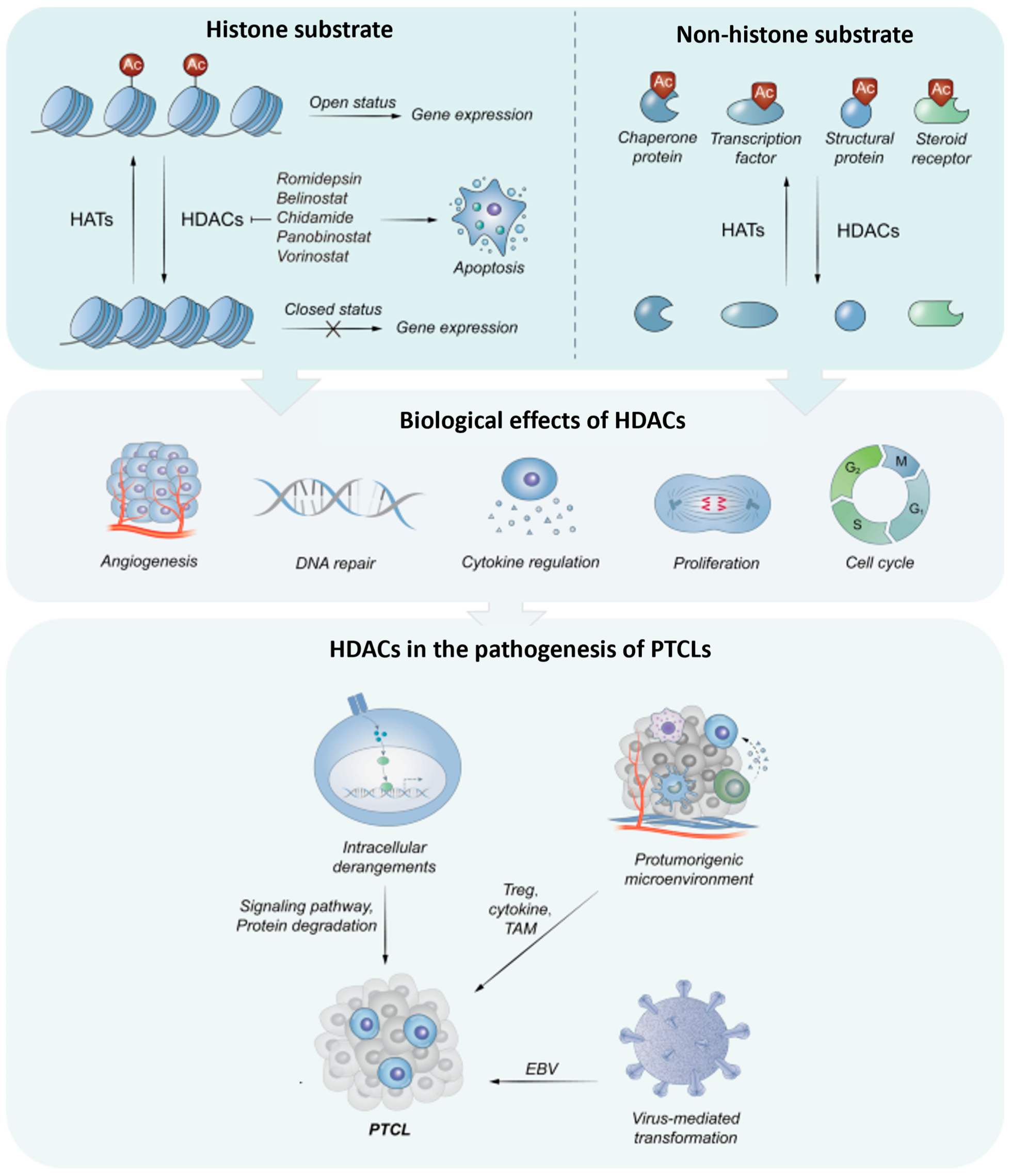
Simple Summary
Abstract

1. Introduction
2. HDAC in Cellular Biology
3. HDACi as Anti-Cancer Treatment
4. Biological Rationale for HDACi Use in Peripheral T-Cell Lymphomas
4.1. Rationale for HDAC Inhibitors in Virus-Induced PTCL
4.2. HDACi as Therapeutic Option in Mature T-Cell Lymphomas
4.2.1. HDACis in First-Line Treatment of PTCL Patients
4.2.2. HDACi in Relapsed/Refractory PTCL Patients
4.2.3. HDACi Efficacy across PTCL Subtypes
5. Real-World Experience with HDAC Inhibitors in PTCL
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Piccaluga, P.P.; Khattab, S.S. A Comparison of the Fifth World Health Organization and the International Consensus Classifications of Mature T-Cell Lymphomas. Int. J. Mol. Sci. 2023, 24, 14170. [Google Scholar] [CrossRef]
- D’Amore, F.; Relander, T.; Lauritzsen, G.F.; Jantunen, E.; Hagberg, H.; Anderson, H.; Holte, H.; Österborg, A.; Merup, M.; Brown, P.; et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J. Clin. Oncol. 2012, 30, 3093–3099. [Google Scholar] [CrossRef] [PubMed]
- Bellei, M.; Foss, F.M.; Shustov, A.R.; Horwitz, S.M.; Marcheselli, L.; Kim, W.S.; Cabrera, M.E.; Dlouhy, I.; Nagler, A.; Advani, R.H.; et al. The outcome of peripheral T-cell lymphoma patients failing first-line therapy: A report from the prospective, International T-Cell Project. Haematologica 2018, 103, 1191–1197. [Google Scholar] [CrossRef]
- Mak, V.; Hamm, J.; Chhanabhai, M.; Shenkier, T.; Klasa, R.; Sehn, L.H.; Villa, D.; Gascoyne, R.D.; Connors, J.M.; Savage, K.J. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: Spectrum of disease and rare long-term survivors. J. Clin. Oncol. 2013, 31, 1970–1976. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Illidge, T.; Fanale, M.; Advani, R.; Bartlett, N.; Christensen, J.H.; Morschhauser, F.; Domenech, E.D.; et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): A global, double-blind, randomised, phase 3 trial. Lancet 2019, 393, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Peng, J.; Huang, Y.; Meng, L.; Li, Q.; Xiong, F.; Li, X. Identification of Histone deacetylase (HDAC)-Associated Proteins with DNA-Programmed Affinity Labeling. Angew. Chem. 2020, 132, 17678–17685. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Chun, P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch. Pharm. Res. 2015, 38, 933–949. [Google Scholar] [CrossRef]
- Carpio, L.R.; Bradley, E.W.; McGee-Lawrence, M.E.; Weivoda, M.M.; Poston, D.D.; Dudakovic, A.; Xu, M.; Tchkonia, T.; Kirkland, J.L.; van Wijnen, A.J.; et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci. Signal. 2016, 9, ra79. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2018, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Olzscha, H.; La Thangue, N.B. HDAC inhibitor-based therapies: Can we interpret the code? Mol. Oncol. 2012, 6, 637–656. [Google Scholar] [CrossRef] [PubMed]
- Haery, L.; Thompson, R.C.; Gilmore, T.D. Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer 2015, 6, 184. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Jin, S.; Lin, S.; Gong, Y.; Zhang, L.; Yang, J.; Mou, W.; Du, J. Update on histone deacetylase inhibitors in peripheral T-cell lymphoma (PTCL). Clin. Epigenet. 2023, 15, 124. [Google Scholar] [CrossRef] [PubMed]
- Waltregny, D.; Glénisson, W.; Tran, S.L.; North, B.J.; Verdin, E.; Colige, A.; Castronovo, V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005, 19, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Nihira, N.T.; Bu, X.; Chu, C.; Zhang, J.; Kolodziejczyk, A.; Fan, Y.; Chan, N.T.; Ma, L.; Liu, J.; et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell Biol. 2020, 22, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Kurasawa, Y.; Wong, J.; Yu-Lee, L.Y. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc. Natl. Acad. Sci. USA 2008, 105, 4179–4184. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, S.; Xia, J.; Zhou, Y.; Peng, L.; Fan, H.; Han, Y.; Duan, L.; Cheng, G.; Yang, H.; et al. Histone deacetylase 3 facilitates TNFα-mediated NF-κB activation through suppressing CTSB induced RIP1 degradation and is required for host defense against bacterial infection. Cell Biosci. 2022, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef]
- Lagger, G.; Doetzlhofer, A.; Schuettengruber, B.; Haidweger, E.; Simboeck, E.; Tischler, J.; Chiocca, S.; Suske, G.; Rotheneder, H.; Wintersberger, E.; et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol. Cell Biol. 2003, 23, 2669–2679. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Juan, L.J.; Shia, W.J.; Chen, M.H.; Yang, W.M.; Seto, E.; Lin, Y.S.; Wu, C.W. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 2000, 275, 20436–20443. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.; Denkert, C.; Budczies, J.; Buckendahl, A.C.; Darb-Esfahani, S.; Noske, A.; Müller, B.M.; Bahra, M.; Neuhaus, P.; Dietel, M.; et al. High class I HDAC activity and expression are associated with RelA/p65 activation in pancreatic cancer in vitro and in vivo. BMC Cancer 2009, 9, 395. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wenzel, P.; Beyer, M.; Schäfer, C.; Brand, P.; Kroemer, A.; Stauber, R.H.; Schmid, R.M.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2017, 36, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Dalla-Favera, R. BCL6: Master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv. Immunol. 2010, 105, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Bereshchenko, O.; Gu, W.; Dalla-Favera, R. Acetylation inactivates the transcriptional repressor BCL6. Nat Genet 2002, 32, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Cortiguera, M.G.; García-Gaipo, L.; Wagner, S.D.; León, J.; Batlle-López, A.; Delgado, M.D. Suppression of BCL6 function by HDAC inhibitor mediated acetylation and chromatin modification enhances BET inhibitor effects in B-cell lymphoma cells. Sci Rep. 2019, 9, 16495. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. Pathological Role of HDAC8: Cancer and Beyond. Cells 2022, 11, 3161. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492, ISSN 0165-6147. [Google Scholar] [CrossRef]
- Wang, Z.T.; Chen, Z.J.; Jiang, G.M.; Wu, Y.M.; Liu, T.; Yi, Y.M.; Zeng, J.; Du, J.; Wang, H.S. Histone deacetylase inhibitors suppress mutant p53 transcription via HDAC8/YY1 signals in triple negative breast cancer cells. Cell Signal. 2016, 28, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Ott, G.; Rosenwald, A.; Campo, E. Understanding MYC-driven aggressive B-cell lymphomas: Pathogenesis and classification. Blood 2013, 122, 3884–3891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.C.; Dalton, W.S.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed. Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, C.; Wei, M.; Wang, C.; Sun, D.; Liu, P.; Zhong, X.; He, Q.; Yu, W. The histone deacetylase HDAC1 activates HIF1α/VEGFA signal pathway in colorectal cancer. Gene 2020, 754, 144851. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Qing, X.; Benda, C.; Huang, Z.; Zhang, M.; Huang, Y.; Zhang, H.; Wang, L.; Lai, Y.; Ward, C.; et al. Nuclear-cytoplasmic shuttling of class IIa histone deacetylases regulates somatic cell reprogramming. Cell Regen. 2019, 8, 21–29. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pulya, S.; Amin, S.A.; Adhikari, N.; Biswas, S.; Jha, T.; Ghosh, B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol. Res. 2021, 163, 105274. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lim, K.H.; Guo, X.; Kawaguchi, Y.; Gao, Y.; Barrientos, T.; Ordentlich, P.; Wang, X.F.; Counter, C.M.; Yao, T.P. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008, 68, 7561–7569. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, A.; Chen, P.; Leng, Y.; Kang, J. Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3–COX2-dependent pathway. Oncogene 2018, 37, 5952–5966. [Google Scholar] [CrossRef] [PubMed]
- Moufarrij, S.; Srivastava, A.; Gomez, S.; Hadley, M.; Palmer, E.; Austin, P.T.; Chisholm, S.; Diab, N.; Roche, K.; Yu, A.; et al. Combining DNMT and HDAC6 inhibitors increases anti-tumor immune signaling and decreases tumor burden in ovarian cancer. Sci. Rep. 2020, 10, 3470. [Google Scholar] [CrossRef]
- Inoue, A.; Yoshida, N.; Omoto, Y.; Oguchi, S.; Yamori, T.; Kiyama, R.; Hayashi, S. Development of cDNA microarray for expression profiling of estrogen-responsive genes. J. Mol. Endocrinol. 2002, 29, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Shinsky, S.A.; Porter, N.J.; Christianson, D.W. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun. 2017, 8, 15368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, J.; Wang, X.; Wang, B.; Zhao, Z.; Fu, J.; Wang, Y.; Zhang, X.; Zhu, P.; Jiang, M.; et al. Degradation of HDAC10 by autophagy promotes IRF3-mediated antiviral innate immune responses. Sci. Signal. 2022, 15, eabo4356. [Google Scholar] [CrossRef] [PubMed]
- Osada, H.; Tatematsu, Y.; Saito, H.; Yatabe, Y.; Mitsudomi, T.; Takahashi, T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer 2004, 112, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, X.; Zhu, S.; Dejene, E.A.; Peng, W.; Sepulveda, A.; Seto, E. HDAC10 Regulates Cancer Stem-Like Cell Properties in KRAS-Driven Lung Adenocarcinoma. Cancer Res. 2020, 80, 3265–3278. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhong, Y.; Huang, T.; Huang, J.; Quan, J.; Su, G.; Xiong, Z.; Lv, Y.; Li, S.; Lai, X.; Xiang, Y.; et al. The HDAC10 instructs macrophage M2 program via deacetylation of STAT3 and promotes allergic airway inflammation. Theranostics 2023, 13, 3568–3581. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, Q.-J.; Zhang, T.-N.; Chen, H.-H.; Yu, X.-F.; Lv, J.-L.; Liu, Y.-Y.; Liu, Y.-S.; Zheng, G.; Zhao, J.-Q.; Wei, Y.-F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, P.Y.; Marshall, G.M. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009, 69, 1702–1705. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Presegué, L.; Vaquero, A. The dual role of sirtuins in cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, P.Y.; Xu, N.; Malyukova, A.; Scarlett, C.J.; Sun, Y.T.; Zhang, X.D.; Ling, D.; Su, S.P.; Nelson, C.; Chang, D.K.; et al. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. 2013, 20, 503–514. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, S.S.; Wu, F.; Jin, Y.M.; Chang, W.Q.; Xu, T.M. HDAC11: A rising star in epigenetics. Biomed. Pharmacother. 2020, 131, 110607. [Google Scholar] [CrossRef]
- Chen, H.; Xie, C.; Chen, Q.; Zhuang, S. HDAC11, an emerging therapeutic target for metabolic disorders. Front. Endocrinol. 2022, 13, 989305. [Google Scholar] [CrossRef] [PubMed]
- Deubzer, H.E.; Schier, M.C.; Oehme, I.; Lodrini, M.; Haendler, B.; Sommer, A.; Witt, O. HDAC11 is a novel drug target in carcinomas. Int. J. Cancer 2013, 132, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wu, X.; Zhao, P.; Xue, K.; Li, J. A pan-cancer analysis identifies HDAC11 as an immunological and prognostic biomarker. FASEB J. 2022, 36, e22326. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Ren, Y.; Feng, M.; Meng, P.; Wang, Q.; Chen, W.; Jiao, Q.; Wang, Y.; Du, L.; Zhou, F.; et al. HDAC11 Regulates Glycolysis through the LKB1/AMPK Signaling Pathway to Maintain Hepatocellular Carcinoma Stemness. Cancer Res. 2021, 81, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fu, L.; Li, S.; Xu, Z.; Li, X. Histone deacetylase 11 suppresses p53 expression in pituitary tumor cells. Cell Biol. Int. 2017, 41, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Zhang, X.; Li, X.; Tian, L.; Shen, S.; Ma, J.; Ai, F. Histone deacetylase (HDAC) 11 inhibits matrix metalloproteinase (MMP) 3 expression to suppress colorectal cancer metastasis. J. Cancer 2022, 13, 1923–1932. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Achachi, A.; Florins, A.; Gillet, N.; Debacq, C.; Urbain, P.; Foutsop, G.M.; Vandermeers, F.; Jasik, A.; Reichert, M.; Kerkhofs, P.; et al. Valproate activates bovine leukemia virus gene expression, triggers apoptosis, and induces leukemia/lymphoma regression in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 10309–10314. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.H.; Tian, X.; Gorgun, G.; Urbano, A.G.; Foss, F.M. Arginine butyrate increases the cytotoxicity of DAB389IL-2 in leukemia and lymphoma cells by upregulation of IL-2Rβ gene. Leuk. Res. 2002, 26, 1077–1083. [Google Scholar] [CrossRef]
- Chen, W.K.; Chen, Y.; Gu, J.X.; Cui, G.H. Effect of trichostatin A on histone acetylation level and apoptosis in HL-60 cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2004, 12, 324–328. (In Chinese) [Google Scholar] [PubMed]
- Liu, T.; Kuljaca, S.; Tee, A.; Marshall, G.M. Histone deacetylase inhibitors: Multifunctional anticancer agents. Cancer Treat Rev. 2006, 32, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.; Jiang, G. The Molecular Mechanism of HDAC Inhibitors in Anticancer Effects. Cell Mol. Immunol. 2006, 3, 285–290. [Google Scholar] [PubMed]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Pal, D.; Raj, K.; Nandi, S.S.; Sinha, S.; Mishra, A.; Mondal, A.; Lagoa, R.; Burcher, J.T.; Bishayee, A. Potential of Synthetic and Natural Compounds as Novel Histone Deacetylase Inhibitors for the Treatment of Hematological Malignancies. Cancers 2023, 15, 2808. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Min, C.; Moore, N.; Shearstone, J.R.; Quayle, S.N.; Huang, P.; van Duzer, J.H.; Jarpe, M.B.; Jones, S.S.; Yang, M. Selective Inhibitors of Histone Deacetylases 1 and 2 Synergize with Azacitidine in Acute Myeloid Leukemia. PLoS ONE 2017, 12, e0169128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bots, M.; Verbrugge, I.; Martin, B.P.; Salmon, J.M.; Ghisi, M.; Baker, A.; Stanley, K.; Shortt, J.; Ossenkoppele, G.J.; Zuber, J.; et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014, 123, 1341–1352. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hao, W.; Wang, L.; Xu, T.; Jia, G.; Jiang, Y.; Qin, C.; Li, X. Marine Cytotoxin Santacruzamate A Derivatives as Potent HDAC1-3 Inhibitors and Their Synergistic Anti-Leukemia Effects with Venetoclax. Mar. Drugs 2024, 22, 250. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, Y.; Xu, J.; Yue, K.; Huang, C.; Qin, M.; Chi, D.; Yu, Q.; Zhu, Y.; Hou, X.; Xu, T.; et al. Potent Hydrazide-Based HDAC Inhibitors with a Superior Pharmacokinetic Profile for Efficient Treatment of Acute Myeloid Leukemia In Vivo. J. Med. Chem. 2022, 65, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.; Coleman, C.; Shah, P.; Yazbeck, V. Advances in Hodgkin’s lymphoma pharmacotherapy: A focus on histone deacetylase inhibitors. Expert Opin. Pharmacother. 2023, 24, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.F.; Chen, L.; Popplewell, L.L.; E Budde, L.; Mei, M.; Armenian, S.H.; Darrah, J.; Nikolaenko, L.; Chen, R.W.; Peters, L.; et al. Preliminary Results from a Phase I Trial of Pembrolizumab Plus Vorinostat in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma, Follicular Lymphoma, and Hodgkin Lymphoma. Blood 2019, 134 (Suppl. S1), 759. [Google Scholar] [CrossRef]
- Wu, C.; Song, Q.; Gao, S.; Wu, S. Targeting HDACs for diffuse large B-cell lymphoma therapy. Sci. Rep. 2024, 14, 289. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Amengual, J.E.; Johannet, P.; Lombardo, M.; Zullo, K.; Hoehn, D.; Bhagat, G.; Scotto, L.; Jirau-Serrano, X.; Radeski, D.; Heinen, J.; et al. Dual Targeting of Protein Degradation Pathways with the Selective HDAC6 Inhibitor ACY-1215 and Bortezomib Is Synergistic in Lymphoma. Clin. Cancer Res. 2015, 21, 4663–4675. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Quintás-Cardama, A.; Santos, F.P.; Garcia-Manero, G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011, 25, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin. Hematol. 2008, 45, 23–30. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Badar, T.; Atallah, E. Do histone deacytelase inhibitors and azacitidine combination hold potential as an effective treatment for high/very-high risk myelodysplastic syndromes? Expert Opin. Investig. Drugs 2021, 30, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Wahaib, K.; Beggs, A.E.; Campbell, H.; Kodali, L.; Ford, P.D. Panobinostat: A histone deacetylase inhibitor for the treatment of relapsed or refractory multiple myeloma. Am. J. Health Syst. Pharm. 2016, 73, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [PubMed]
- El Omari, N.; Bakrim, S.; Khalid, A.; Abdalla, A.N.; Almalki, W.H.; Lee, L.H.; Ardianto, C.; Ming, L.C.; Bouyahya, A. Molecular mechanisms underlying the clinical efficacy of panobinostat involve Stochasticity of epigenetic signaling, sensitization to anticancer drugs, and induction of cellular cell death related to cellular stresses. Biomed. Pharmacother. 2023, 164, 114886. [Google Scholar] [CrossRef] [PubMed]
- Bat-Erdene, A.; Miki, H.; Oda, A.; Nakamura, S.; Teramachi, J.; Amachi, R.; Tenshin, H.; Hiasa, M.; Iwasa, M.; Harada, T.; et al. Synergistic targeting of Sp1, a critical transcription factor for myeloma cell growth and survival, by panobinostat and proteasome inhibitors. Oncotarget 2016, 7, 79064–79075. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pizzi, M.; Margolskee, E.; Inghirami, G. Pathogenesis of Peripheral T Cell Lymphoma. Annu. Rev. Pathol. 2018, 13, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Navari, M.; Etebari, M.; Pileri, S.A. Molecular Genetics of Peripheral T-cell Lymphomas. Int. J. Hematol. 2015, 99, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Van Arnam, J.S.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Elenitoba-Johnson; Novel insights into the pathogenesis of T-cell lymphomas. Blood 2018, 131, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Agostinelli, C.; Califano, A.; Rossi, M.; Basso, K.; Zupo, S.; Went, P.; Klein, U.; Zinzani, P.L.; Baccarani, M.; et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J. Clin. Investig. 2007, 117, 823–834. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Piccaluga, P.P.; Agostinelli, C.; Califano, A.; Carbone, A.; Fantoni, L.; Ferrari, S.; Gazzola, A.; Gloghini, A.; Righi, S.; Rossi, M.; et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res. 2007, 67, 10703–10710. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Fuligni, F.; De Leo, A.; Bertuzzi, C.; Rossi, M.; Bacci, F.; Sabattini, E.; Agostinelli, C.; Gazzola, A.; Laginestra, M.A.; et al. Molecular profiling improves classification and prognostication of nodal peripheral T-cell lymphomas: Results of a phase III diagnostic accuracy study. J. Clin. Oncol. 2013, 31, 3019–3025. [Google Scholar] [CrossRef] [PubMed]
- Fiore, D.; Cappelli, L.V.; Broccoli, A.; Zinzani, P.L.; Chan, W.C.; Inghirami, G. Peripheral T cell lymphomas: From the bench to the clinic. Nat. Rev. Cancer. 2020, 20, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.-M.; Huang, Y.-H.; Huang, J.-Y.; Wang, Z.-F.; Fu, D.; Liu, H.; Liu, F.; Leboeuf, C.; Wang, L.; Ye, J.; et al. Histone modifier gene mutations in peripheral T-cell lymphoma not otherwise specified. Haematologica 2018, 103, 679–687. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef]
- Yu, X.; Li, H.; Zhu, M.; Hu, P.; Liu, X.; Qing, Y.; Wang, X.; Wang, H.; Wang, Z.; Xu, J.; et al. Involvement of p53 Acetylation in Growth Suppression of Cutaneous T-Cell Lymphomas Induced by HDAC Inhibition. J. Investig. Dermatol. 2020, 140, 2009–2022.e4. [Google Scholar] [CrossRef]
- Rosato, R.R.; Grant, S. Histone deacetylase inhibitors: Insights into mechanisms of lethality. Expert Opin. Ther. Targets 2005, 9, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.P.; Matsuoka, M. Natural history of adult T-cell leukemia/lymphoma and approaches to therapy. Oncogene 2005, 24, 6047–6057. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.; Amador, C.; Chadburn, A.; Hsi, E.D.; Slack, G.; Medeiros, L.J.; Feldman, A.L. Genetic profiling and biomarkers in peripheral T-cell lymphomas: Current role in the diagnostic work-up. Mod. Pathol. 2022, 35, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Li, X.; Zeng, H.; Qian, W. Molecular insights into pathogenesis and targeted therapy of peripheral T cell lymphoma. Exp. Hematol. Oncol. 2020, 9, 30. [Google Scholar] [CrossRef]
- Nakhoul, H.; Lin, Z.; Wang, X.; Roberts, C.; Dong, Y.; Flemington, E. High-Throughput Sequence Analysis of Peripheral T-Cell Lymphomas Indicates Subtype-Specific Viral Gene Expression Patterns and Immune Cell Microenvironments. mSphere 2019, 4, e00248-19. [Google Scholar] [CrossRef] [PubMed]
- Haverkos, B.M.; Alpdogan, O.; Baiocchi, R.A.; Brammer, J.E.; Feldman, T.A.; Capra, M.; Brem, E.A.; Nair, S.; Scheinberg, P.; Pereira, J.; et al. Targeted therapy with nanatinostat and valganciclovir in recurrent EBV-positive lymphoid malignancies: A phase 1b/2 study. Blood Adv. 2023, 7, 6339–6350. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-Y.; Min, G.-J.; Jeon, Y.-W.; Park, S.-S.; Park, S.; Shin, S.-H.; Yahng, S.-A.; Yoon, J.-H.; Lee, S.-E.; Cho, B.-S.; et al. Impact of Epstein-Barr Virus on Peripheral T-Cell Lymphoma Not Otherwise Specified and Angioimmunoblastic T-Cell Lymphoma. Front. Oncol. 2022, 11, 797028. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Cheung, A.K.L.; Choi, C.K.; Yeung, P.L.; Middeldorp, J.M.; Lung, M.L.; Tsao, S.W.; Chiang, A.K.S. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int. J. Cancer 2016, 138, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, Z.; Fan, W.; Liu, Y.; Liu, F.; Wan, X.; Liu, M.; Wang, X.; Zeng, D.; Wang, Y.; et al. Targeting cancer cell plasticity by HDAC inhibition to reverse EBV-induced dedifferentiation in nasopharyngeal carcinoma. Signal Transduct. Target. Ther. 2021, 6, 333. [Google Scholar] [CrossRef] [PubMed]
- Ego, T.; Ariumi, Y.; Shimotohno, K. The interaction of HTLV-1 Tax with HDAC1 negatively regulates the viral gene expression. Oncogene 2002, 21, 7241–7246. [Google Scholar] [CrossRef] [PubMed]
- Schnell, A.P.; Kohrt, S.; Thoma-Kress, A.K. Latency Reversing Agents: Kick and Kill of HTLV-1? Int. J. Mol. Sci. 2021, 22, 5545. [Google Scholar] [CrossRef]
- Dupuis, J.; Morschhauser, F.; Ghesquières, H.; Tilly, H.; Casasnovas, O.; Thieblemont, C.; Ribrag, V.; Bossard, C.; Le Bras, F.; Bachy, E.; et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: A non-randomised, phase 1b/2 study. Lancet Haematol. 2015, 2, e160–e165. [Google Scholar] [CrossRef] [PubMed]
- Bachy, E.; Camus, V.; Thieblemont, C.; Sibon, D.; Casasnovas, R.-O.; Ysebaert, L.; Damaj, G.; Guidez, S.; Pica, G.M.; Kim, W.S.; et al. Romidepsin Plus CHOP Versus CHOP in Patients with Previously Untreated Peripheral T-Cell Lymphoma: Results of the Ro-CHOP Phase III Study (Conducted by LYSA). J. Clin. Oncol. 2022, 40, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.B.; Cashen, A.F.; Nikolinakos, P.G.; Beaven, A.W.; Barta, S.K.; Bhat, G.; Hasal, S.J.; De Vos, S.; Oki, Y.; Deng, C.; et al. Belinostat in combination with standard cyclophosphamide, doxorubicin, vincristine and prednisone as first-line treatment for patients with newly diagnosed peripheral T-cell lymphoma. Exp. Hematol. Oncol. 2021, 10, 15. [Google Scholar] [CrossRef]
- Gui, L.; Cao, J.; Ji, D.; Zhang, H.; Fan, Q.; Zhu, J.; Song, Y.; Jiang, S.; Ning, Z.; Yu, J.; et al. Chidamide combined with cyclophosphamide, doxorubicin, vincristine and prednisone in previously untreated patients with peripheral T-cell lymphoma. Chin. J. Cancer Res. 2021, 33, 616–626. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Y.; Zhang, M.; Song, W.; Cai, Q.; Zhang, L.; Sun, X.; Zou, L.; Zhang, H.; Wang, L.; Xue, H. Chidamide plus prednisone, etoposide, and thalidomide for untreated angioimmunoblastic T-cell lymphoma in a Chinese population: A multicenter phase II trial. Am. J. Hematol. 2022, 97, 623–629. [Google Scholar] [CrossRef]
- Zhang, W.; Su, L.; Liu, L.; Gao, Y.; Wang, Q.; Su, H.; Song, Y.; Zhang, H.; Shen, J.; Jing, H.; et al. The combination of chidamide with the CHOEP regimen in previously untreated patients with peripheral T-cell lymphoma: A prospective, multicenter, single arm, phase 1b/2 study. Cancer Biol. Med. 2021, 18, 841. [Google Scholar] [CrossRef]
- Falchi, L.; Ma, H.; Klein, S.; Lue, J.K.; Montanari, F.; Marchi, E.; Deng, C.; Kim, H.A.; Rada, A.M.; Jacob, A.T.; et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: A multicenter phase 2 study. Blood 2021, 137, 2161–2170. [Google Scholar] [CrossRef] [PubMed]
- Bachy, E.; Camus, V.; Thieblemont, C.; Casasnovas, R.-O.; Ysebaert, L.; Damaj, G.L.; Guidez, S.; Pica, G.-M.; Kim, W.S.; Lim, S.T.; et al. Final Analysis of the Ro-CHOP Phase III Study (Conducted by LYSA): Romidepsin Plus CHOP in Patients with Peripheral T-Cell Lymphoma. Blood 2020, 136 (Suppl. S1), 32–33. [Google Scholar] [CrossRef]
- Yang, P.; Tao, Y.; Zhao, A.; Shen, K.; Li, H.; Wang, J.; Zhou, H.; Wang, Z.; Wang, M.; Qu, Y.; et al. Efficacy and safety of histone deacetylase inhibitors in peripheral T-cell lymphoma: A systematic review and meta-analysis on prospective clinical trials. Front. Oncol. 2023, 13, 1127112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, S.; Chen, J.; Yu, Z. Histone Deacetylases (HDACs) Guided Novel Therapies for T-cell lymphomas. Int. J. Med. Sci. 2019, 16, 424. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef] [PubMed]
- Shimony, S.; Horowitz, N.; Ribakovsky, E.; Rozovski, U.; Avigdor, A.; Zloto, K.; Berger, T.; Avivi, I.; Perry, C.; Abadi, U.; et al. Romidepsin treatment for relapsed or refractory peripheral and cutaneous T-cell lymphoma: Real-life data from a national multicenter observational study. Hematol. Oncol. 2019, 37, 569–577. [Google Scholar] [CrossRef]
- Kalac, M.; Jain, S.; Tam, C.S.; Xiao, Z.; Montanari, F.; Kanakry, J.; Huber, B.D.; Goldfinger, M.; O’connor, O.A.; Marchi, E. Real-world experience of combined treatment with azacitidine and romidepsin in patients with peripheral T-cell lymphoma. Blood Adv. 2023, 7, 3760–3763. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jia, B.; Xu, W.; Li, W.; Liu, T.; Liu, P.; Zhao, W.; Zhang, H.; Sun, X.; Yang, H.; et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: A multicenter real-world study in China. J. Hematol. Oncol. 2017, 10, 69. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, W.; Zhao, D.; Liu, T.; Niu, T.; Song, Y.; Xu, W.; Jin, J.; Cai, Q.; Huang, H.; Li, Z.; et al. A Multi-Center, Real-World Study of Chidamide for Patients with Relapsed or Refractory Peripheral T-Cell Lymphomas in China. Front. Oncol. 2021, 11, 750323. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, C.; Zhao, D.; Zhang, Y.; Wang, W.; Zhou, D.; Zhang, W. Long-time follow-up of patients with untreated peripheral T cell lymphoma following chidamide combined with cyclophosphamide, epirubicin, vindesine, prednisone, and etoposide therapy: A single-center propensity score-matching study. Clin. Transl. Oncol. 2023, 25, 2514–2522. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, W.; Wang, X.; Li, J.; Yin, X.; Zhao, Y.; Tang, Y.; Wang, A.; Bai, O. Chidamide Maintenance Therapy Following Induction Therapy in Patients with Peripheral T-Cell Lymphoma Who Are Ineligible for Autologous Stem Cell Transplantation: Case Series from China. Front. Oncol. 2022, 12, 875469. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

{kind=link}
{kind=link}
| HDAC | Class | Cellular Localization | Substrate Specificity | Substrates | Function | Expression Pattern | Associated Diseases |
|---|---|---|---|---|---|---|---|
| HDAC1 | I | Nuclear | Histone proteins | Androgen receptor, SHP, TP53, MyoD, SMC4, E2F1, STAT3 | Gene regulation, cell cycle control | Ubiquitous | Cancer, neurodegenerative disorders |
| HDAC2 | I | Nuclear | Histone proteins | Glucocorticoid receptor, YY1, BCL6, STAT3 | Gene regulation, cell cycle control | Ubiquitous | Cancer, neurodegenerative disorders |
| HDAC3 | I | Nuclear, cytoplasmic, membrane | Histone proteins | SHP, YY1, GATA1, RELA, STAT3, MEF2D | Gene regulation, cell cycle control | Ubiquitous | Cancer, metabolic diseases |
| HDAC4 | II A | Nuclear, cytoplasmic | Histone and nonhistone | GCMA, GATA1, HP1 | Muscle differentiation, development | Tissue-specific (heart, skeletal muscle, brain) | Muscular disorders, neurodegenerative diseases |
| HDAC5 | II A | Nuclear, cytoplasmic | Histone and nonhistone | GCMA, SMAD7, HP1 | Muscle differentiation, development | Tissue-specific (heart, skeletal muscle, brain) | Muscular disorders, neurodegenerative diseases |
| HDAC6 | IIB | Cytoplasmic | Cytoplasmic proteins | α-Tubulin, HSP90, SHP, SMAD7 | Aggresome formation, protein degradation | Tissue-specific (heart, liver, kidney) | Neurodegenerative disorders, cancer |
| HDAC7 | II A | Nuclear | Histone and nonhistone | PLAG1, PLAG2 | Vascular development, immune response | Tissue-specific (endothelium, heart, skeletal muscle, pancreas, placenta, thymus) | Cardiovascular diseases, cancer |
| HDAC8 | I | Nuclear | Histone proteins | - | Cell cycle | Ubiquitous | Cancer |
| HDAC9 | II A | Nuclear | Histone and nonhistone | - | Development, cardiac function | Tissue-specific (brain, heart, skeletal muscle) | Cardiovascular diseases, cancer |
| HDAC 10 | II B | Cytoplasmic, nuclear | Cytoplasmic proteins | - | Cellular proliferation, apoptosis | Tissue-specific (liver, spleen, kidney) | Cancer, neurodegenerative diseases |
| HDAC 11 | IV | Nuclear | Histone proteins | - | Gene regulation | Tissue-specific (brain, heart, kidney, testis) | Cancer, inflammatory diseases |
| SIRT1 | III | Nuclear, cytoplasmic | Histone and nonhistone, NAD-dependent | - | Metabolic, stress response | Ubiquitous | Aging-related diseases, metabolism disorders |
| SIRT2 | III | Cytoplasmic, nuclear | Histone and nonhistone, NAD-dependent | - | Cell cycle, homeostasis | Ubiquitous | Metabolic diseases, cancer |
| SIRT3 | III | Mitochondrial | NAD-dependent | - | Mitochondrial function, energy metabolism | Ubiquitous | Metabolic diseases, cancer |
| SIRT4 | III | Mitochondrial | NAD-dependent | - | Metabolism | Tissue-specific (pancreas) | Metabolic diseases, cancer |
| SIRT5 | III | Mitochondrial | NAD-dependent | - | Metabolism | Ubiquitous | Metabolic diseases, cancer |
| SIRT6 | III | Nuclear | Histone and nonhistone, NAD-dependent | - | DNA repair, genome stability | Ubiquitous | Aging-related disease, cancer |
| SIRT7 | III | Nucleolar | Histone and nonhistone, NAD-dependent | - | Ribosomal DNA transcription, cell growth | Ubiquitous | Cancer |
| Study | Design | Treatment | ASCT Permitted | Subtype Efficacy | Toxicity Profile (Grade ¾ Events) |
|---|---|---|---|---|---|
| Dupuis, 2015 [103] | Phase I/II single-arm | Ro-CHOP | No | N/A | Cardiac toxicity Febrile neutropenia Hematologic toxicities |
| Bachy, 2022 [104] | Phase III, RCT | Ro-CHOP | No | AITL (PFS 19.5 mo vs. 10.6 mo) | >10% difference in ≥grade 3 hematologic toxicities |
| Johnston, 2021 [105] | Phase I, single arm | Bel-CHOP | No | Hematologic toxicity Febrile Neutropenia Nausea SAE rate 43% | |
| Guy, 2021 [106] | Phase I, single arm | Chidamide-CHOP | No | N/A | Hematologic toxicity Vomiting |
| Wang, 2022 [107] | Phase II, single arm | Chidamide+ Prednison+ Etoposide + Thalidomide | No | AITL (90.2%/ 54.9%) | Hematologic toxicity |
| Zhang, 2021 [108] | Phase Ib/II, single arm | Chidamide + CHOEP | No | ALK+ AITL (65.9%/41.5%) | Hematologic toxicity |
| Falchi, 2021 [109] | Phase II, single arm | R-Azacytidine | Yes (4 patients/3 in remission) | AITL (80%/60%) | Hematologic toxicity |
| Article | Country of Experience | Number of Patients | Subtype of PTCL | Therapeutic Approach | Stem Cell Transplant Included | Results ORR |
|---|---|---|---|---|---|---|
| Shi, Y. [117] | China | 256 | PTCL | Chidamide monotherapy | No | 39.06% |
| Shi, Y. [117] | China | 32 | AITL | Chidamide monotherapy | No | 49.23% |
| Shi, Y. [117] | China | 13 | ALK+ ALCL | Chidamide monotherapy | No | 66.67% |
| Shi, Y. [117] | China | 127 | ALK+ ALCL | Chidamide + Chemotherapy | No | 51.18% |
| Shimony, S. [115] | Israel | 42 | PTCL | Romidepsin monotherapy | No | 33% |
| Kalac, M. [116] | USA, Australia | 26 | PTCL | Romidepsin–azacitidine | Yes (1 Allo, 7 auto) | 76.9% |
| Kalac, M. [116] | USA, Australia | 19 | AITL | Romidepsin–azacitidine | Yes (1 Allo, 7 auto) | 69.5% |
| Liu, W. [118] | China | 261 | PTCL | Chidamine monotherapy | No | 58.6% |
| Liu, W. [118] | China | 287 | PTCL | Chidamide + Chemotherapy | No | 73.2% |
| Liu, W. [118] | China | 177 | AITL | Chidamine monotherapy/Chidamide + Chemotherapy | No | 75.1% |
| Wei, C. [119] | China | 32 | PTCL | Chidamide + CHOEP | Yes | 68.8% |
| Guo, W. [120] | China | 48 | PTCL | Chidamide maintenance | No | 93.8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irimia, R.; Piccaluga, P.P. Histone Deacetylase Inhibitors for Peripheral T-Cell Lymphomas. Cancers 2024, 16, 3359. https://doi.org/10.3390/cancers16193359
Irimia R, Piccaluga PP. Histone Deacetylase Inhibitors for Peripheral T-Cell Lymphomas. Cancers. 2024; 16(19):3359. https://doi.org/10.3390/cancers16193359
Chicago/Turabian StyleIrimia, Ruxandra, and Pier Paolo Piccaluga. 2024. "Histone Deacetylase Inhibitors for Peripheral T-Cell Lymphomas" Cancers 16, no. 19: 3359. https://doi.org/10.3390/cancers16193359
APA StyleIrimia, R., & Piccaluga, P. P. (2024). Histone Deacetylase Inhibitors for Peripheral T-Cell Lymphomas. Cancers, 16(19), 3359. https://doi.org/10.3390/cancers16193359