
CELF2 Sustains a Proliferating/OLIG2+ Glioblastoma Cell Phenotype via the Epigenetic Repression of SOX3

 , , ,
, , ,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. siRNAs and shRNAs
2.3. Orthotopic Xenografts
2.4. RNA Sequencing Analysis
2.5. ChIP Sequencing (ChIP-Seq) and Bioinformatics Analysis
2.6. Spheroid Formation Assays
2.7. Western Blot Analysis
2.8. Immunofluorescence
2.9. Immunohistochemistry and Immunohistofluorescence
2.10. RNA Immunoprecipitation PCR Assay
2.11. Statistical Analysis
3. Results
3.1. Characterization of CELF2 Expression in GBM Samples
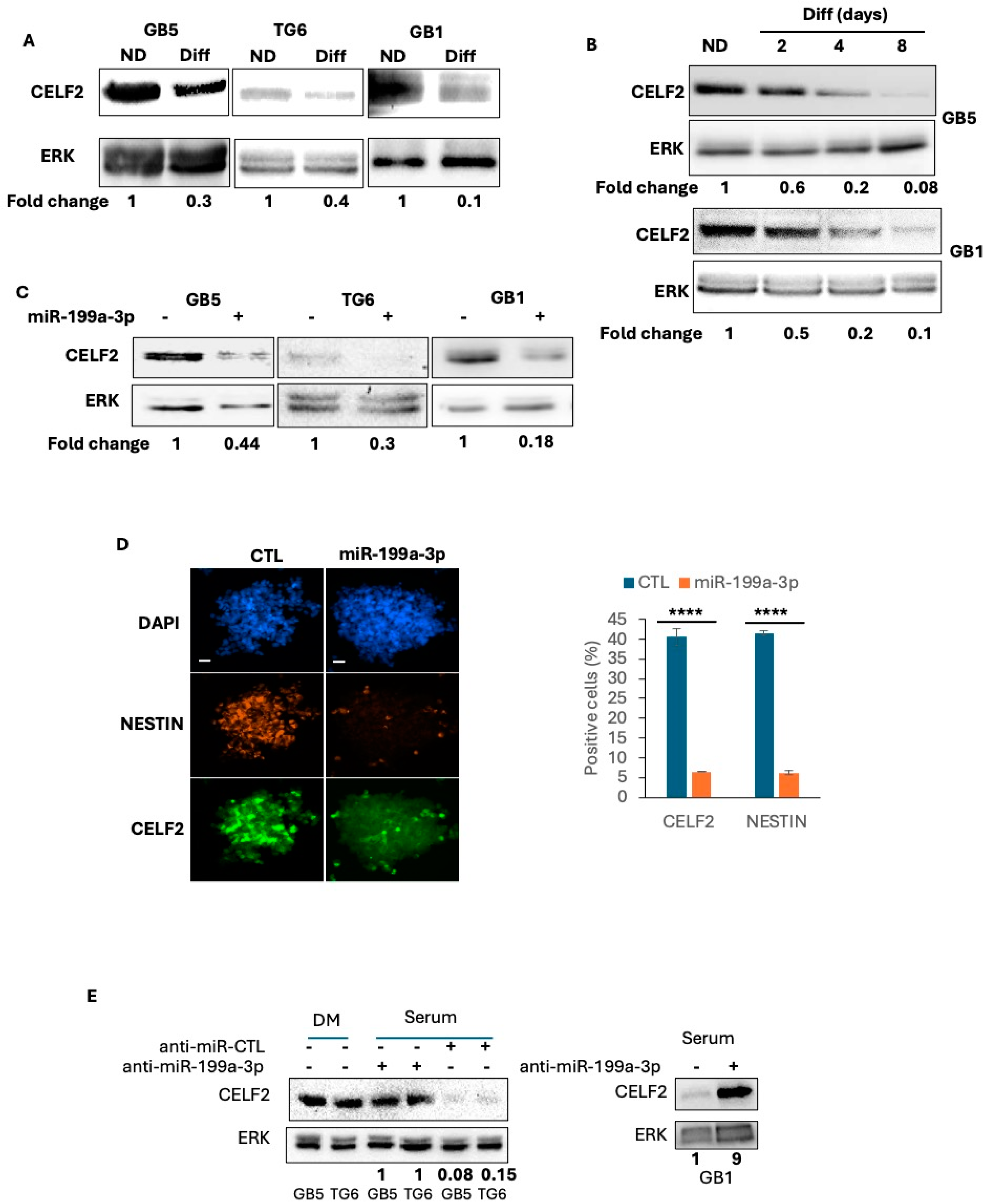
3.2. CELF2 Is Expressed in GSCs and Is Downregulated upon Cell Differentiation
3.3. CELF2 Is Required for GSC Maintenance and Tumorigenic Potential
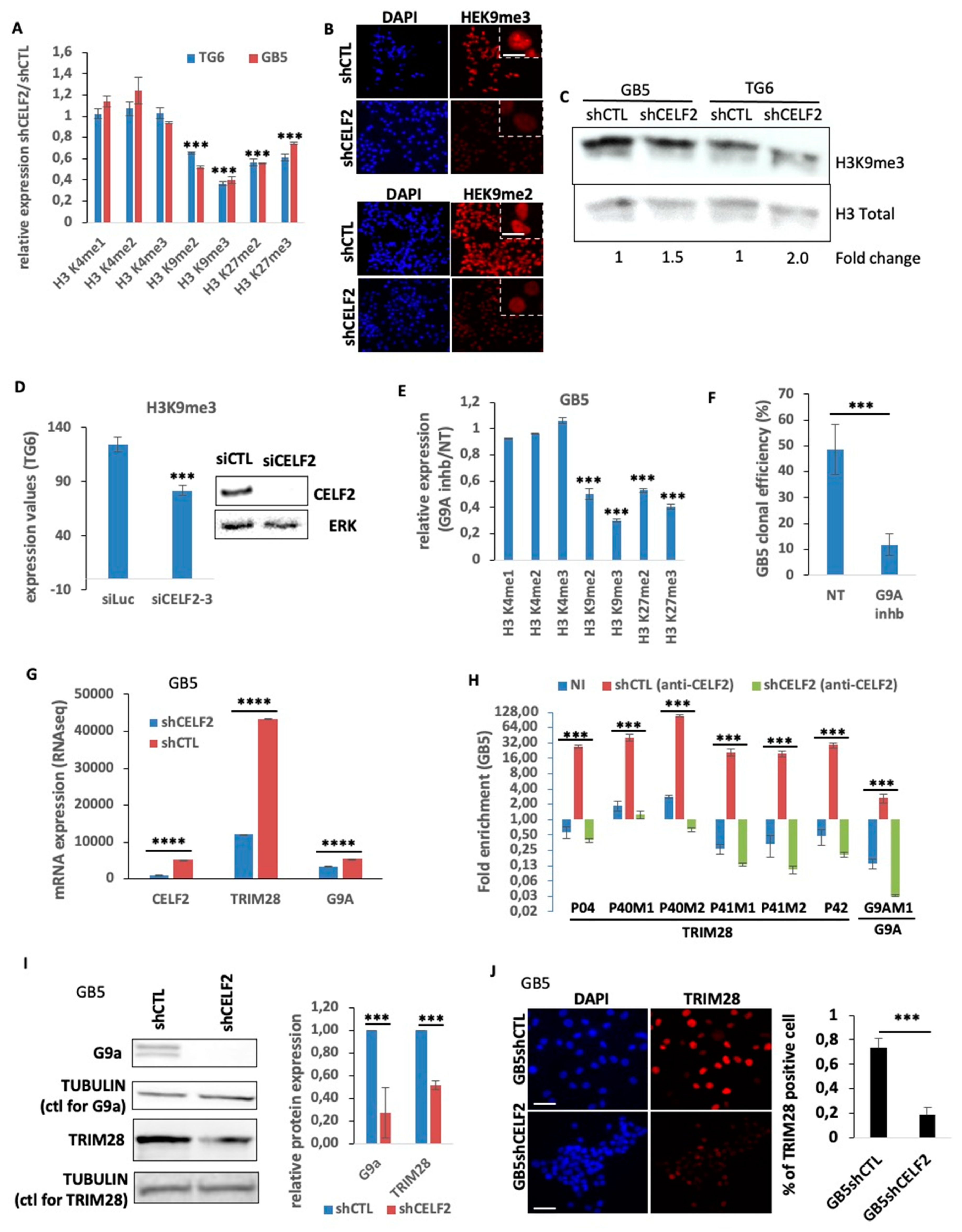
3.4. CELF2 Controls the Level and Distribution of H3K9me3 in Proliferating GSCs
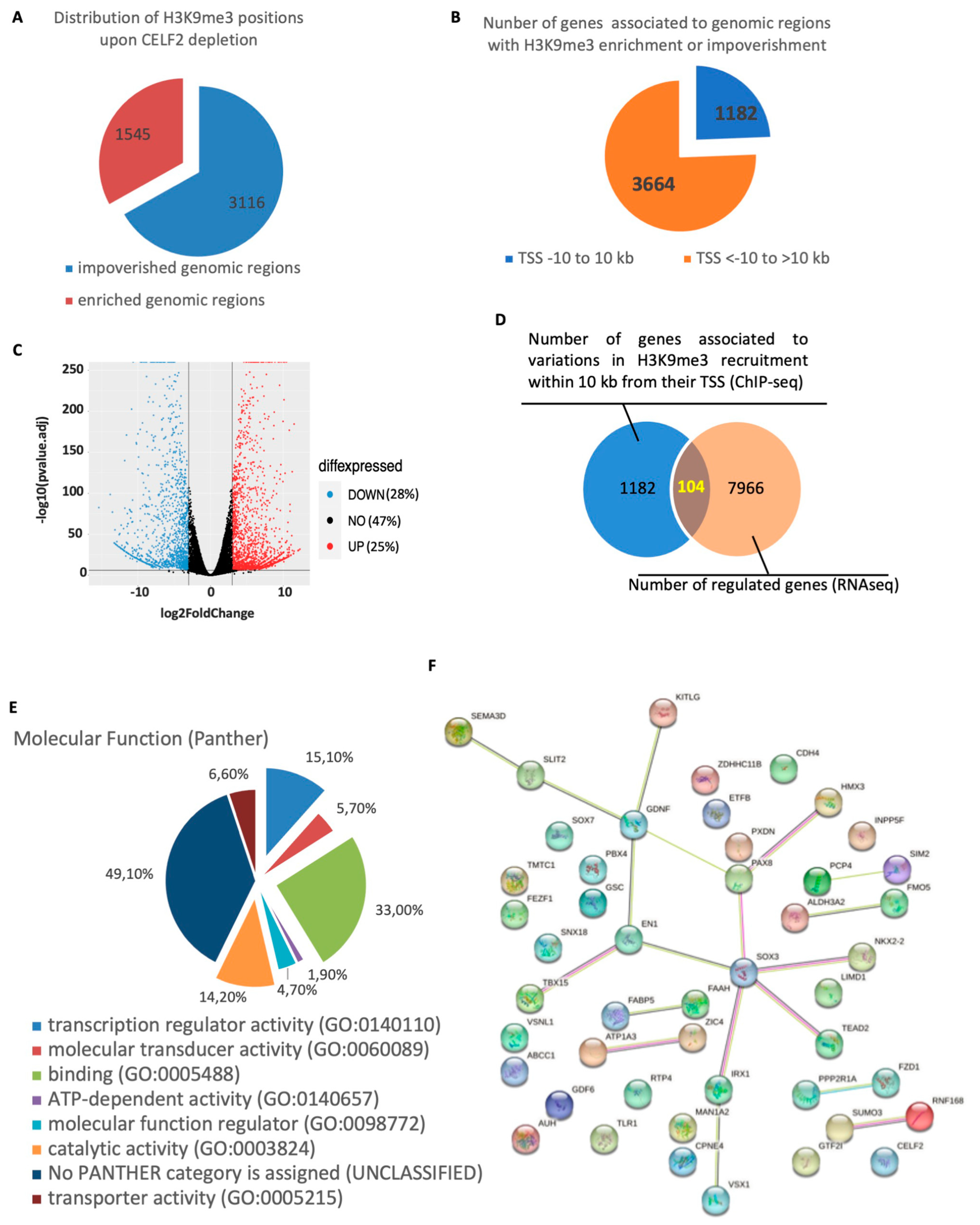
3.5. A CELF2-Dependent Model of H3K9me3 Distribution in the GSCs’ Chromatin Landscape
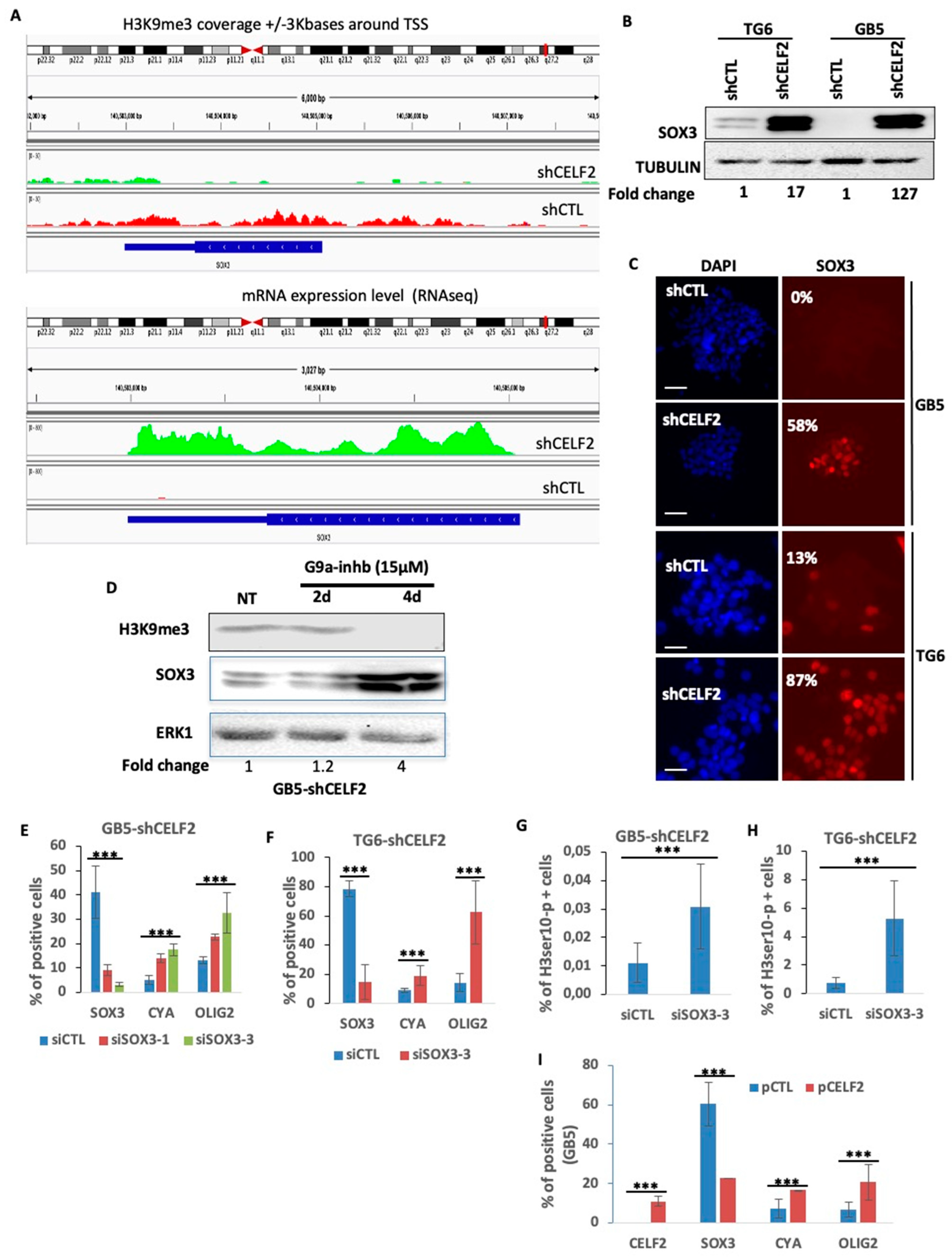
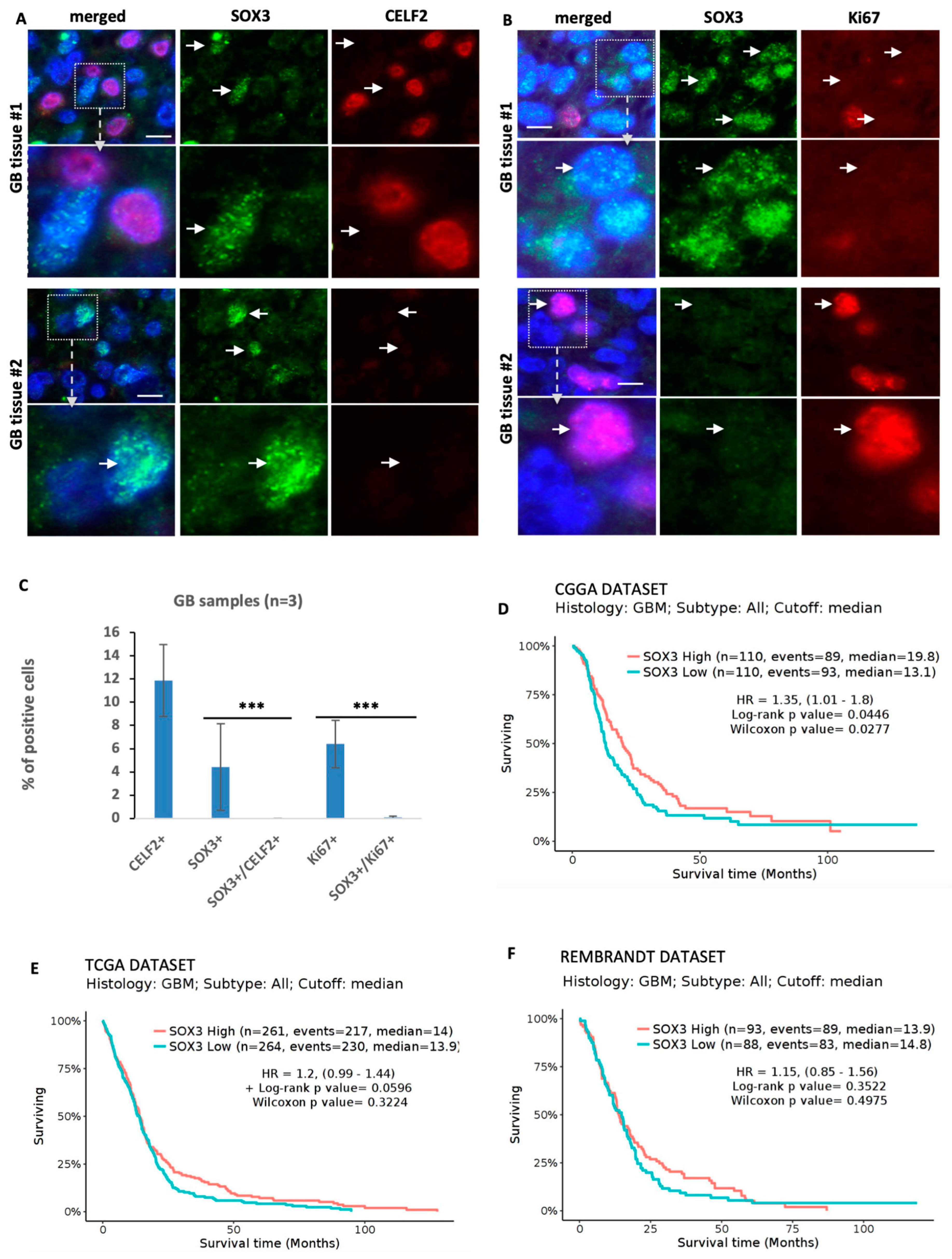
3.6. SOX3 Is a Major Player in the Repression of the OLIG2+ Proliferating Phenotype of GBM Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Aum, D.J.; Kim, D.H.; Beaumont, T.L.; Leuthardt, E.C.; Dunn, G.P.; Kim, A.H. Molecular and cellular heterogeneity: The hallmark of glioblastoma. Neurosurg. Focus 2014, 37, E11. [Google Scholar] [CrossRef]
- Chen, J.; McKay, R.M.; Parada, L.F. Malignant glioma: Lessons from genomics, mouse models, and stem cells. Cell 2012, 149, 36–47. [Google Scholar] [CrossRef]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.; Iwama, T.; Park, D.M. The evidence of glioblastoma heterogeneity. Sci. Rep. 2015, 5, 7979. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Turchi, L.; Burel-Vandenbos, F.; Fareh, M.; Almairac, F.; Virolle, V.; Figarella-Branger, D.; Baeza-Kallee, N.; Lagadec, P.; Kubiniek, V.; et al. DOCK4 promotes loss of proliferation in glioblastoma progenitor cells through nuclear beta-catenin accumulation and subsequent miR-302-367 cluster expression. Oncogene 2018, 37, 241–254. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Turchi, L.; Debruyne, D.N.; Almairac, F.; Virolle, V.; Fareh, M.; Neirijnck, Y.; Burel-Vandenbos, F.; Paquis, P.; Junier, M.P.; Van Obberghen-Schilling, E.; et al. Tumorigenic Potential of miR-18A* in Glioma Initiating Cells Requires NOTCH-1 Signaling. Stem Cells 2013, 31, 1252–1265. [Google Scholar] [CrossRef] [PubMed]
- Fareh, M.; Turchi, L.; Virolle, V.; Debruyne, D.; Almairac, F.; Divonne, S.D.-L.-F.; Paquis, P.; Preynat-Seauve, O.; Krause, K.H.; Chneiweiss, H.; et al. The miR 302-367 cluster drastically affects self-renewal and infiltration properties of glioma-initiating cells through CXCR4 repression and consequent disruption of the SHH-GLI-NANOG network. Cell Death Differ. 2012, 19, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Almairac, F.; Turchi, L.; Sakakini, N.; Debruyne, D.N.; Elkeurti, S.; Gjernes, E.; Polo, B.; Bianchini, L.; Fontaine, D.; Paquis, P.; et al. ERK-Mediated Loss of miR-199a-3p and Induction of EGR1 Act as a “Toggle Switch” of GBM Cell Dedifferentiation into NANOG- and OCT4-Positive Cells. Cancer Res. 2020, 80, 3236–3250. [Google Scholar] [CrossRef]
- El-Habr, E.A.; Dubois, L.G.; Burel-Vandenbos, F.; Bogeas, A.; Lipecka, J.; Turchi, L.; Lejeune, F.X.; Coehlo, P.L.; Yamaki, T.; Wittmann, B.M.; et al. A driver role for GABA metabolism in controlling stem and proliferative cell state through GHB production in glioma. Acta Neuropathol. 2017, 133, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Jovcevska, I.; Kocevar, N.; Komel, R. Glioma and glioblastoma—How much do we (not) know? Mol. Clin. Oncol. 2013, 1, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.C.; Bjerkvig, R. Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert Rev. Anticancer. Ther. 2012, 12, 635–642. [Google Scholar] [CrossRef]
- Barreau, C.; Paillard, L.; Méreau, A.; Osborne, H.B. Mammalian CELF/Bruno-like RNA-binding proteins: Molecular characteristics and biological functions. Biochimie 2006, 88, 515–525. [Google Scholar] [CrossRef]
- Ladd, A.N.; Cooper, T.A. Multiple domains control the subcellular localization and activity of ETR-3, a regulator of nuclear and cytoplasmic RNA processing events. J. Cell Sci. 2004, 117, 3519–3529. [Google Scholar] [CrossRef]
- Dasgupta, T.; Ladd, A.N. The importance of CELF control: Molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins, Wiley interdisciplinary reviews. RNA 2012, 3, 104–121. [Google Scholar]
- Guo, Q.; Wu, Y.; Guo, X.; Cao, L.; Xu, F.; Zhao, H.; Zhu, J.; Wen, H.; Ju, X.; Wu, X. The RNA-Binding Protein CELF2 Inhibits Ovarian Cancer Progression by Stabilizing FAM198B. Mol. Ther. Nucleic Acids 2021, 23, 169–184. [Google Scholar] [CrossRef]
- Piqué, L.; de Paz, A.M.; Piñeyro, D.; Martínez-Cardús, A.; de Moura, M.C.; Llinàs-Arias, P.; Setien, F.; Gomez-Miragaya, J.; Gonzalez-Suarez, E.; Sigurdsson, S.; et al. Epigenetic inactivation of the splicing RNA-binding protein CELF2 in human breast cancer. Oncogene 2019, 38, 7106–7112. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.T.; Fan, S.; Lu, B.; Yin, S.; Yang, S.; Nie, W.; Wang, M.; Zhou, L.; Li, T.; Li, X.; et al. CELF2 suppresses non-small cell lung carcinoma growth by inhibiting the PREX2-PTEN interaction. Carcinogenesis 2020, 41, 377–389. [Google Scholar] [CrossRef]
- Liao, C.; Chen, W.; Wang, J. MicroRNA-20a Regulates Glioma Cell Proliferation, Invasion, and Apoptosis by Targeting CUGBP Elav-Like Family Member 2. World Neurosurg. 2019, 121, e519–e527. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Jiao, B.H.; Fan, F.S.; Lu, S.K.; Song, J.; Guo, C.Y.; Yang, J.K.; Yang, L. Downregulation of miR-95-3p inhibits proliferation, and invasion promoting apoptosis of glioma cells by targeting CELF2. Int. J. Oncol. 2015, 47, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Su, B.; Song, G.; Liu, X.; Yan, Z.; Wang, S.; Hu, F.; Yang, J. miR-363-3p induces EMT via the Wnt/β-catenin pathway in glioma cells by targeting CELF2. J. Cell Mol. Med. 2021, 25, 10418–10429. [Google Scholar] [CrossRef]
- Patru, C.; Romao, L.; Varlet, P.; Coulombel, L.; Raponi, E.; Cadusseau, J.; Renault-Mihara, F.; Thirant, C.; Leonard, N.; Berhneim, A.; et al. CD133, CD15/SSEA-1, CD34 or side populations do not resume tumor-initiating properties of long-term cultured cancer stem cells from human malignant glio-neuronal tumors. BMC Cancer 2010, 10, 66. [Google Scholar] [CrossRef]
- Keene, J.D.; Komisarow, J.M.; Friedersdorf, M.B. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. 2006, 1, 302–307. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Leseva, M.; Knowles, B.B.; Messerschmidt, D.M.; Solter, D. Erase-Maintain-Establish: Natural Reprogramming of the Mammalian Epigenome. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 155–163. [Google Scholar] [CrossRef]
- Shinkai, Y.; Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Genes. Dev. 2011, 25, 781–788. [Google Scholar] [CrossRef]
- Yokochi, T.; Poduch, K.; Ryba, T.; Lu, J.; Hiratani, I.; Tachibana, M.; Shinkai, Y.; Gilbert, D.M. G9a selectively represses a class of late-replicating genes at the nuclear periphery. Proc. Natl. Acad. Sci. USA 2009, 106, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.R.; Brand, O.J.; Pasini, A.; Jenkins, G.; Knox, A.J.; Pang, L. Interplay between EZH2 and G9a Regulates CXCL10 Gene Repression in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Källström, M.; Muhr, J. Sox21 promotes the progression of vertebrate neurogenesis. Nat. Neurosci. 2005, 8, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Bylund, M.; Andersson, E.; Novitch, B.G.; Muhr, J. Vertebrate neurogenesis is counteracted by Sox1-3 activity. Nat. Neurosci. 2003, 6, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Oron, E.; Nelson, B.; Razis, S.; Ivanova, N. Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell 2012, 10, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Klum, S.; Zaouter, C.; Alekseenko, Z.; Björklund, Å.; Hagey, D.W.; Ericson, J.; Muhr, J.; Bergsland, M. Sequentially acting SOX proteins orchestrate astrocyte- and oligodendrocyte-specific gene expression. EMBO Rep. 2018, 19, e46635. [Google Scholar] [CrossRef]
- Vicentic, J.M.; Drakulic, D.; Garcia, I.; Vukovic, V.; Aldaz, P.; Puskas, N.; Nikolic, I.; Tasic, G.; Raicevic, S.; Garros-Regulez, L.; et al. SOX3 can promote the malignant behavior of glioblastoma cells. Cell Oncol. 2019, 42, 41–54. [Google Scholar] [CrossRef]
- Shen, J.; Zhai, J.; Wu, X.; Xie, G.; Shen, L. Serum proteome profiling reveals SOX3 as a candidate prognostic marker for gastric cancer. J. Cell Mol. Med. 2020, 24, 6750–6761. [Google Scholar] [CrossRef]
- Ajith, S.; Gazzara, M.R.; Cole, B.S.; Shankarling, G.; Martinez, N.M.; Mallory, M.J.; Lynch, K.W. Position-dependent activity of CELF2 in the regulation of splicing and implications for signal-responsive regulation in T cells. RNA Biol. 2016, 13, 569–581. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turchi, L.; Sakakini, N.; Saviane, G.; Polo, B.; Saurty-Seerunghen, M.S.; Gabut, M.; Gouillou, C.A.; Guerlais, V.; Pasquier, C.; Vignais, M.L.; et al. CELF2 Sustains a Proliferating/OLIG2+ Glioblastoma Cell Phenotype via the Epigenetic Repression of SOX3. Cancers 2023, 15, 5038. https://doi.org/10.3390/cancers15205038
Turchi L, Sakakini N, Saviane G, Polo B, Saurty-Seerunghen MS, Gabut M, Gouillou CA, Guerlais V, Pasquier C, Vignais ML, et al. CELF2 Sustains a Proliferating/OLIG2+ Glioblastoma Cell Phenotype via the Epigenetic Repression of SOX3. Cancers. 2023; 15(20):5038. https://doi.org/10.3390/cancers15205038
Chicago/Turabian StyleTurchi, Laurent, Nathalie Sakakini, Gaelle Saviane, Béatrice Polo, Mirca Saras Saurty-Seerunghen, Mathieu Gabut, Corine Auge Gouillou, Vincent Guerlais, Claude Pasquier, Marie Luce Vignais, and et al. 2023. "CELF2 Sustains a Proliferating/OLIG2+ Glioblastoma Cell Phenotype via the Epigenetic Repression of SOX3" Cancers 15, no. 20: 5038. https://doi.org/10.3390/cancers15205038
APA StyleTurchi, L., Sakakini, N., Saviane, G., Polo, B., Saurty-Seerunghen, M. S., Gabut, M., Gouillou, C. A., Guerlais, V., Pasquier, C., Vignais, M. L., Almairac, F., Chneiweiss, H., Junier, M.-P., Burel-Vandenbos, F., & Virolle, T. (2023). CELF2 Sustains a Proliferating/OLIG2+ Glioblastoma Cell Phenotype via the Epigenetic Repression of SOX3. Cancers, 15(20), 5038. https://doi.org/10.3390/cancers15205038