

USP53 Exerts Tumor-Promoting Effects in Triple-Negative Breast Cancer by Deubiquitinating CRKL

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Plasmids and siRNA
2.3. Reagents and Primary Antibodies
2.4. Western Blotting
2.5. Immunoprecipitation
2.6. Immunofluorescence Analysis
2.7. Immunohistochemistry
2.8. Quantitative Real-Time PCR
2.9. Cell Proliferation Assay
2.10. Migration and Invasion Assay
2.11. CHX Chase and Ubiquitination Assay
2.12. Statistical Analysis
3. Results
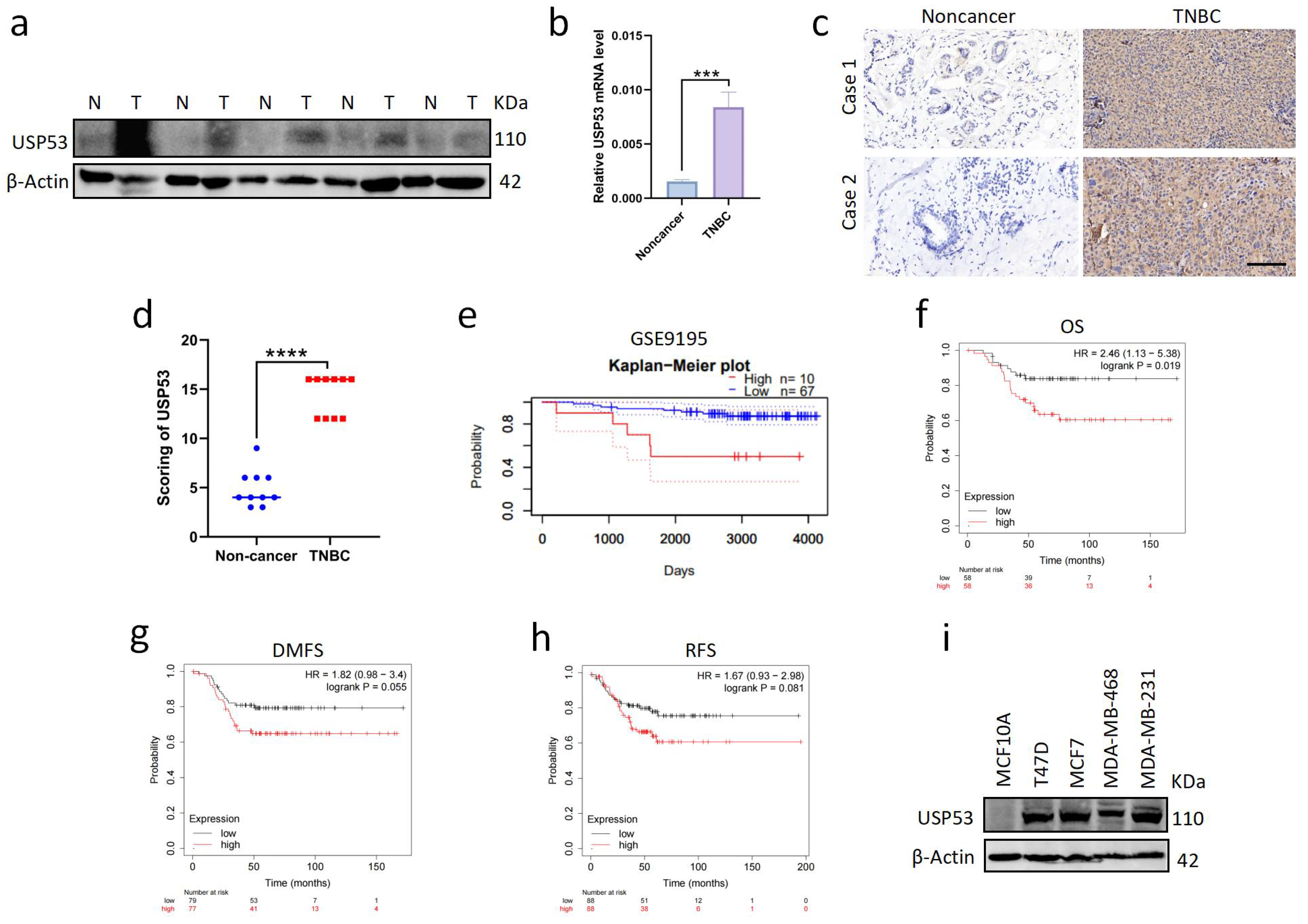
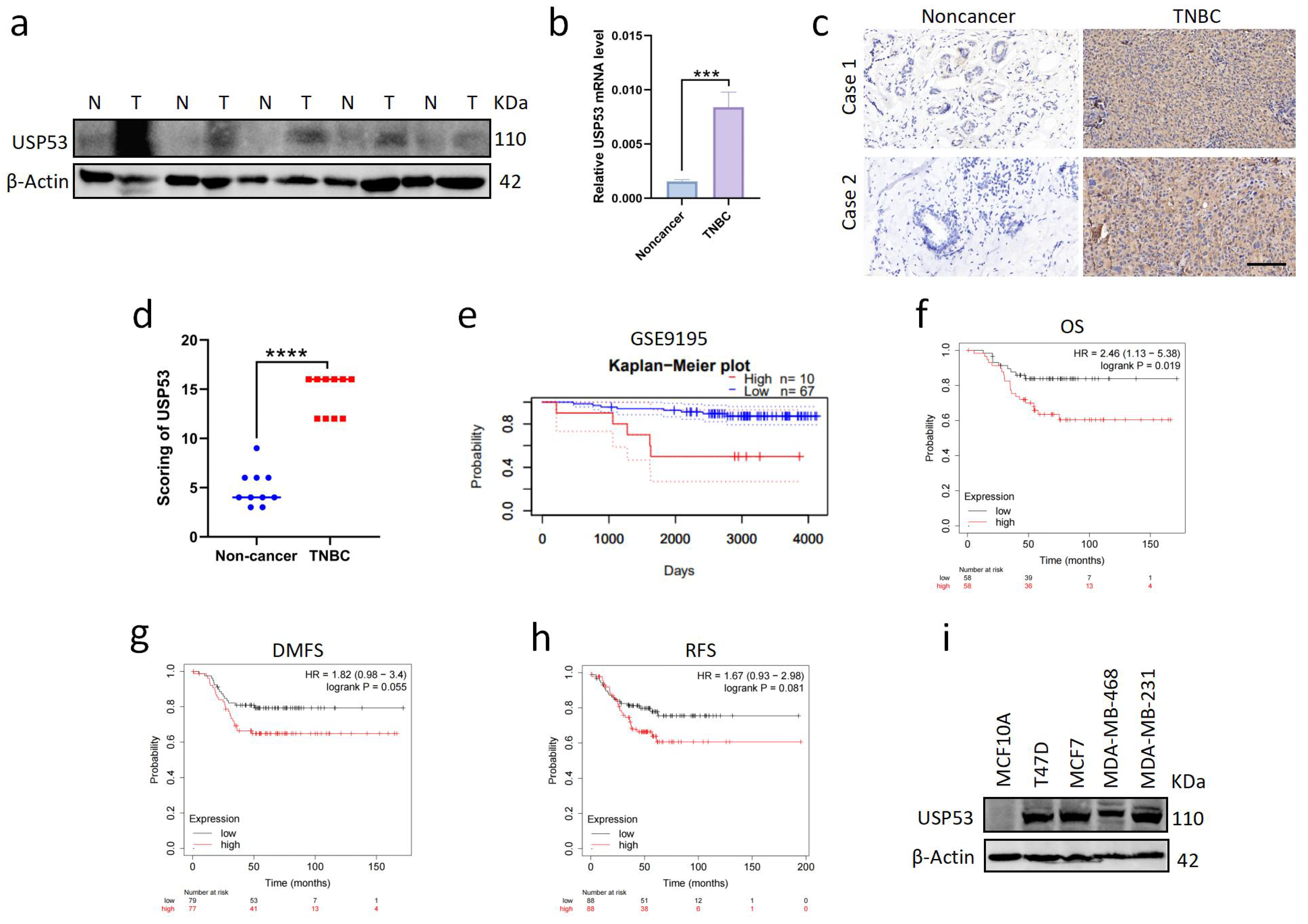
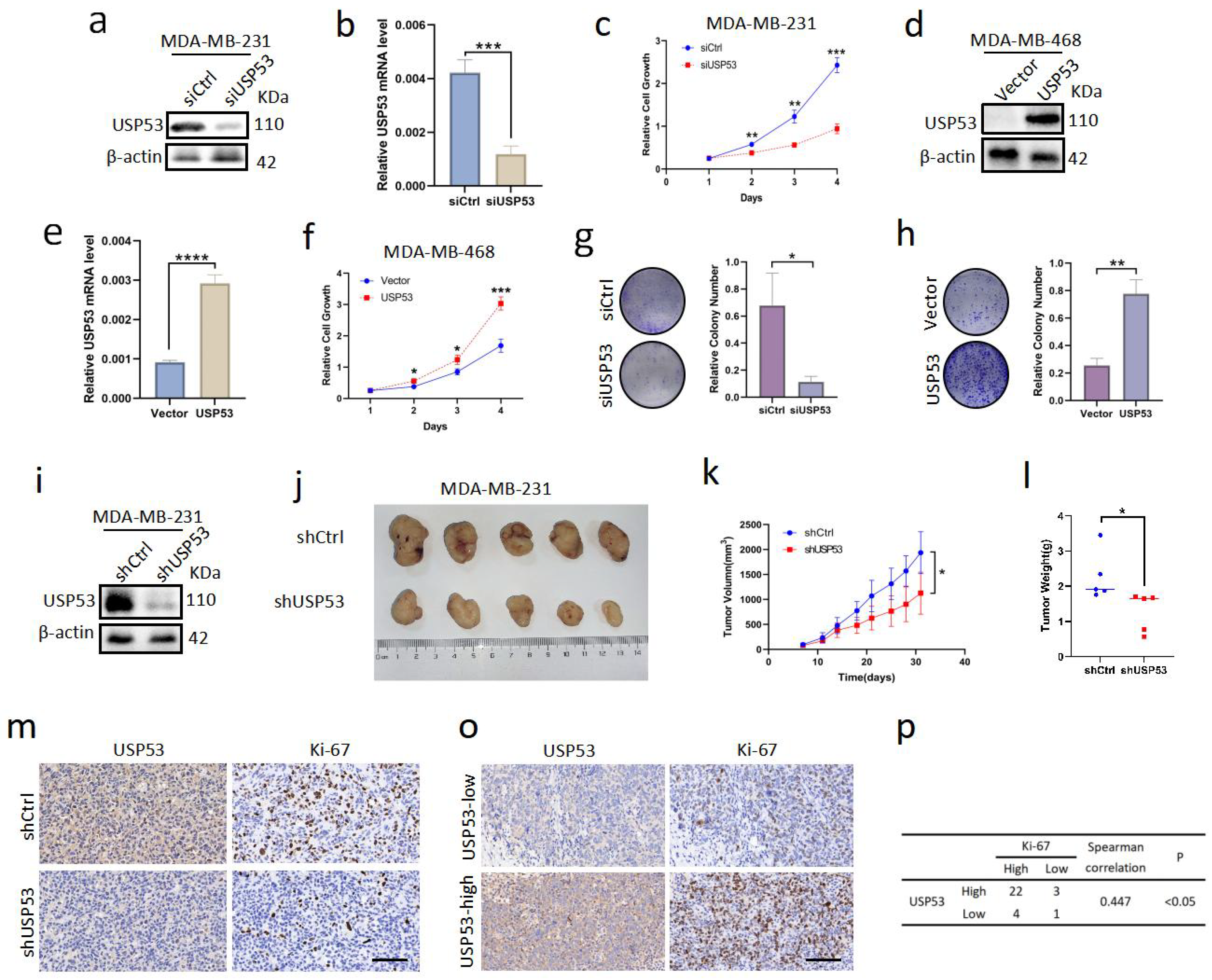
3.1. USP53 Is Overexpressed in TNBC and Is Related to Poor Prognosis
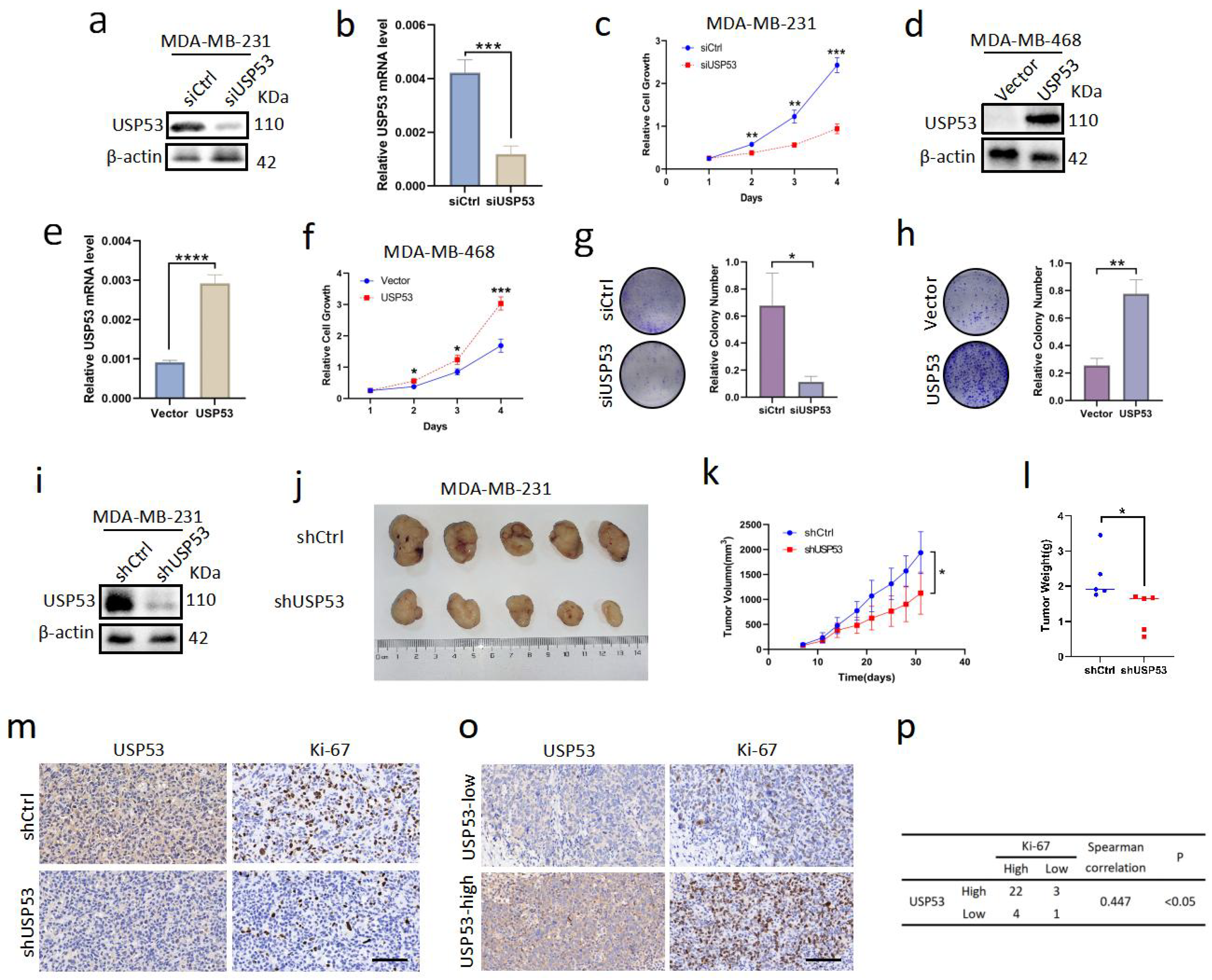
3.2. USP53 Promotes Cell Proliferation in TNBC
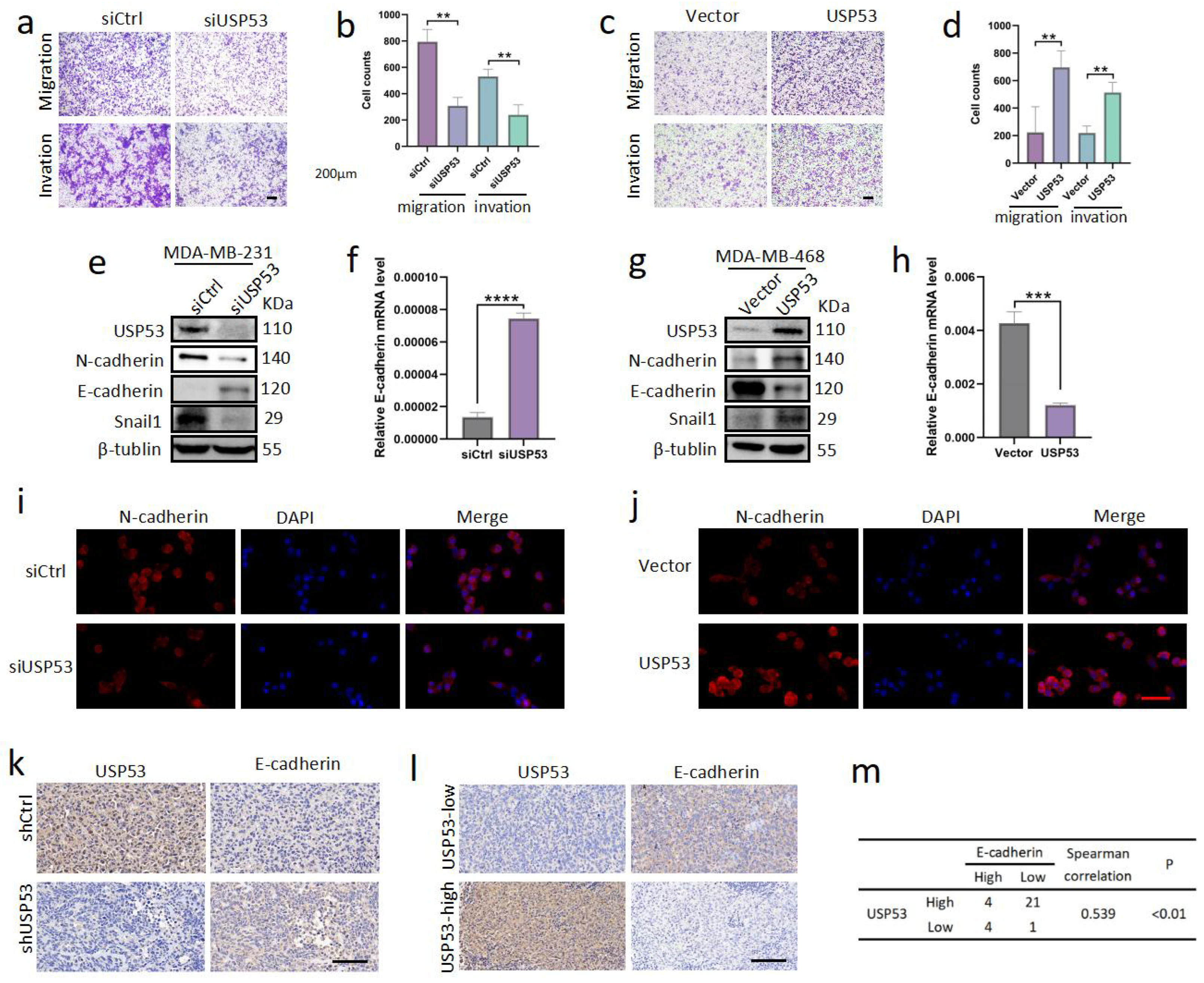
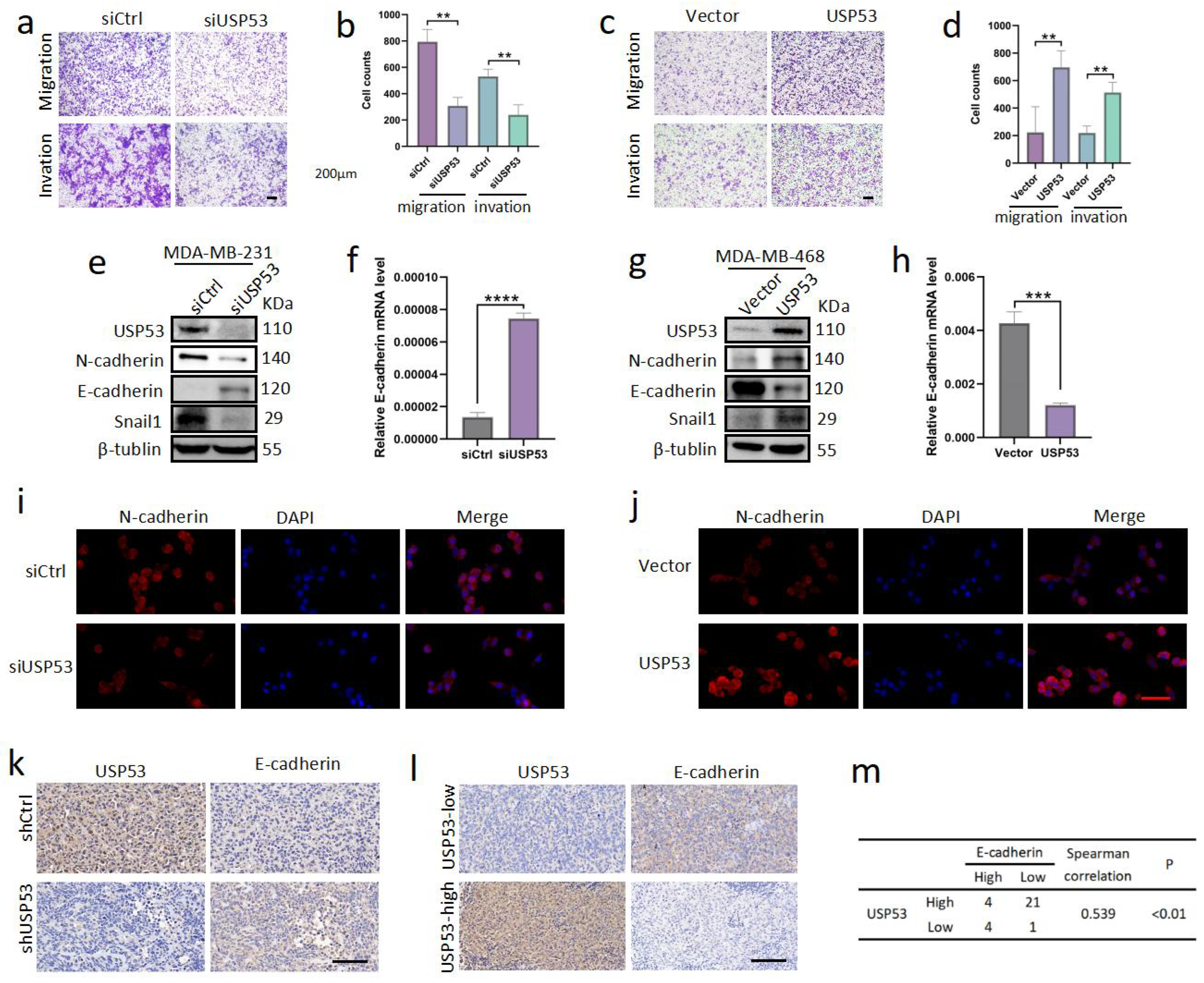
3.3. USP53 Promotes TNBC Cell Metastasis
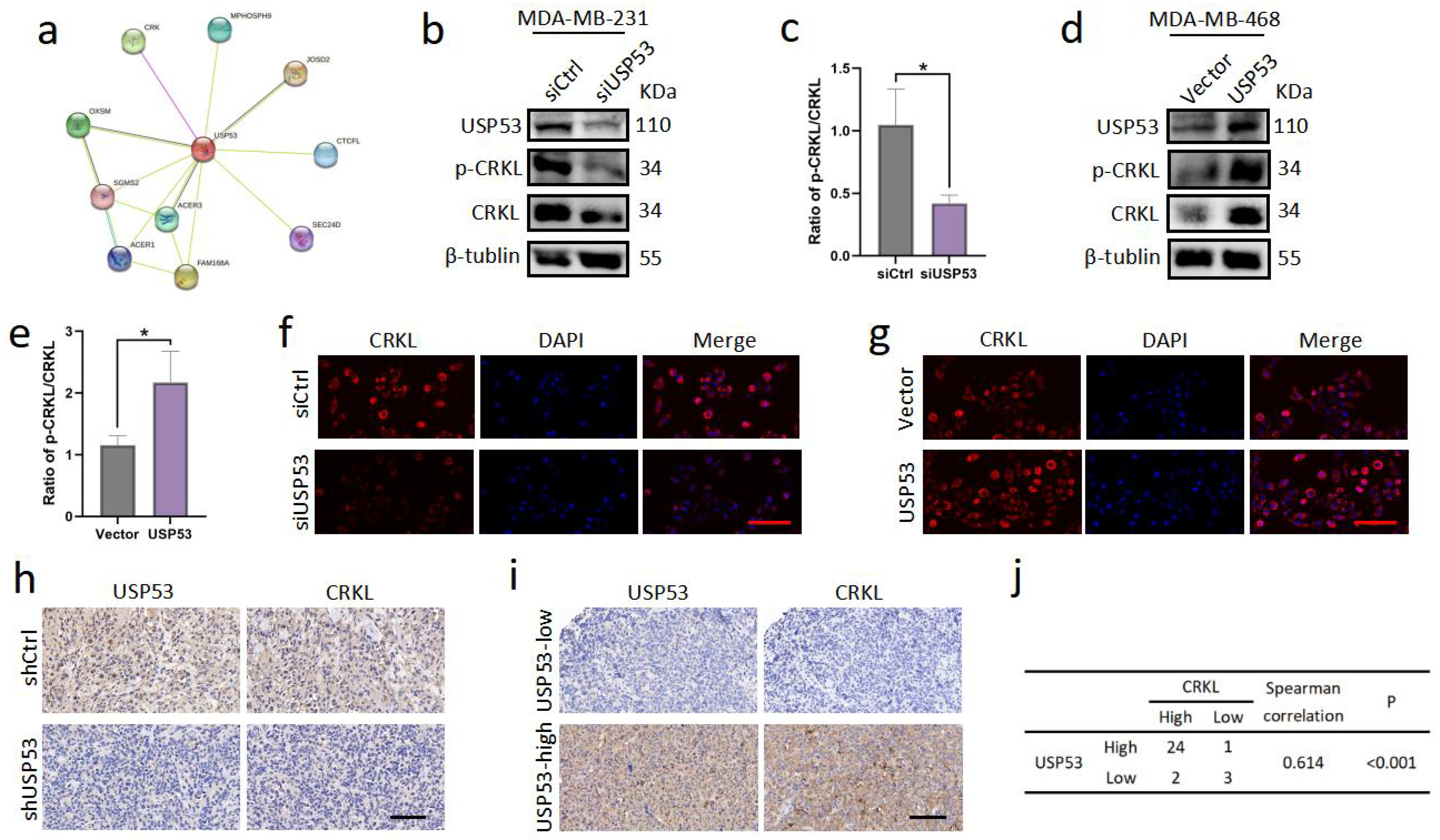
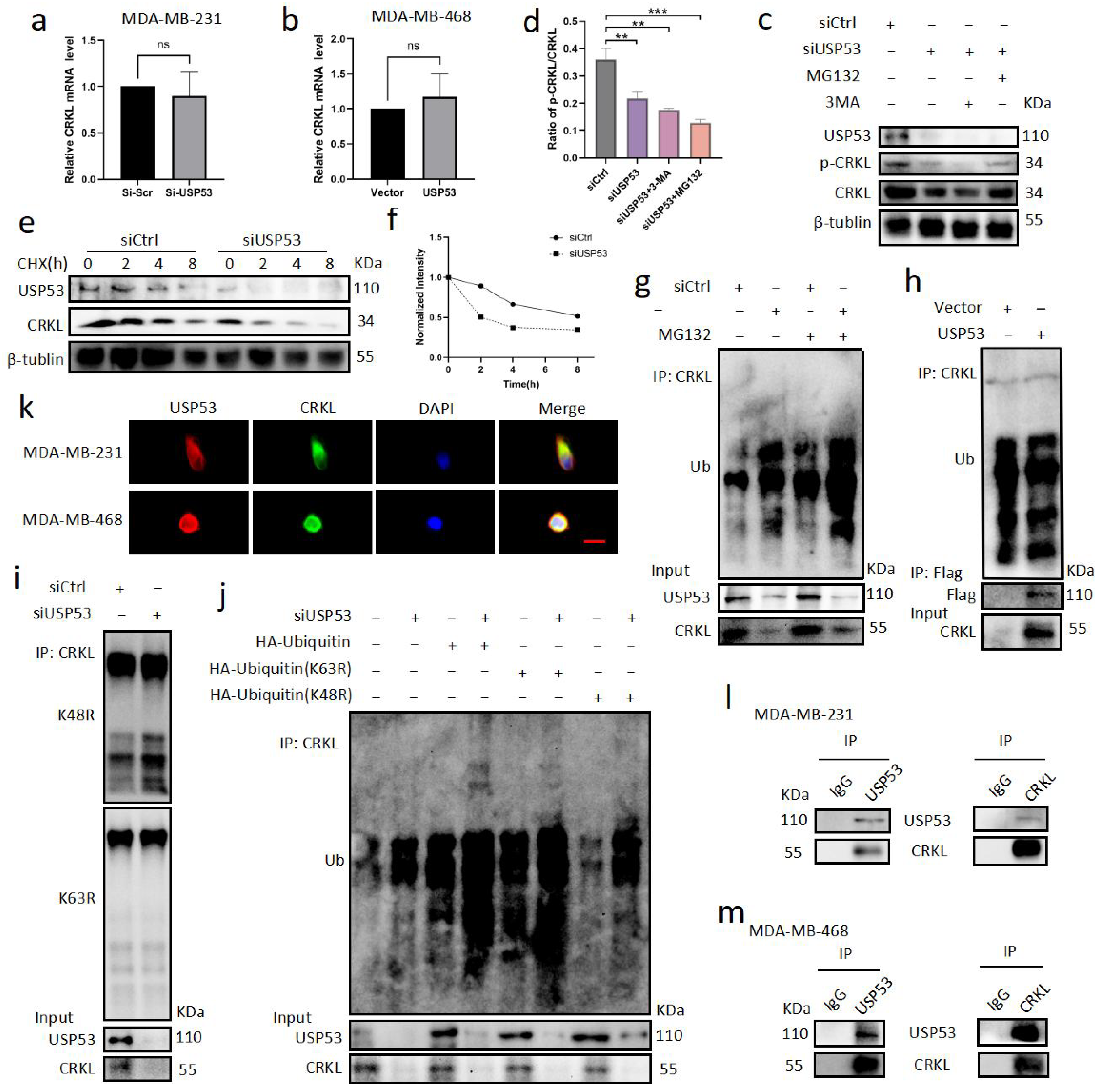
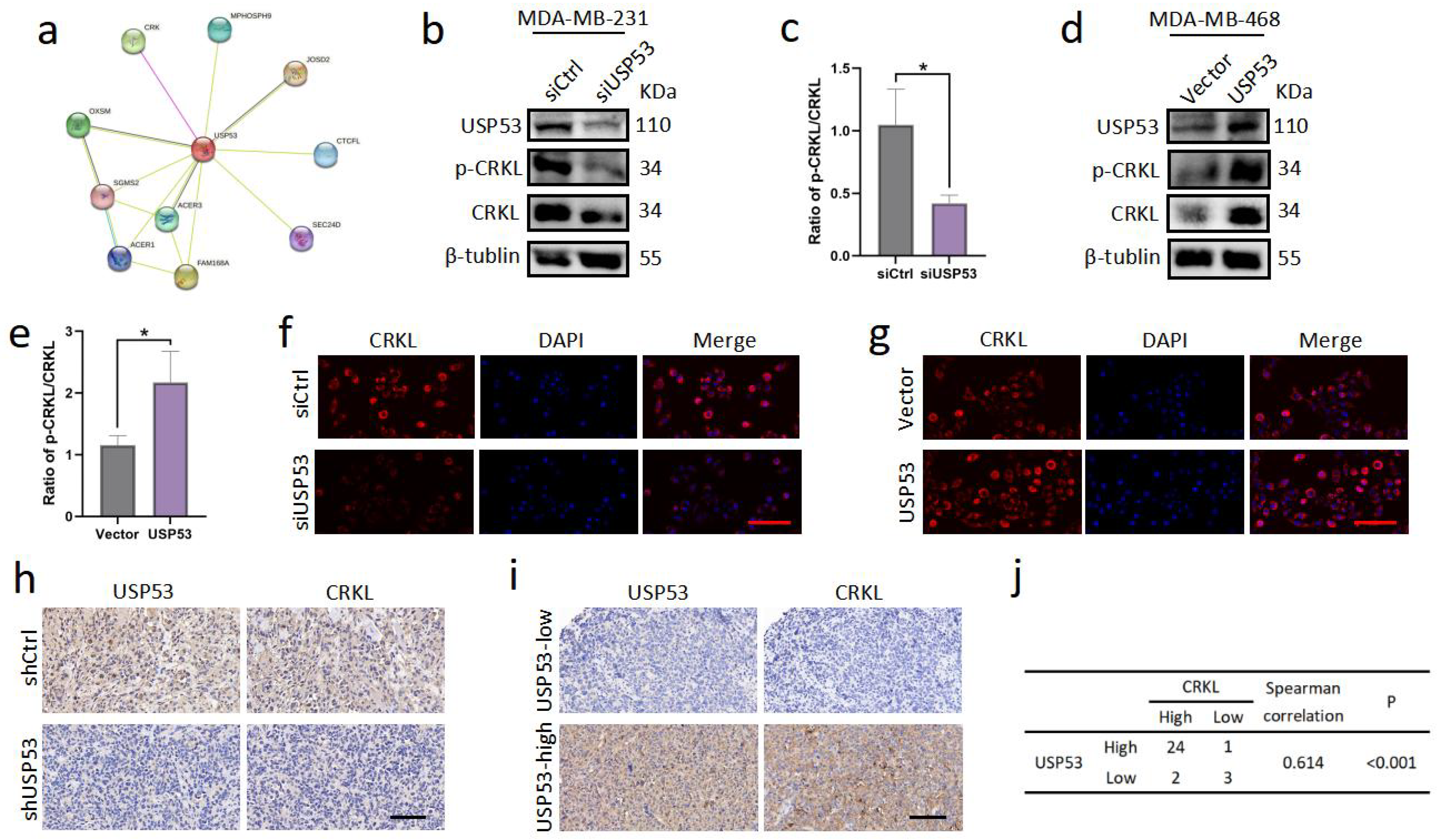
3.4. USP53 Regulates the Protein Level of CRKL in TNBC
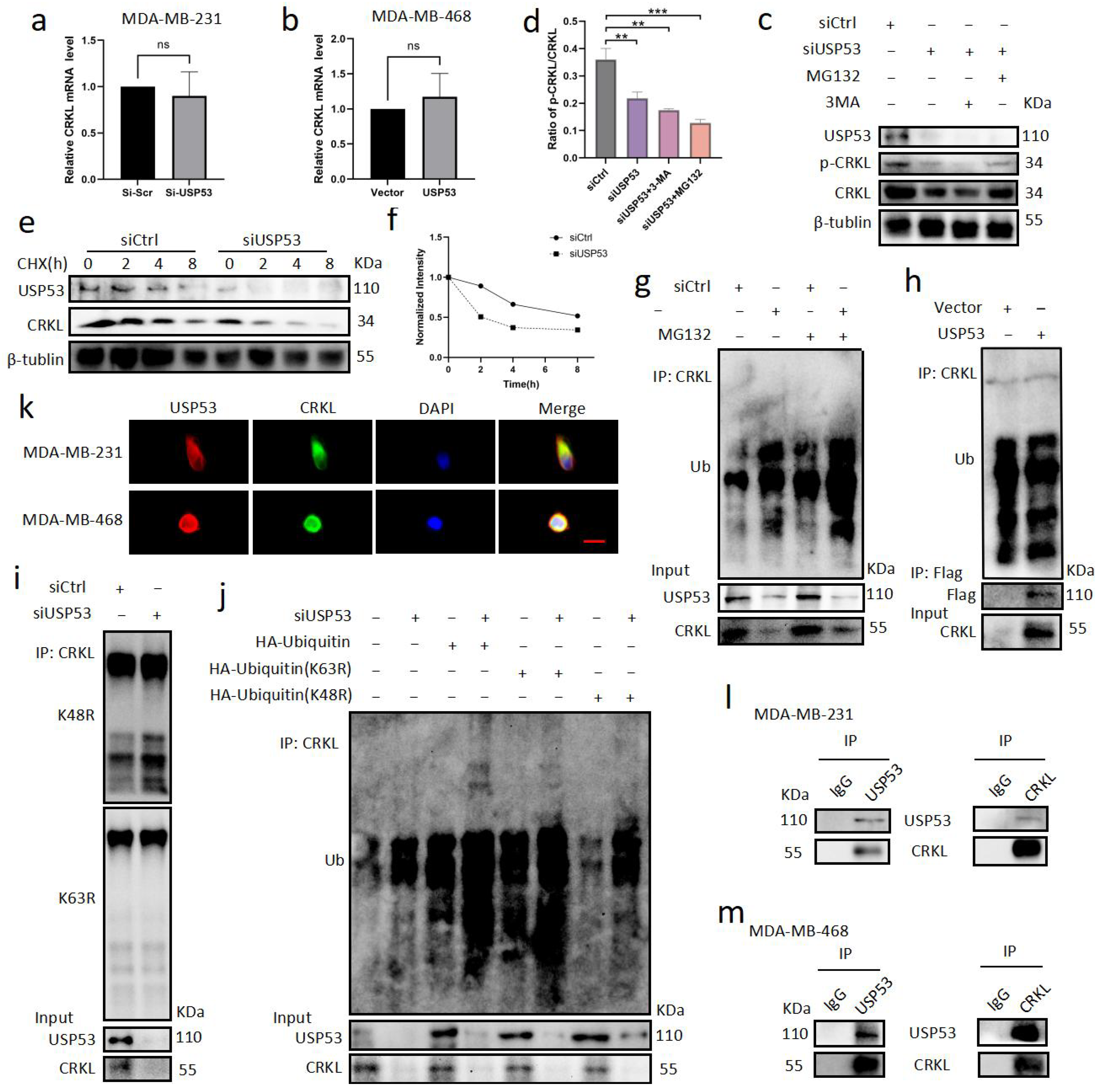
3.5. USP53 Binds, Stabilizes, and Deubiquitinates CRKL
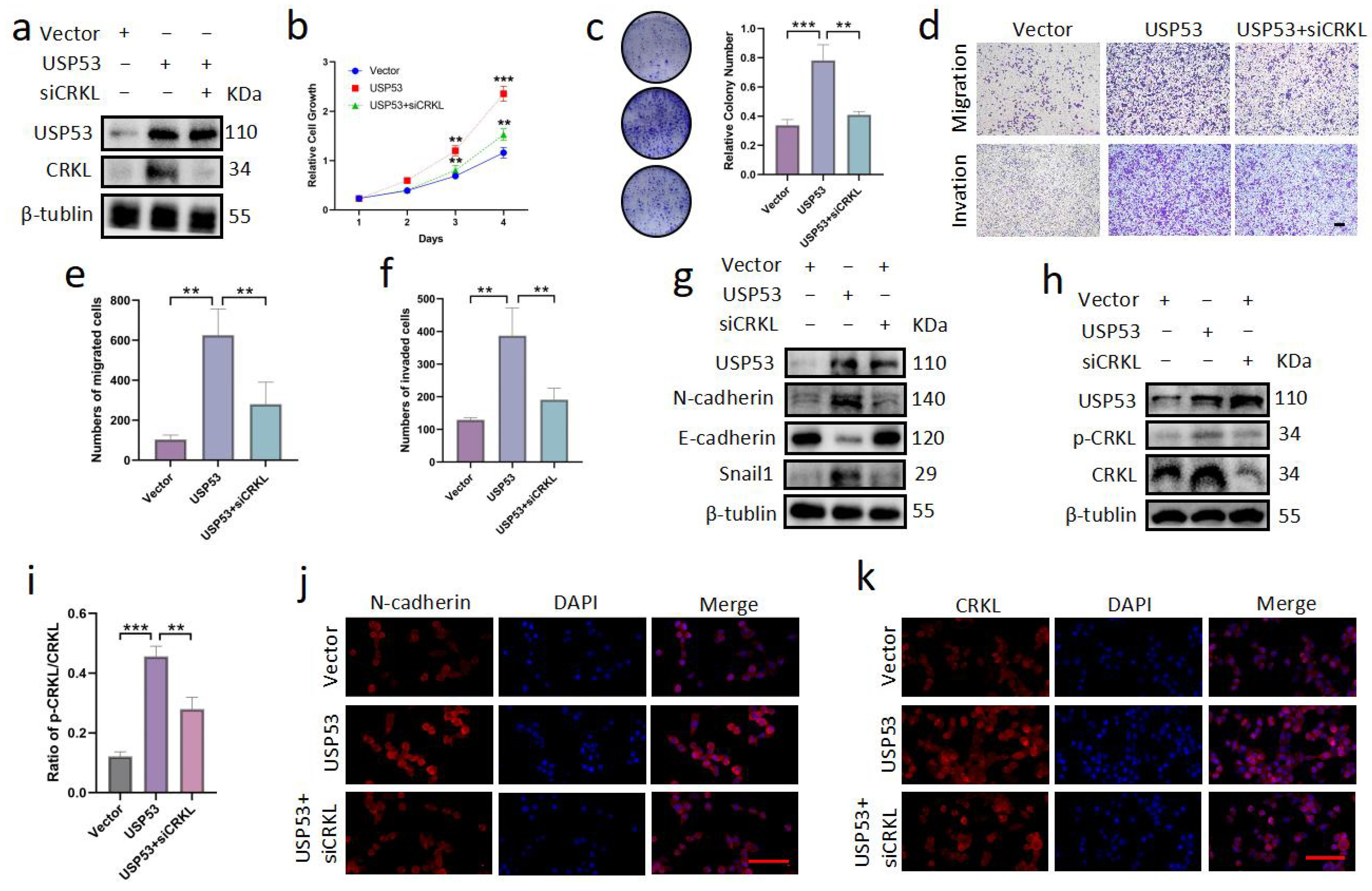
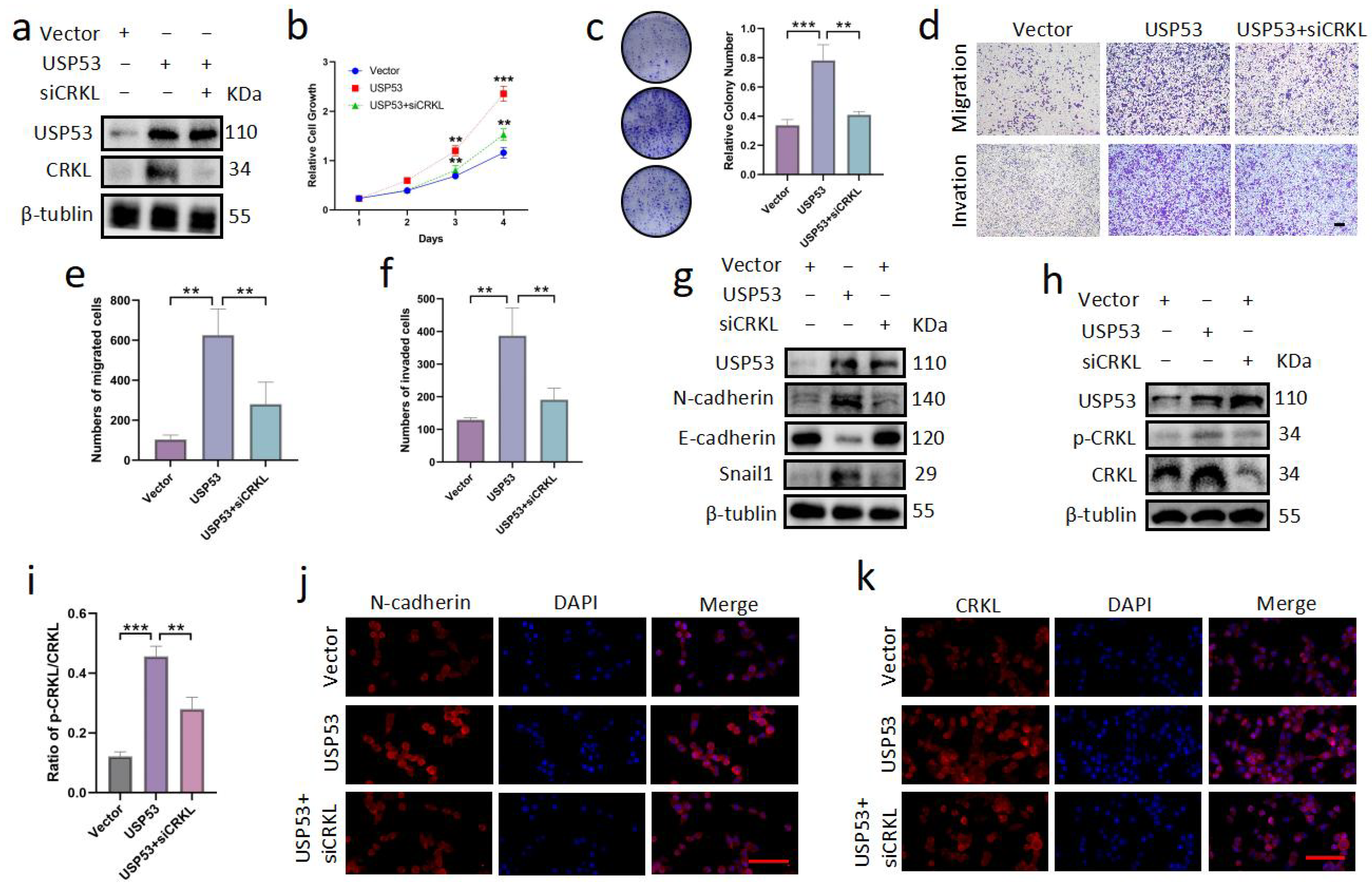
3.6. Knockdown of CRKL Can Reverse Cell Proliferation, Migration, and Invasion Induced by USP53 Upregulation
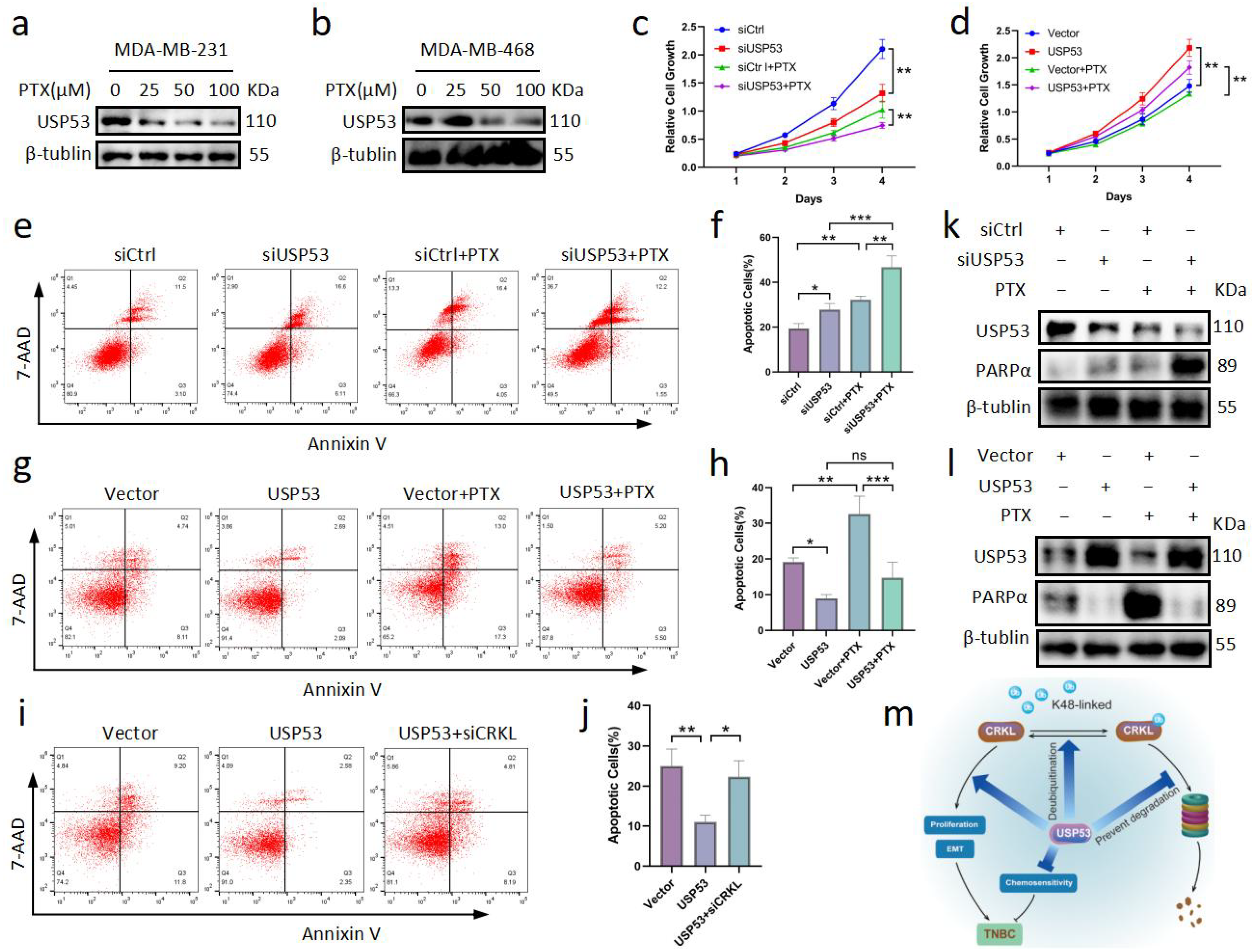
3.7. USP53 Reduces Chemosensitivity in TNBC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zagami, P.; Carey, A.L. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef]
- Baek, D.; Park, H.K.; Lee, K.; Jung, S.; Joung, S.; Kim, J.; Lee, J.W. Ubiquitin-specific protease 53 promotes osteogenic differentiation of human bone marrow-derived mesenchymal stem cells. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bull, N.L.; Ellmers, R.; Foskett, P.; Strautnieks, S.; Sambrotta, M.; Czubkowski, P.; Jankowska, I.; Wagner, B.; Deheragoda, M.; Thompson, R.J. Cholestasis due to usp53 deficiency. J. Pediatr. Gastroenterol. Nutr. 2021, 72, 667. [Google Scholar] [CrossRef] [PubMed]
- Maddirevula, S.; Alhebbi, H.; Alqahtani, A.; Algoufi, T.; Alsaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Barr, M.; Alzaidan, H.; Almehaideb, A.; et al. Identification of novel loci for pediatric cholestatic liver disease defined by kif12, ppm1f, usp53, lsr, and wdr83os pathogenic variants. Genet. Med. 2019, 21, 1164–1172. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Gong, J.Y.; Li, L.; Li, J.; Zhang, M.; Lu, Y.; Xie, X.; Hong, Y.; Yu, Z.; et al. Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: Clinical, histological and ultrastructural characterization. Liver Int. 2020, 40, 1142–1150. [Google Scholar] [CrossRef]
- Kazmierczak, M.; Harris, S.L.; Kazmierczak, P.; Shah, P.; Starovoytov, V.; Ohlemiller, K.K.; Schwander, M. Progressive hearing loss in mice carrying a mutation in usp53. J. Neurosci. 2015, 35, 15582–15598. [Google Scholar] [CrossRef]
- Yao, Y.; Ma, W.; Guo, H.; Liu, Y.; Xia, P.; Wu, X.; Chen, Y.; Wang, K.; Mei, C.; Wang, G.; et al. Usp53 plays an antitumor role in hepatocellular carcinoma through deubiquitination of cytochrome c. Oncogenesis 2022, 11, 31. [Google Scholar] [CrossRef]
- Cheng, W.; Tang, Y.; Tong, X.; Zhou, Q.; Xie, J.; Wang, J.; Han, Y.; Ta, N.; Ye, Z. Usp53 activated by h3k27 acetylation regulates cell viability, apoptosis and metabolism in esophageal carcinoma via the ampk signaling pathway. Carcinogenesis 2022, 43, 349–359. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, X.; Wang, H.; Zhou, Q.; Xie, J.; Wang, J.; Han, Y.; Ta, N.; Ye, Z. Usp53 promotes apoptosis and inhibits glycolysis in lung adenocarcinoma through fkbp51-akt1 signaling. Mol. Carcinog. 2020, 59, 1000–1011. [Google Scholar] [CrossRef]
- Gui, D.; Dong, Z.; Peng, W.; Jiang, W.; Huang, G.; Liu, G.; Ye, Z.; Wang, Y.; Xu, Z.; Fu, J.; et al. Ubiquitin-specific peptidase 53 inhibits the occurrence and development of clear cell renal cell carcinoma through NF-κB pathway inactivation. Cancer Med. 2021, 10, 3674–3688. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yao, X.; Wu, C.; Chen, S.; Fan, D. Knockdown of ubiquitin-specific protease 53 enhances the radiosensitivity of human cervical squamous cell carcinoma by regulating DNA damage-binding protein 2. Technol. Cancer Res. Treat. 2020, 19. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Fan, Q.; Dlugos, C.P.; Soofi, A.A.; Zhang, J.; Verma, R.; Park, T.; Wong, H.; Curran, T.; Nihalani, D.; et al. Crk1/2 and CrkL form a hetero-oligomer and functionally complement each other during podocyte morphogenesis. Kidney Int. 2014, 85, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Senechal, K.; Halpern, J.; Sawyers, C.L. The CrkL adaptor protein transforms fibroblasts and functions in transformation by the BCR-ABL oncogene. J. Biol. Chem. 1996, 271, 23255–23261. [Google Scholar] [CrossRef] [PubMed]
- Hallock, P.T.; Chin, S.; Blais, S.; Neubert, T.; Glass, D.J. Sorbs1 and-2 interact with CrkL and are required for acetylcholine receptor cluster formation. Mol. Cell. Biol. 2016, 36, 262–270. [Google Scholar] [CrossRef]
- Matsuki, T.; Pramatarova, A.; Howell, B.W. Reduction of Crk and CrkL expression blocks reelin-induced dendritogenesis. J. Cell Sci. 2008, 121, 1869–1875. [Google Scholar] [CrossRef]
- Segovis, C.M.; Schoon, R.A.; Dick, C.J.; Nacusi, L.P.; Leibson, P.J.; Billadeau, D.D. PI3K links NKG2D signaling to a CrkL pathway involved in natural killer cell adhesion, polarity, and granule secretion. J. Immunol. 2009, 182, 6933–6942. [Google Scholar] [CrossRef]
- Yeung, C.L.; Ngo, V.N.; Grohar, P.J.; Arnaldez, F.; Asante, A.; Wan, X.; Khan, J.; Hewitt, S.; Khanna, C.; Staudt, L. Loss-of-function screen in rhabdomyosarcoma identifies CrkL-Yes as a critical signal for tumor growth. Oncogene 2013, 32, 5429–5438. [Google Scholar] [CrossRef]
- Nishihara, H.; Maeda, M.; Oda, A.; Tsuda, M.; Sawa, H.; Nagashima, K.; Tanaka, S. Dock2 associates with CrkL and regulates Rac1 in human leukemia cell lines. Blood J. Am. Soc. Hematol. 2002, 100, 3968–3974. [Google Scholar] [CrossRef]
- Yanagi, H.; Wang, L.; Nishihara, H.; Kimura, T.; Tanino, M.; Yanagi, T.; Fukuda, S.; Tanaka, S. CrkL plays a pivotal role in tumorigenesis of head and neck squamous cell carcinoma through the regulation of cell adhesion. Biochem. Biophys. Res. Commun. 2012, 418, 104–109. [Google Scholar] [CrossRef]
- Guo, C.; Zhao, D.; Zhang, Q.; Kimura, T.; Tanino, M.; Yanagi, T.; Fukuda, S.; Tanaka, S. miR-429 suppresses tumor migration and invasion by targeting CrkL in hepatocellular carcinoma via inhibiting RAF/MEK/ERK pathway and epithelial-mesenchymal transition. Sci. Rep. 2018, 8, 2375. [Google Scholar] [CrossRef]
- Han, G.; Wu, D.; Yang, Y.; Li, Z.; Zhang, J.; Li, C. CrkL mediates CCL20/CCR6-induced EMT in gastric cancer. Cytokine 2015, 76, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, X.; Hu, B.; Wang, X.; Wang, Q.; Wang, W. The effects of micro-429 on inhibition of cervical cancer cells through targeting ZEB1 and CrkL. Biomed. Pharmacother. 2016, 80, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Miao, Z.; Wang, Z.; Xu, Y.; Wu, J.; Liu, X.; You, Y.; Li, J. Overexpression of CrkL correlates with malignant cell proliferation in breast cancer. Tumor Biol. 2013, 34, 2891–2897. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Jiao, Y.; Yang, Y.; Wang, Z.; Xuan, Q.; Liu, H.; Lu, S.; Wang, Z.; Liu, Y.; Li, S. CrkL regulates SDF-1-induced breast cancer biology through balancing ERK1/2 and PI3K/AKT pathways. Med. Oncol. 2015, 32, 411. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Godin, B. Increased soluble CrkL in serum of breast cancer patients is associated with advanced disease. Cancers 2019, 11, 961. [Google Scholar] [CrossRef]
- Sun, H.; Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Bartholomeusz, G.; Talpaz, M.; Donato, N.J. BCR-ABL ubiquitination and USP9X inhibition block kinase signaling and promote CML cell apoptosis. Blood J. Am. Soc. Hematol. 2011, 117, 3151–3162. [Google Scholar] [CrossRef]
- Ahmad, S.; Alsayed, Y.M.; Druker, B.J.; Platanias, L.C. The type I interferon receptor mediates tyrosine phosphorylation of the CrkL adaptor protein. J. Biol. Chem. 1997, 272, 29991–29994. [Google Scholar] [CrossRef]
- Mueller, D.L. E3 ubiquitin ligases as T cell anergy factors. Nat. Immunol. 2004, 5, 883–890. [Google Scholar] [CrossRef]
- Huang, C. Roles of E3 ubiquitin ligases in cell adhesion and migration. Cell Adhes. Migr. 2010, 4, 10–18. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Weinberg, R.A. EMT and cancer: More than meets the eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Xie, K.; Lan, T.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; et al. TXNDC12 promotes EMT and metastasis of hepatocellular carcinoma cells via activation of β-catenin. Cell Death Differ. 2020, 27, 1355–1368. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, A.R.; Ban, Y.; Crowley, M.J.; Lee, S.B.; Ramchandani, D.; Du, W.; Elemento, O.; George, J.T.; Jolly, M.K.; Levine, H. Differential contributions of pre- and post-EMT tumor cells in breast cancer metastasis—Contribution of EMT mechanism in breast tumor metastasis. Cancer Res. 2020, 80, 163–169. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-mesenchymal transition in cancer: A historical overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Rouzier, R.; Rajan, R.; Wagner, P.; Hess, K.R.; Gold, D.L.; Stec, J.; Ayers, M.; Ross, J.S.; Zhang, P.; Buchholz, T.A.; et al. Microtubule-associated protein tau: A marker of paclitaxel sensitivity in breast cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 8315–8320. [Google Scholar] [CrossRef]
- Zheng, H.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Ge, F.; Chen, W.; Qin, J.; Zhou, Z.; Liu, R.; Liu, L.; Tan, J.; Zou, T.; Li, H.; Ren, G.; et al. Ataxin-3 like (ATXN3L), a member of the Josephin family of deubiquitinating enzymes, promotes breast cancer proliferation by deubiquitinating Krüppel-like factor 5 (KLF5). Oncotarget 2015, 6, 21369. [Google Scholar] [CrossRef]
- Wu, Y.; Qin, J.; Li, F.; Yang, C.; Li, Z.; Zhou, Z.; Zhang, H.; Li, Y.; Wang, X.; Liu, R.; et al. USP3 promotes breast cancer cell proliferation by deubiquitinating KLF5. J. Biol. Chem. 2019, 294, 17837–17847. [Google Scholar] [CrossRef]
- Lambies, G.; Miceli, M.; Martínez-Guillamon, C.; Olivera-Salguero, R.; Carolina-Paola, F.; Irene, C.; Boyko, S.A.; Sharon, Y.R.; Joaquín, A.; Antonio, G.; et al. TGFβ-activated USP27X deubiquitinase regulates cell migration and chemoresistance via stabilization of Snail1. USP27X deubiquitinates Snail1 in tumor cells. Cancer Res. 2019, 79, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Kalodimos, C.; Inagaki, F.; Tanaka, S. Crk and CrkL adaptor proteins: Networks for physiological and pathological signaling. Cell Commun. Signal. 2009, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liu, S.; Sun, M. The role of CT10 regulation of kinase-like in cancer. Future Oncol. 2014, 10, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Abdul, S.; Majid, A.; Wang, J.; Liu, Q.; Sun, M.; Liu, S. Bidirectional interaction of lncRNA AFAP1-AS1 and CrkL accelerates the proliferative and metastatic abilities of hepatocarcinoma cells. J. Adv. Res. 2020, 24, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Gao, C.; Zhao, D.; Li, J.; Wang, J.; Sun, X.; Liu, Q.; Hao, L.; Greenaway, F.T.; Tian, Y.; et al. A novel ETV6-miR-429-CrkL regulatory circuitry contributes to aggressiveness of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 70. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.W.; Du, J.; Boehm, J.S.; He, F.; Weir, B.; Wang, X.; Butaney, M.; Sequist, L.; Luo, B.; Engelman, J.; et al. Amplification of CrkL induces transformation and epidermal growth factor receptor inhibitor resistance in human non–small cell lung cancers. Cancer Discov. 2011, 1, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Xie, C.; Li, Q.; Qianze, D.; Enhua, W.; Yan, W. Crk promotes lung cancer cell invasion through ERK-MMP9 pathway. Mol. Carcinog. 2015, 54, E35–E44. [Google Scholar]
- Fathers, K.E.; Bell, E.S.; Rajadurai, C.V.; Cory, S.; Zhao, H.; Mourskaia, A.; Zuo, D.; Madore, J.; Monast, A.; Mes-Masson, A.; et al. Crk adaptor proteins act as key signaling integrators for breast tumorigenesis. Breast Cancer Res. 2012, 14, R74. [Google Scholar] [CrossRef]
- Bian, X.; Liang, Z.; Feng, A.; Salgado, E. and Shim, H. HDAC inhibitor suppresses proliferation and invasion of breast cancer cells through regulation of miR-200c targeting CrkL. Biochem. Pharmacol. 2018, 147, 30–37. [Google Scholar] [CrossRef]
- Sattler, M.; Salgia, R. Role of the adapter protein CrkL in signal transduction of normal hematopoietic and Bcr/Abl-transformed cells. Leukemia 1998, 12, 637–644. [Google Scholar] [CrossRef]
- Franke, F.C.; Müller, J.; Abal, M.; Eduardo, D.; Ulrich, N.; Henri, W.; Solenne, C.; Ewa, N.; Klaus-Peter, J. The tumor suppressor SASH1 interacts with the signal adaptor CrkL to inhibit epithelial–mesenchymal transition and metastasis in colorectal cancer. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.K.; Chen, D.; Li, X. miR-335 suppresses cell proliferation and migration by upregulating CrkL in bladder cancer. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2399–2408. [Google Scholar] [PubMed]
- Zhang, P.; Xiao, Z.; Wang, S.; Zhang, M.; Wei, Y.; Hang, Q.; Kim, J.; Yao, F.; Rodriguez-Aguayo, C.; Ton, B.; et al. ZRANB1 is an EZH2 deubiquitinase and a potential therapeutic target in breast cancer. Cell Rep. 2018, 23, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Chen, H.; Zhou, Z.; Wan, Y.; Liu, Z. ATXN3 promotes breast cancer metastasis by deubiquitinating KLF4. Cancer Lett. 2019, 467, 19–28. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Tang, W.; Yao, F. USP53 Exerts Tumor-Promoting Effects in Triple-Negative Breast Cancer by Deubiquitinating CRKL. Cancers 2023, 15, 5033. https://doi.org/10.3390/cancers15205033
Liu Y, Tang W, Yao F. USP53 Exerts Tumor-Promoting Effects in Triple-Negative Breast Cancer by Deubiquitinating CRKL. Cancers. 2023; 15(20):5033. https://doi.org/10.3390/cancers15205033
Chicago/Turabian StyleLiu, Yi, Wei Tang, and Feng Yao. 2023. "USP53 Exerts Tumor-Promoting Effects in Triple-Negative Breast Cancer by Deubiquitinating CRKL" Cancers 15, no. 20: 5033. https://doi.org/10.3390/cancers15205033
APA StyleLiu, Y., Tang, W., & Yao, F. (2023). USP53 Exerts Tumor-Promoting Effects in Triple-Negative Breast Cancer by Deubiquitinating CRKL. Cancers, 15(20), 5033. https://doi.org/10.3390/cancers15205033