
Artificial Intelligence Reveals Distinct Prognostic Subgroups of Muscle-Invasive Bladder Cancer on Histology Images

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Survival Modeling
2.1.1. Data
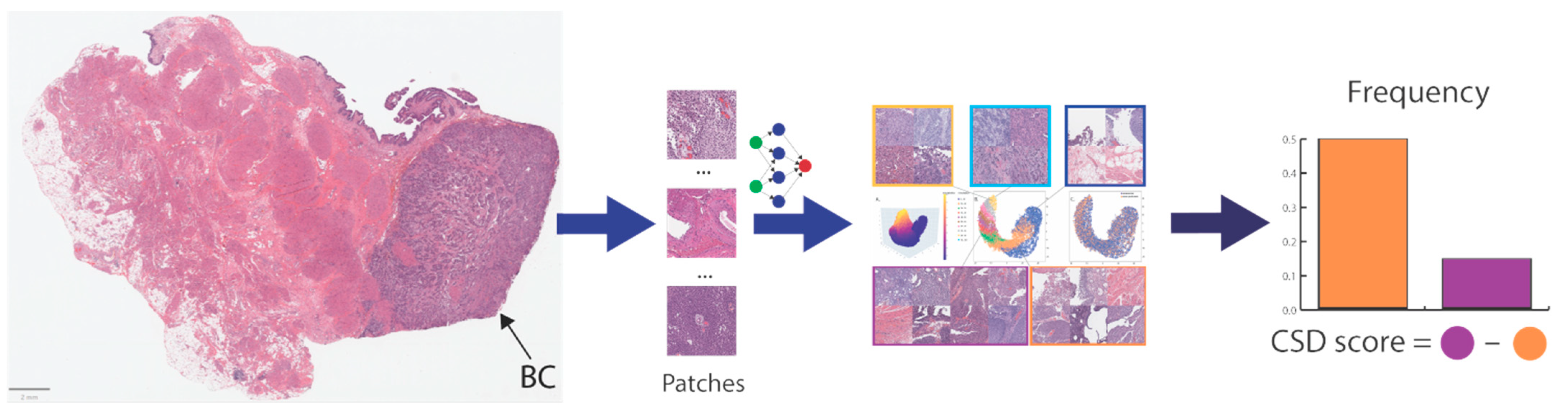
2.1.2. Image Preprocessing
2.1.3. Model Development
- (1)
- We first calculated the patch frequency for 10 bins with equal width (histogram) at the case level. The bin width was calculated for each case using Equation (1).
- (2)
- Secondly, we applied the maximum normalization to the patch frequencies, including Dr and Dm, to achieve a value range between 0 and 1 for all bins. Third, we estimated the unadjusted CSD score (Su) using the following equation:
- (3)
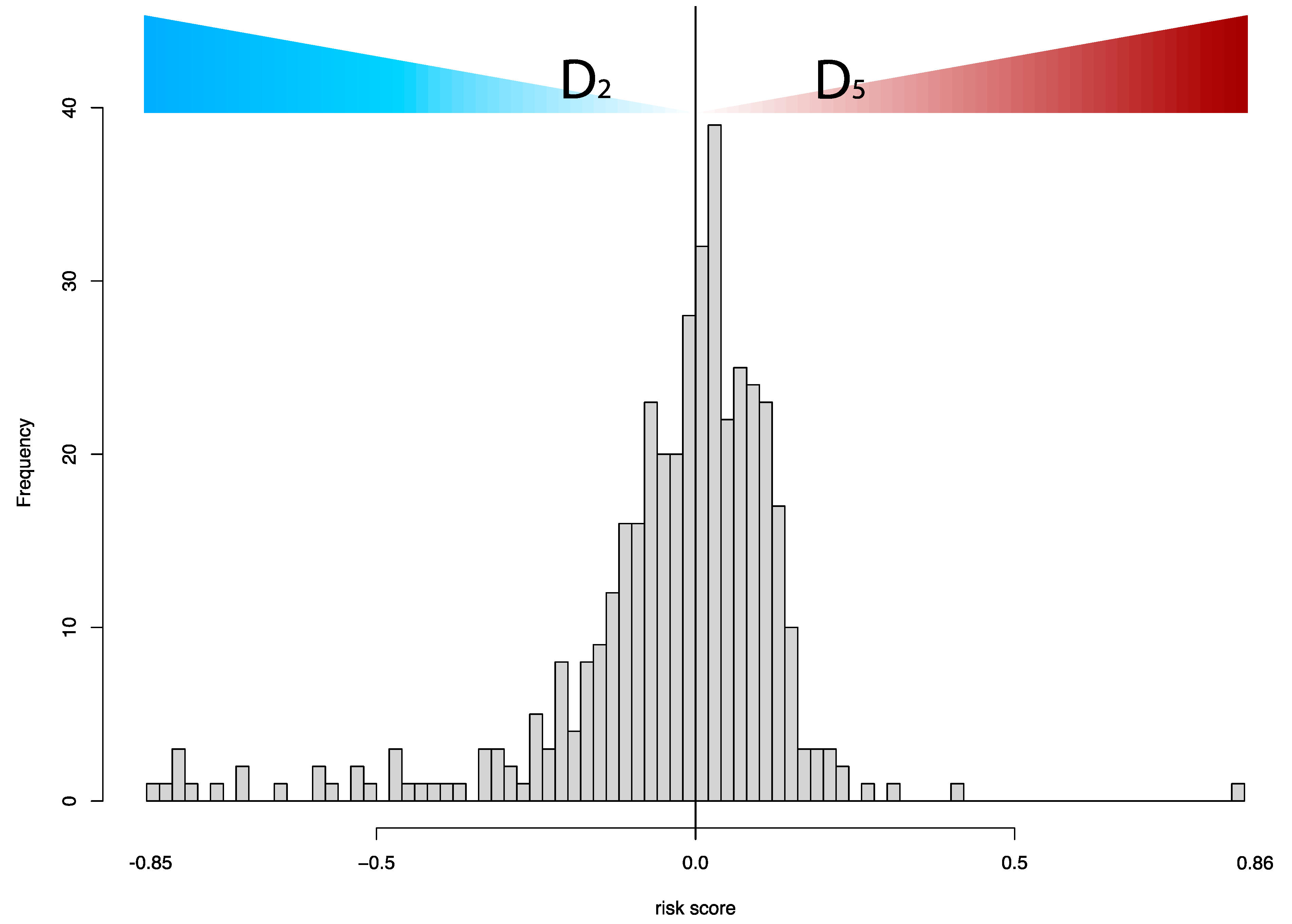
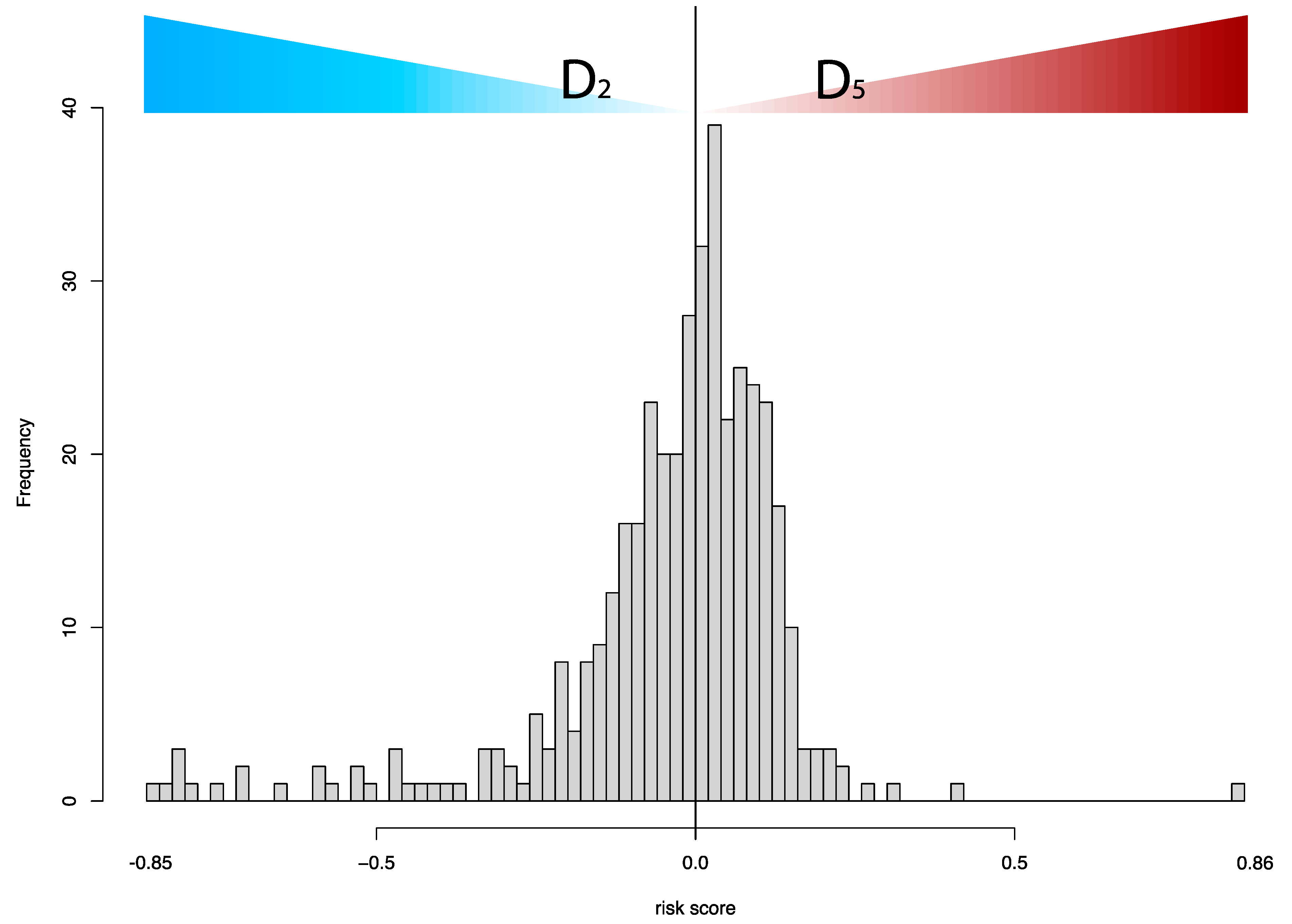
- Since out-of-distribution data may have a different frequency distribution than the development set, we introduced the following algorithm to adjust the CSD score estimation without having the ground truth:
- (a)
- Calculate the mean µ of Su;
- (b)
- Calculate the median μ1/2 of (Su − µ);
- (c)
- Adjust the scores by µ and μ1/2 according to the equation:
2.2. Evaluation
2.2.1. Data
2.2.2. Prognosis
2.2.3. Association with Familiar Molecular Signatures of Bladder Cancer
2.3. Metrics, Statistics and Software
3. Results
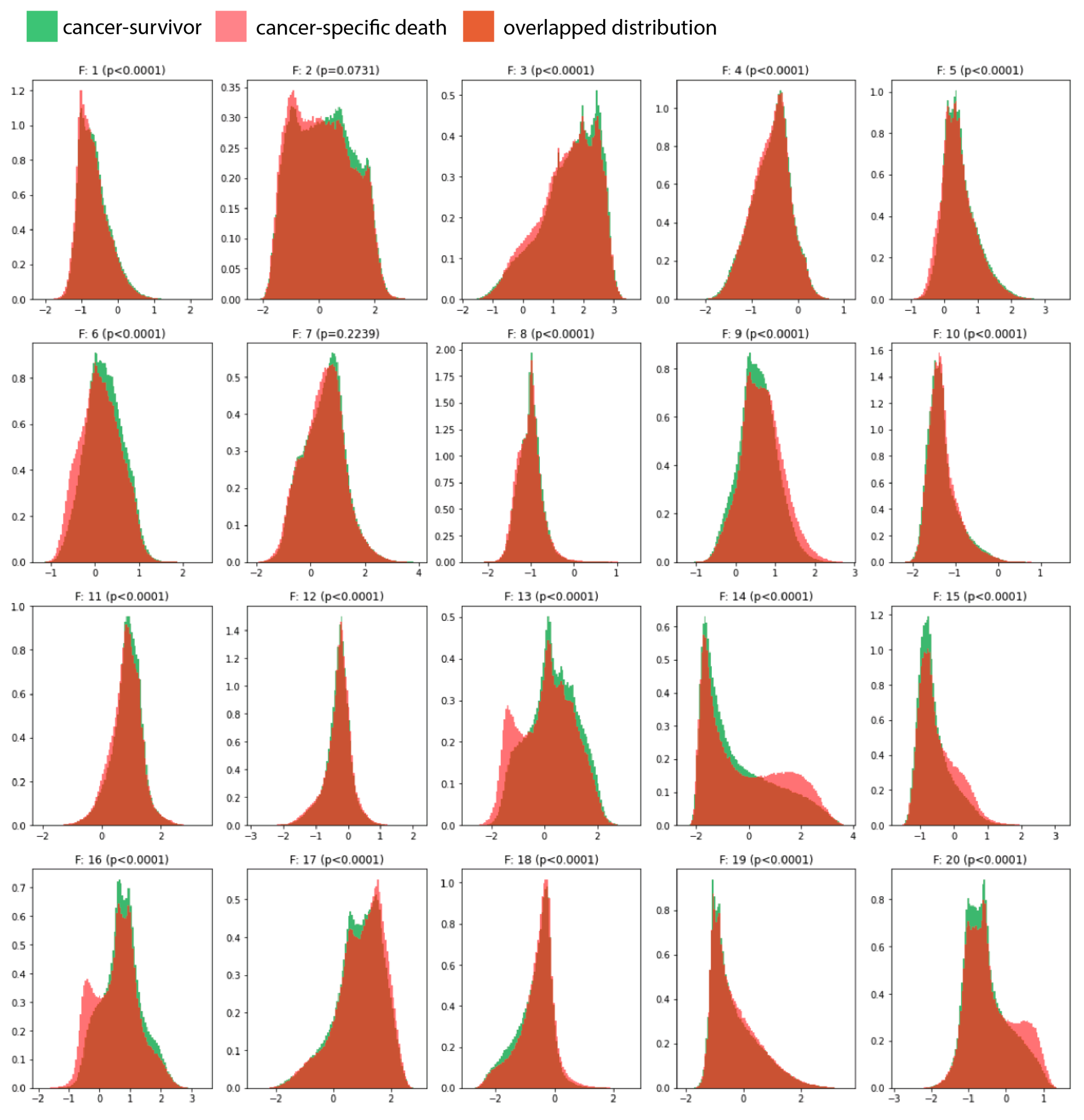
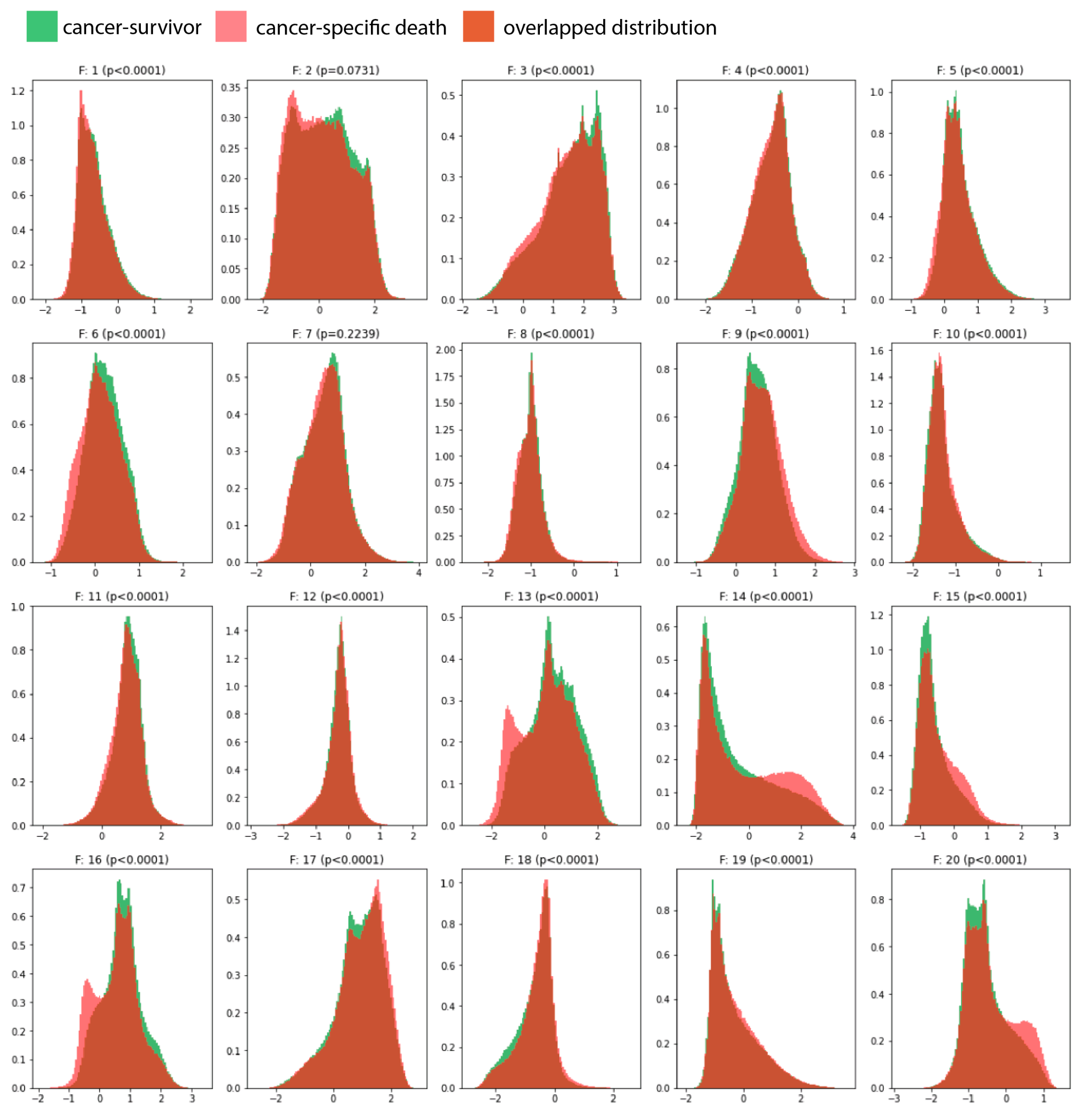
3.1. Survival Modeling
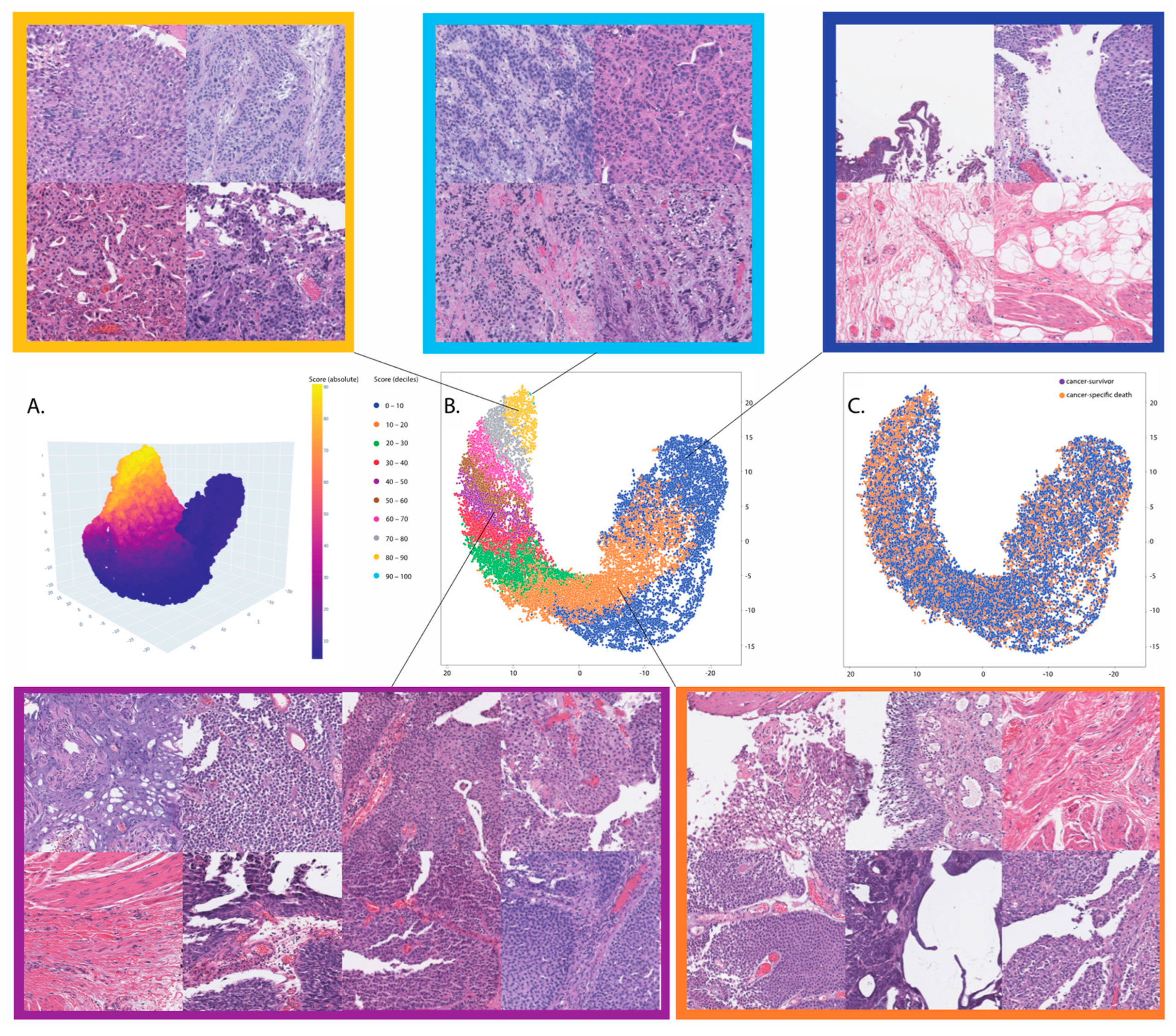
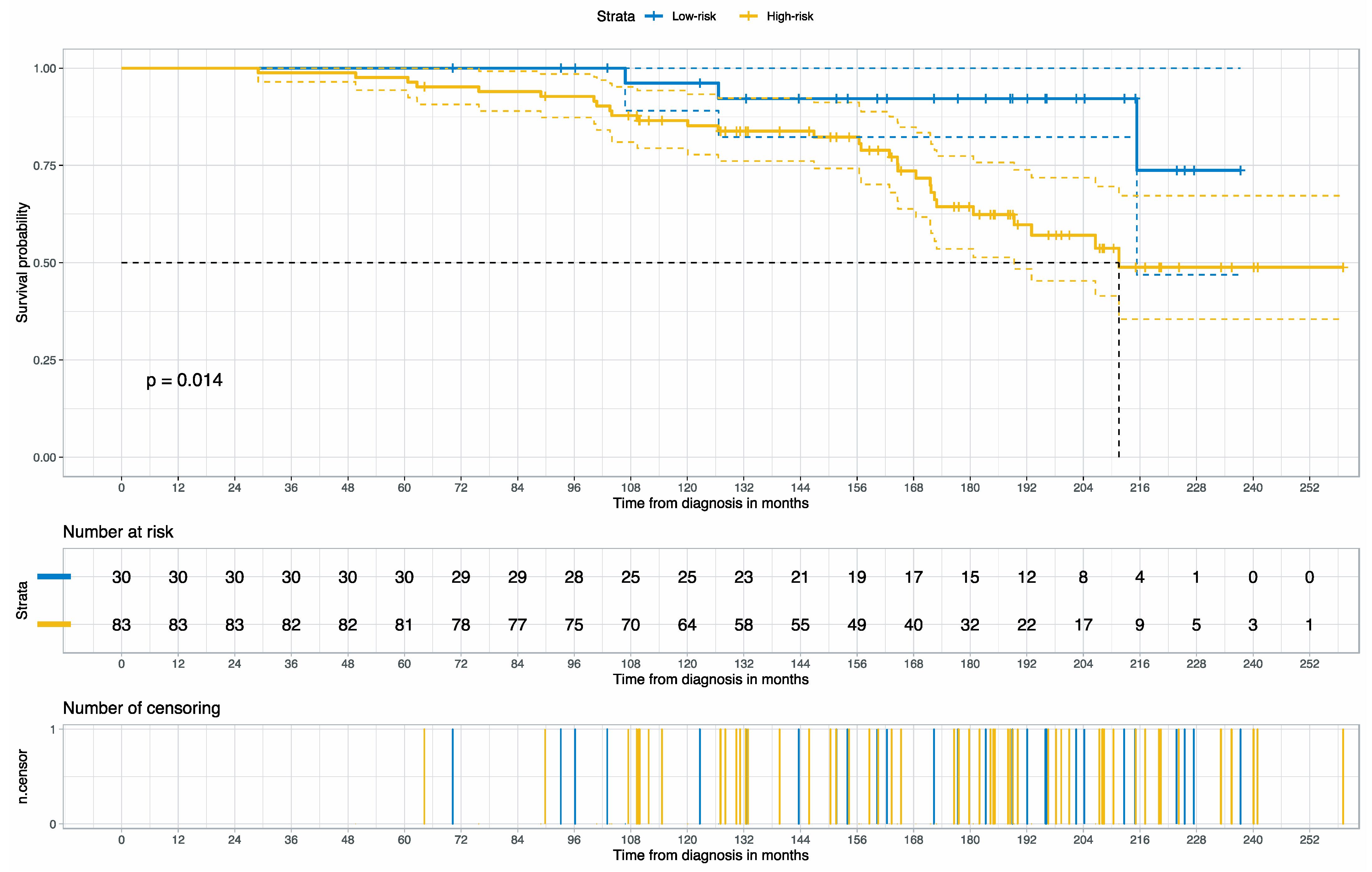
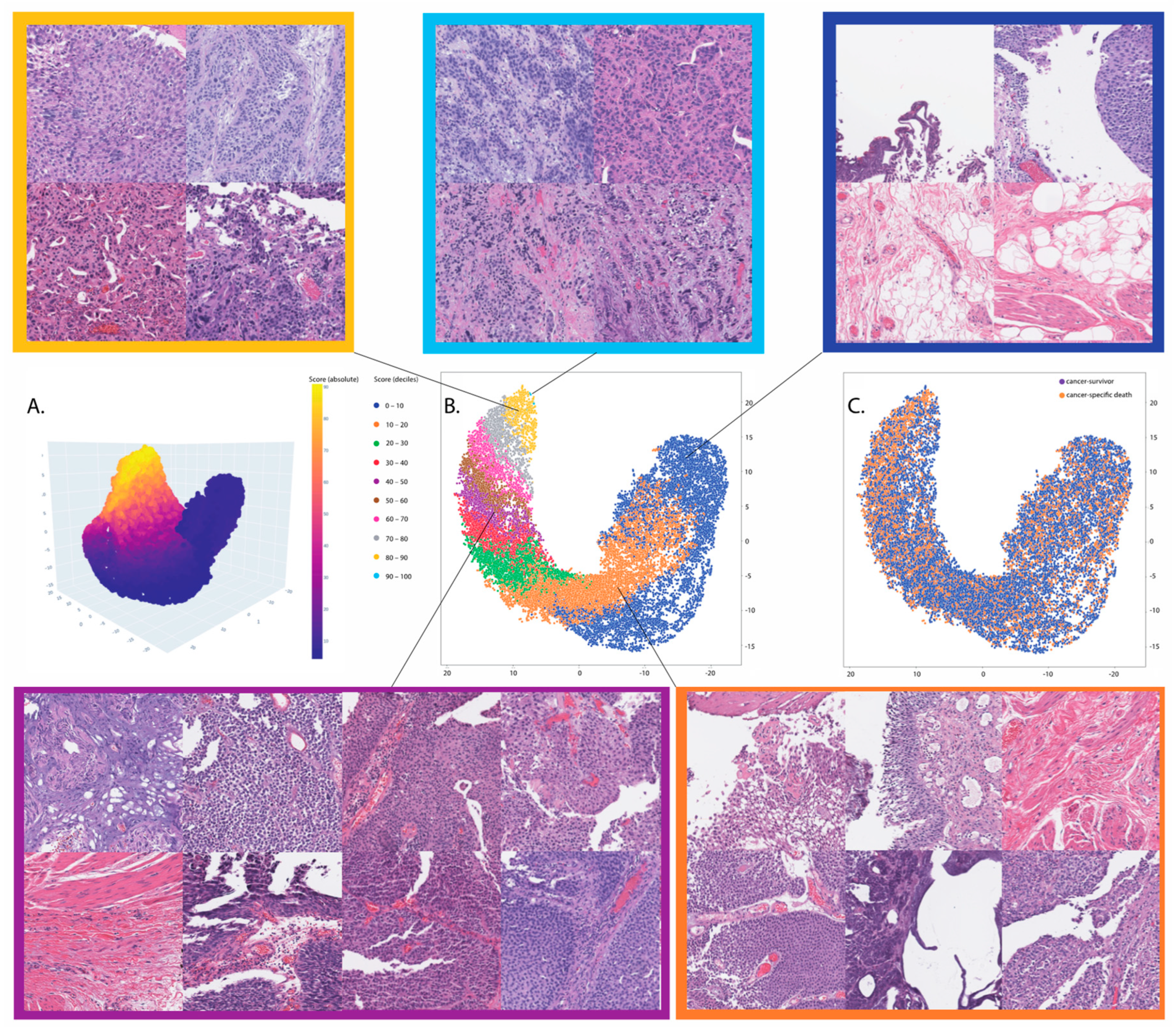
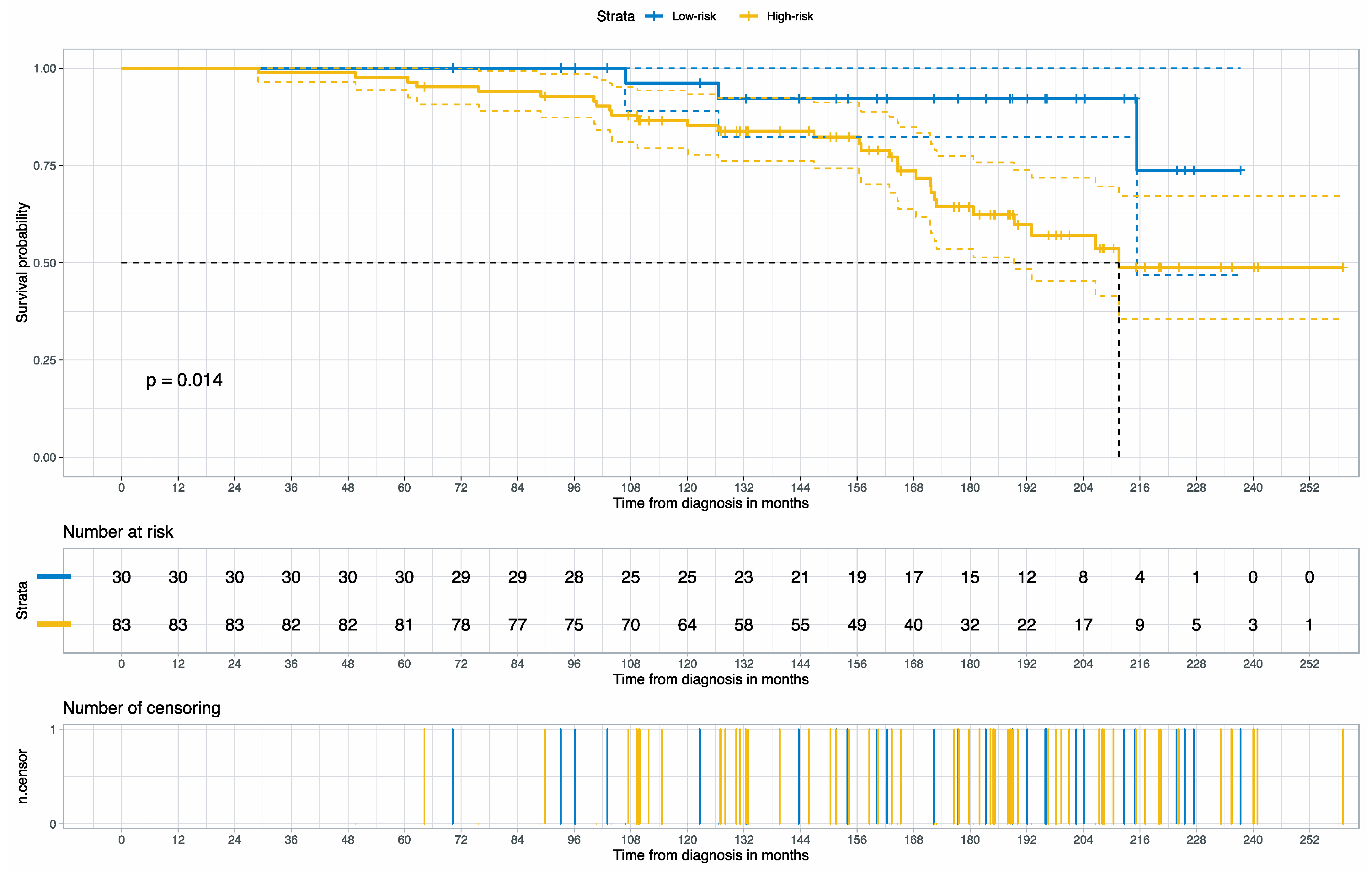
3.2. Prognosis for Muscle-Invasive Bladder Cancer
3.3. Association with Molecular Signatures of Bladder Cancer
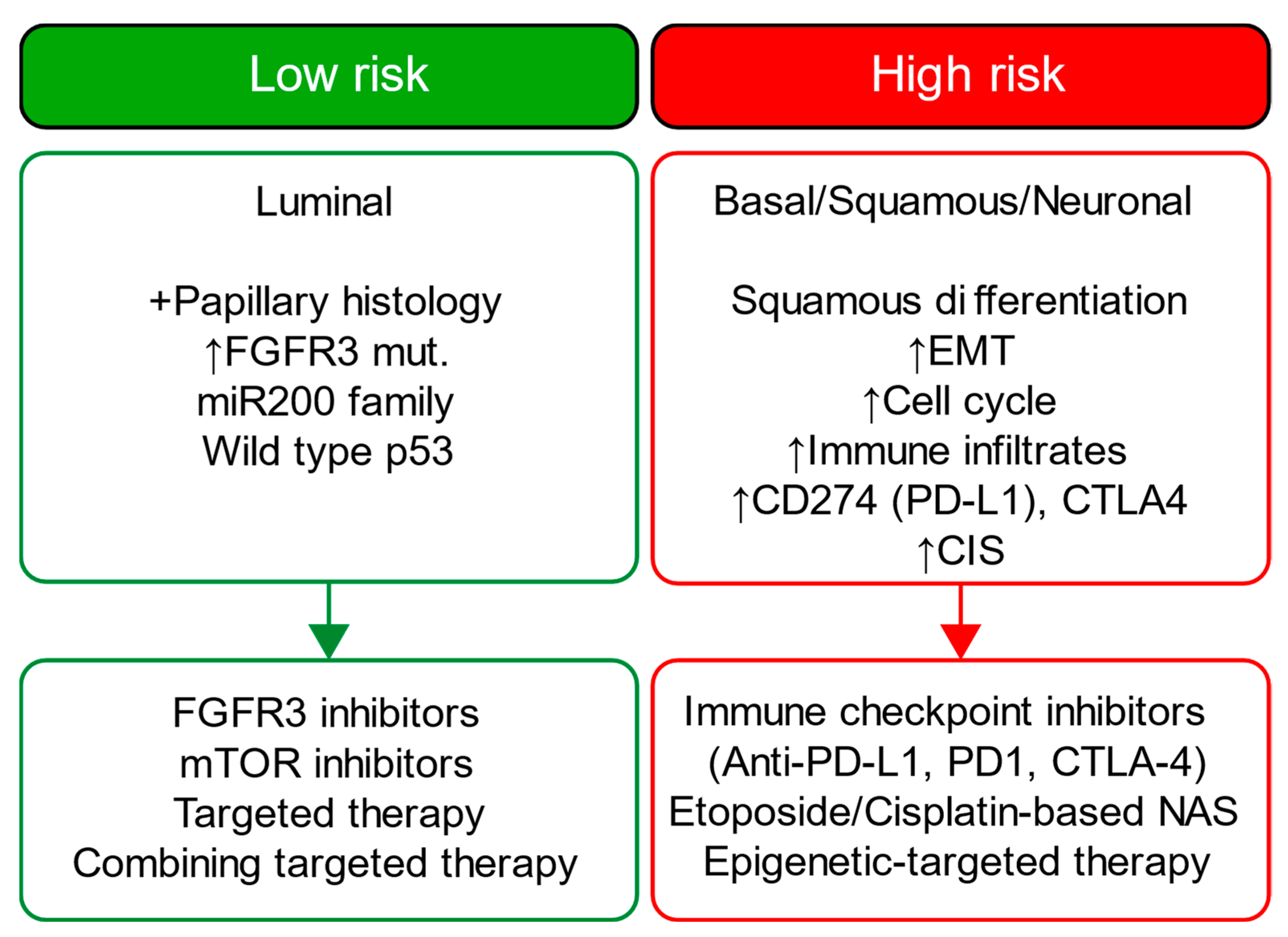
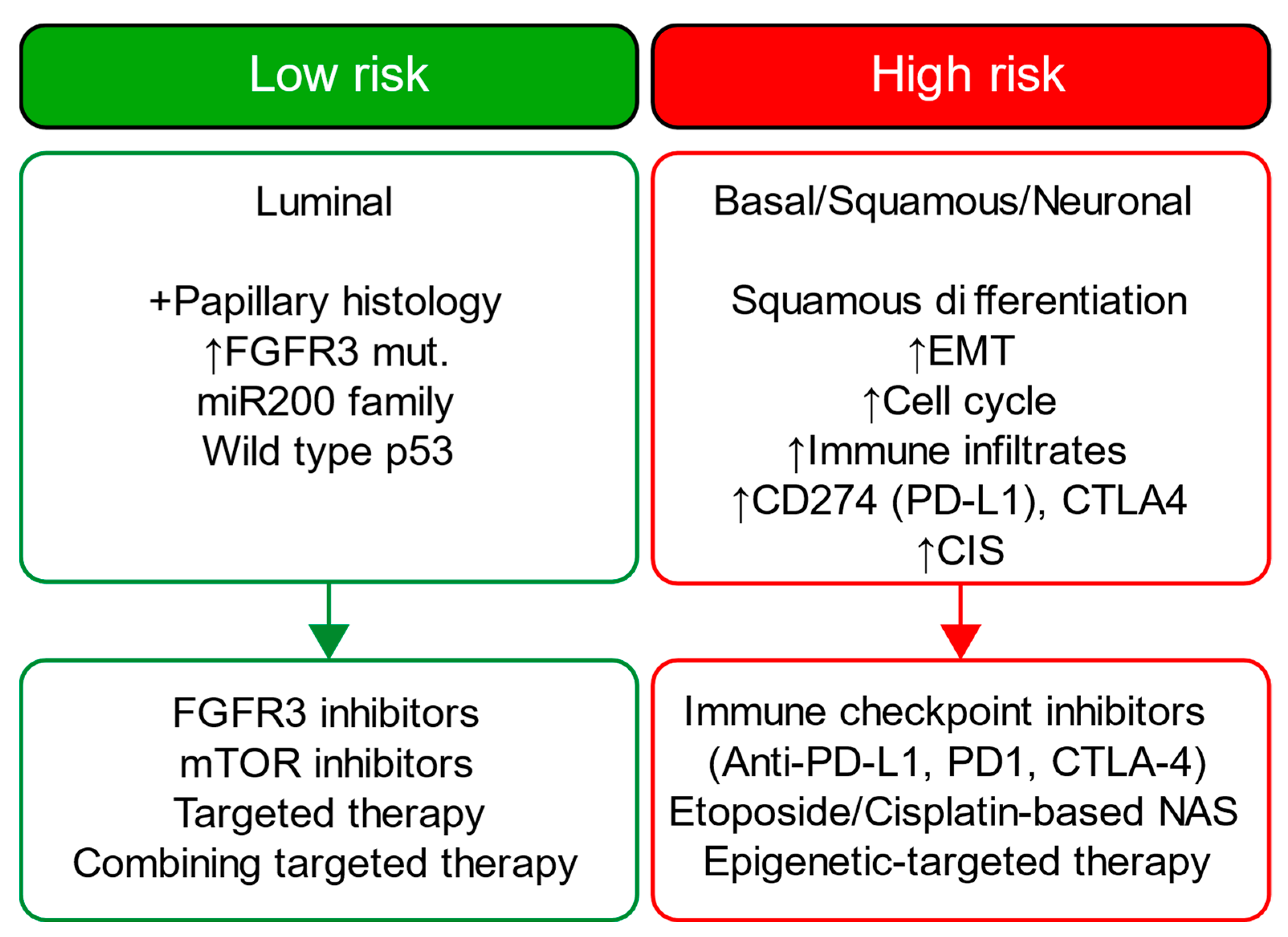
4. Discussion
Challenges and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lobo, N.; Afferi, L.; Moschini, M.; Mostafid, H.; Porten, S.; Psutka, S.P.; Gupta, S.; Smith, A.B.; Williams, S.B.; Lotan, Y. Epidemiology, Screening, and Prevention of Bladder Cancer. Eur. Urol. Oncol. 2022, 5, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J. GLOBOCAN 2008 v1. 2, Cancer Incidence and Mortality World-Wide: IARC Cancer Base No. 10. Available online: https://gco.iarc.fr (accessed on 10 May 2023).
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Echle, A.; Rindtorff, N.T.; Brinker, T.J.; Luedde, T.; Pearson, A.T.; Kather, J.N. Deep learning in cancer pathology: A new generation of clinical biomarkers. Br. J. Cancer 2021, 124, 686–696. [Google Scholar] [PubMed]
- Lafarge, M.W.; Koelzer, V.H. Towards computationally efficient prediction of molecular signatures from routine histology images. Lancet Digit. Health 2021, 3, e752–e753. [Google Scholar] [CrossRef]
- Nayak, T.; Chadaga, K.; Sampathila, N.; Mayrose, H.; Gokulkrishnan, N.; Bairy, G.M.; Prabhu, S.; Swathi, K.S.; Umakanth, S. Deep learning based detection of monkeypox virus using skin lesion images. Med. Nov. Technol. Devices 2023, 18, 100243. [Google Scholar] [CrossRef]
- Woerl, A.C.; Eckstein, M.; Geiger, J.; Wagner, D.C.; Daher, T.; Stenzel, P.; Fernandez, A.; Hartmann, A.; Wand, M.; Roth, W.; et al. Deep Learning Predicts Molecular Subtype of Muscle-invasive Bladder Cancer from Conventional Histopathological Slides. Eur. Urol. 2020, 78, 256–264. [Google Scholar] [CrossRef]
- Mundhada, A.; Sundaram, S.; Swaminathan, R.; D’Cruze, L.; Govindarajan, S.; Makaram, N. Differentiation of urothelial carcinoma in histopathology images using deep learning and visualization. J. Pathol. Inform. 2023, 14, 100155. [Google Scholar] [CrossRef]
- Zheng, Q.; Yang, R.; Ni, X.; Yang, S.; Xiong, L.; Yan, D.; Xia, L.; Yuan, J.; Wang, J.; Jiao, P.; et al. Accurate Diagnosis and Survival Prediction of Bladder Cancer Using Deep Learning on Histological Slides. Cancers 2022, 14, 5807. [Google Scholar] [CrossRef]
- Nguyen, A.; Yosinski, J.; Clune, J. Deep neural networks are easily fooled: High confidence predictions for unrecognizable images. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Boston, MA, USA, 7–12 June 2015; pp. 427–436. [Google Scholar]
- Guo, C.; Pleiss, G.; Sun, Y.; Weinberger, K.Q. On calibration of modern neural networks. In Proceedings of the International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 1321–1330. [Google Scholar]
- Team, P.P.; Gohagan, J.K.; Prorok, P.C.; Hayes, R.B.; Kramer, B.-S. The prostate, lung, colorectal and ovarian (PLCO) cancer screening trial of the National Cancer Institute: History, organization, and status. Control. Clin. Trials 2000, 21, 251S–272S. [Google Scholar]
- Andriole, G.L.; Crawford, E.D.; Grubb III, R.L.; Buys, S.S.; Chia, D.; Church, T.R.; Fouad, M.N.; Gelmann, E.P.; Kvale, P.A.; Reding, D.J. Mortality results from a randomized prostate-cancer screening trial. N. Engl. J. Med. 2009, 360, 1310–1319. [Google Scholar] [CrossRef]
- Hasson, M.A.; Fagerstrom, R.M.; Kahane, D.C.; Walsh, J.H.; Myers, M.H.; Caughman, C.; Wenzel, B.; Haralson, J.C.; Flickinger, L.M.; Turner, L.M.; et al. Design and evolution of the data management systems in the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control. Clin. Trials 2000, 21, 329S–348S. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, P.F.; Prorok, P.C.; Yu, K.; Kramer, B.S.; Black, A.; Gohagan, J.K.; Crawford, E.D.; Grubb, R.L.; Andriole, G.L. Extended mortality results for prostate cancer screening in the PLCO trial with median follow-up of 15 years. Cancer 2017, 123, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Eminaga, O.; Abbas, M.; Shen, J.; Laurie, M.; Brooks, J.D.; Liao, J.C.; Rubin, D.L. PlexusNet: A neural network architectural concept for medical image classification. Comput. Biol. Med. 2023, 154, 106594. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2018, 174, 1033. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [PubMed]
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000, 56, 337–344. [Google Scholar] [CrossRef]
- Heller, G.; Mo, Q. Estimating the concordance probability in a survival analysis with a discrete number of risk groups. Lifetime Data Anal. 2016, 22, 263–279. [Google Scholar] [CrossRef]
- Uno, H.; Cai, T.; Pencina, M.J.; D’Agostino, R.B.; Wei, L.J. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat. Med. 2011, 30, 1105–1117. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Ishiguro, M.; Kitagawa, G. Akaike information criterion statistics. J. Am. Stat. Assoc. 1986, 81, 26853. [Google Scholar]
- Vrieze, S.I. Model selection and psychological theory: A discussion of the differences between the Akaike information criterion (AIC) and the Bayesian information criterion (BIC). Psychol. Methods 2012, 17, 228. [Google Scholar] [CrossRef]
- Neath, A.A.; Cavanaugh, J.E. The Bayesian information criterion: Background, derivation, and applications. Wiley Interdiscip. Rev. Comput. Stat. 2012, 4, 199–203. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Schober, P.; Boer, C.; Schwarte, L.A. Correlation coefficients: Appropriate use and interpretation. Anesth. Analg. 2018, 126, 1763–1768. [Google Scholar] [CrossRef]
- Mukaka, M.M. Statistics corner: A guide to appropriate use of correlation coefficient in medical research. Malawi. Med. J. 2012, 24, 69–71. [Google Scholar] [PubMed]
- Craney, T.A.; Surles, J.G. Model-dependent variance inflation factor cutoff values. Qual. Eng. 2002, 14, 391–403. [Google Scholar]
- Gulli, A.; Pal, S. Deep Learning with Keras; Packt Publishing Ltd.: Birmingham, UK, 2017. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M. Tensorflow: A system for large-scale machine learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation ({OSDI} 16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Fuster, S.; Khoraminia, F.; Kiraz, U.; Kanwal, N.; Kvikstad, V.; Eftestøl, T.; Zuiverloon, T.C.; Janssen, E.A.; Engan, K. Invasive cancerous area detection in Non-Muscle invasive bladder cancer whole slide images. In Proceedings of the 2022 IEEE 14th Image, Video, and Multidimensional Signal Processing Workshop (IVMSP), Nafplio, Greece, 26–29 June 2022; pp. 1–5. [Google Scholar]
- Wenger, K.; Tirdad, K.; Cruz, A.D.; Mari, A.; Basheer, M.; Kuk, C.; van Rhijn, B.W.; Zlotta, A.R.; van der Kwast, T.H.; Sadeghian, A. A semi-supervised learning approach for bladder cancer grading. Mach. Learn. Appl. 2022, 9, 100347. [Google Scholar]
- Zhang, Z.; Chen, P.; McGough, M.; Xing, F.; Wang, C.; Bui, M.; Xie, Y.; Sapkota, M.; Cui, L.; Dhillon, J. Pathologist-level interpretable whole-slide cancer diagnosis with deep learning. Nat. Mach. Intell. 2019, 1, 236–245. [Google Scholar]
- Lucas, M.; Jansen, I.; van Leeuwen, T.G.; Oddens, J.R.; de Bruin, D.M.; Marquering, H.A. Deep Learning-based Recurrence Prediction in Patients with Non-muscle-invasive Bladder Cancer. Eur. Urol. Focus 2022, 8, 165–172. [Google Scholar] [CrossRef]
- Loeffler, C.M.L.; Ortiz Bruechle, N.; Jung, M.; Seillier, L.; Rose, M.; Laleh, N.G.; Knuechel, R.; Brinker, T.J.; Trautwein, C.; Gaisa, N.T.; et al. Artificial Intelligence-based Detection of FGFR3 Mutational Status Directly from Routine Histology in Bladder Cancer: A Possible Preselection for Molecular Testing? Eur. Urol. Focus 2022, 8, 472–479. [Google Scholar] [CrossRef]
- Yu, K.-H.; Berry, G.J.; Rubin, D.L.; Re, C.; Altman, R.B.; Snyder, M. Association of omics features with histopathology patterns in lung adenocarcinoma. Cell Syst. 2017, 5, 620–627.e3. [Google Scholar]
- Kather, J.N.; Heij, L.R.; Grabsch, H.I.; Loeffler, C.; Echle, A.; Muti, H.S.; Krause, J.; Niehues, J.M.; Sommer, K.A.; Bankhead, P. Pan-cancer image-based detection of clinically actionable genetic alterations. Nat. Cancer 2020, 1, 789–799. [Google Scholar] [PubMed]
- Sirinukunwattana, K.; Domingo, E.; Richman, S.D.; Redmond, K.L.; Blake, A.; Verrill, C.; Leedham, S.J.; Chatzipli, A.; Hardy, C.; Whalley, C.M. Image-based consensus molecular subtype (imCMS) classification of colorectal cancer using deep learning. Gut 2021, 70, 544–554. [Google Scholar] [PubMed]
- Coudray, N.; Tsirigos, A. Deep learning links histology, molecular signatures and prognosis in cancer. Nat. Cancer 2020, 1, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.A.; Wang, J.K.; Chui, W.F.; Mountain, V.; Gullapally, S.C.; Srinivasan, R.; Mitchell, R.N.; Glass, B.; Hoffman, S.; Rao, S.K. Human-interpretable image features derived from densely mapped cancer pathology slides predict diverse molecular phenotypes. Nat. Commun. 2021, 12, 1–15. [Google Scholar]
- Hong, R.; Liu, W.; DeLair, D.; Razavian, N.; Fenyö, D. Predicting endometrial cancer subtypes and molecular features from histopathology images using multi-resolution deep learning models. Cell Rep. Med. 2021, 2, 100400. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Choudhury, N.J.; Campanile, A.; Antic, T.; Yap, K.L.; Fitzpatrick, C.A.; Wade, J.L., 3rd; Karrison, T.; Stadler, W.M.; Nakamura, Y.; O’Donnell, P.H. Afatinib Activity in Platinum-Refractory Metastatic Urothelial Carcinoma in Patients With ERBB Alterations. J. Clin. Oncol. 2016, 34, 2165–2171. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; Andre, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef]
- Ching, C.B.; Hansel, D.E. Expanding therapeutic targets in bladder cancer: The PI3K/Akt/mTOR pathway. Lab. Investig. 2010, 90, 1406–1414. [Google Scholar] [CrossRef]
- Hurst, C.D.; Alder, O.; Platt, F.M.; Droop, A.; Stead, L.F.; Burns, J.E.; Burghel, G.J.; Jain, S.; Klimczak, L.J.; Lindsay, H.; et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell 2017, 32, 701–715.e707. [Google Scholar] [CrossRef]
- Saporta, A.; Gui, X.; Agrawal, A.; Pareek, A.; Truong, S.Q.; Nguyen, C.D.; Ngo, V.-D.; Seekins, J.; Blankenberg, F.G.; Ng, A.Y. Benchmarking saliency methods for chest X-ray interpretation. Nat. Mach. Intell. 2022, 4, 867–878. [Google Scholar]
- Bokadia, H.; Yang, S.C.H.; Li, Z.; Folke, T.; Shafto, P. Evaluating perceptual and semantic interpretability of saliency methods: A case study of melanoma. Appl. AI Lett. 2022, 3, e77. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Desciak, E.B.; Maloney, M.E. Artifacts in frozen section preparation. Dermatol. Surg. 2000, 26, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Pech, P.; Bergström, K.; Rauschning, W.; Haughton, V.M. Attenuation values, volume changes and artifacts in tissue due to freezing. Acta Radiol. 1987, 28, 779–782. [Google Scholar] [CrossRef]
- Rolls, G.O.; Farmer, N.J.; Hall, J.B. Artifacts in Histological and Cytological Preparations; Leica Microsystems: Wetzlar, Germany, 2008. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Options |
|---|---|
| Block architecture (microarchitecture design) | Inception block (Inception) Residual block (ResNet) Conventional block (VGG) Attention block (soft_att) |
| Width | 2, 4, 6 |
| Depth | 3, 4, 5 |
| Length (pathways) | 2, 3, 4 |
| Junctions (interconnection between pathways) | 1, 2, 3 |
| Global pooling | Average vs. Maximum |
| Addition of transformer | Yes vs. No |
| Training Set | Optimization Set | Validation Set | ||
|---|---|---|---|---|
| Characteristic | N = 26 | N = 6 | N = 81 | p-Value 1 |
| Age at diagnosis in year, median (IQR) | 65.0 (62.2–68.8) | 69.0 (65.8–70.0) | 65.0 (61.0–68.0) | 0.50 |
| Sex, n (%) | 0.13 | |||
| Male | 25 (96%) | 5 (83%) | 64 (79%) | |
| Female | 1 (3.8%) | 1 (17%) | 17 (21%) | |
| WHO Grade 1973, n (%)+ | 0.26 | |||
| G1 | 5 (19%) | 1 (17%) | 15 (19%) | |
| G2 | 10 (38%) | 3 (50%) | 15 (19%) | |
| G3 | 11 (42%) | 2 (33%) | 47 (58%) | |
| Unknown | 0 (0%) | 0 (0%) | 4 (4.9%) | |
| AJCC tumor staging | ||||
| T stage, n (%) | 0.35 | |||
| Ta | 4 (15%) | 0 (0%) | 1 (1.2%) | |
| Tis | 11 (42%) | 5 (83%) | 31 (38%) | |
| T1 | 5 (19%) | 1 (17%) | 19 (23%) | |
| T2 | 3 (12%) | 0 (0%) | 22 (27%) | |
| T3 | 2 (7.7%) | 0 (0%) | 4 (4.9%) | |
| T4 | 0 (0%) | 0 (0%) | 1 (1.2%) | |
| Unknown | 1 (3.8%) | 0 (0%) | 3 (3.7%) | |
| N stage, n (%) | 0.46 | |||
| Nx/N0 | 25 (96%) | 6 (100%) | 74 (91%) | |
| N1 | 0 (0%) | 0 (0%) | 4 (4.9%) | |
| Unknown | 1 (3.8%) | 0 (0%) | 3 (3.7%) | |
| M stage, n (%) | 0.47 | |||
| Mx/M0 | 25 (96%) | 6 (100%) | 75 (93%) | |
| M1 | 0 (0%) | 0 (0%) | 3 (3.7%) | |
| Unknown | 1 (3.8%) | 0 (0%) | 3 (3.7%) | |
| Follow-up duration in months, median (IQR) | 172 (130–201) | 151 (87–192) | 168 (130–197) | 0.80 |
| Cancer-specific death, n (%) | 6 (23%) | 1 (17%) | 25 (31%) | 0.60 |
| Whole-slide images, n (%) | 46 (23.5%) | 8 (4.1%) | 142 (72.4%) | - |
| Patches, n (%) | 26,949 (16.5%) | 7574 (4.6%) | 129,122 (78.9%) | - |
| Variable | HR | 95% CI | z | p |
|---|---|---|---|---|
| Age at diagnosis | 1.03 | (0.96–1.11) | 0.87 | 0.39 |
| Grading (WHO 1973) | ||||
| G1 (ref.) | – | – | – | – |
| G2 | 2.21 | (0.20–24.48) | 0.64 | 0.52 |
| G3 | 11.99 | (1.61–89.21) | 2.43 | 0.02 |
| Unknown | 11.72 | (1.03–133.02) | 1.99 | 0.05 |
| Risk score | 8.39 | (1.53–46.12) | 2.45 | 0.01 |
| Characteristic | N = 412 |
|---|---|
| Age at diagnosis in years, median (IQR) | 68 (60–76) |
| Sex, n (%) | |
| Female | 107 (26%) |
| Male | 305 (74%) |
| pM | |
| M0/x | 398 (97%) |
| M1 | 11 (2.7%) |
| Unknown | 3 (0.7%) |
| pN | |
| N0x | 282 (68%) |
| M1 | 123 (30%) |
| Unknown | 7 (1.7%) |
| pT | |
| T1 | 2 (0.5%) |
| T2 | 112 (27%) |
| T3 | 190 (46%) |
| T4 | 54 (13%) |
| Unknown | 54 (13%) |
| Grade, n (%) | |
| Unknown | 1 (0.2%) |
| High grade | 390 (95%) |
| Low grade | 21 (5.1%) |
| History of non-muscle invasive bladder cancer, n (%) | |
| Unknown | 127 (31%) |
| NO | 227 (55%) |
| YES | 58 (14%) |
| Bladder cancer pathologic stage, n (%) | |
| I–II | 151 (36.7%) |
| III | 130 (31.6%) |
| IV | 130 (31.6%) |
| Unknown | 1 (0.2%) |
| Death, n (%) | 185 (45%) |
| Follow-up duration in month, median (IQR) | 19 (12–33) |
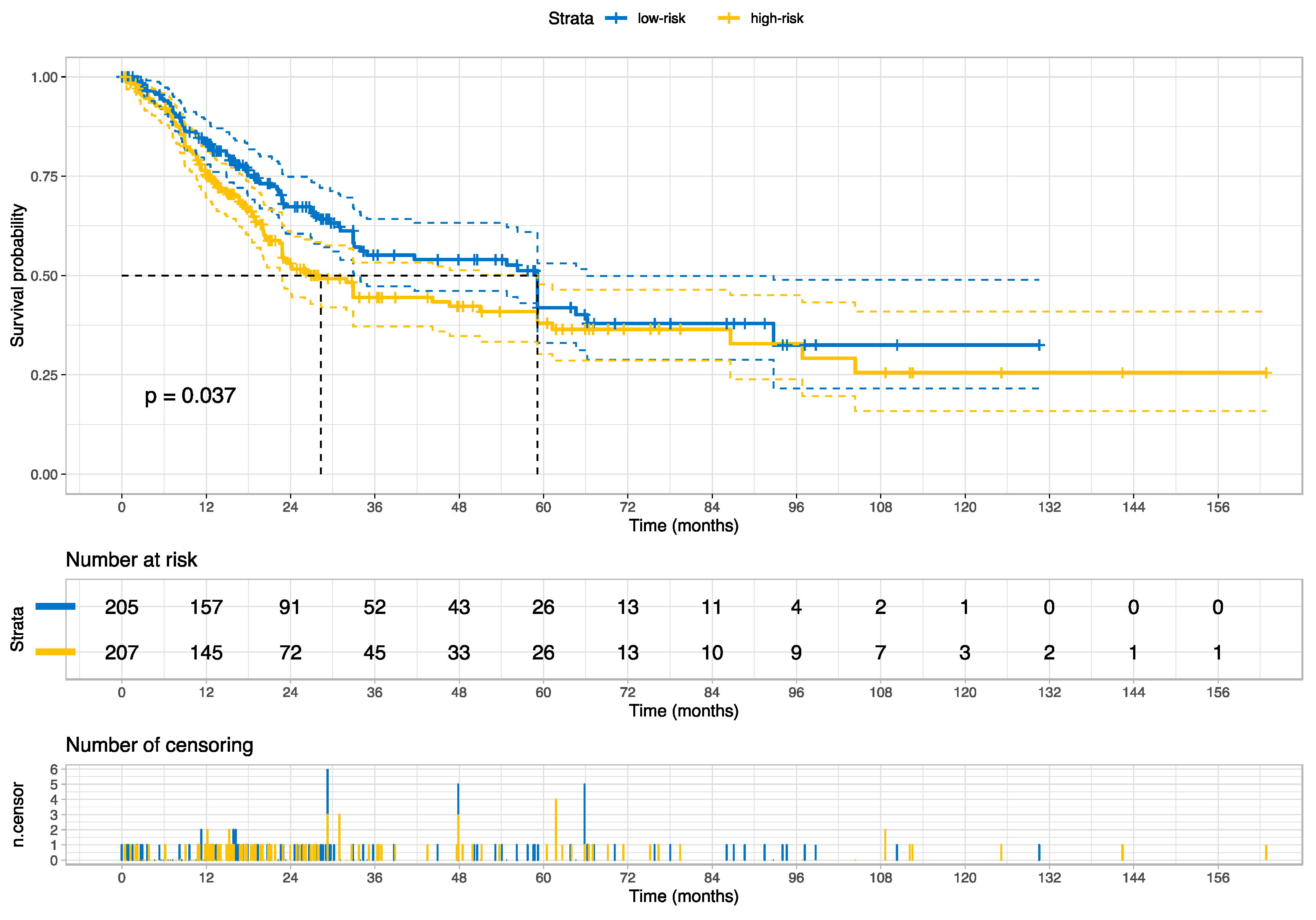
| Variable | HR | 95% CI | z | p |
|---|---|---|---|---|
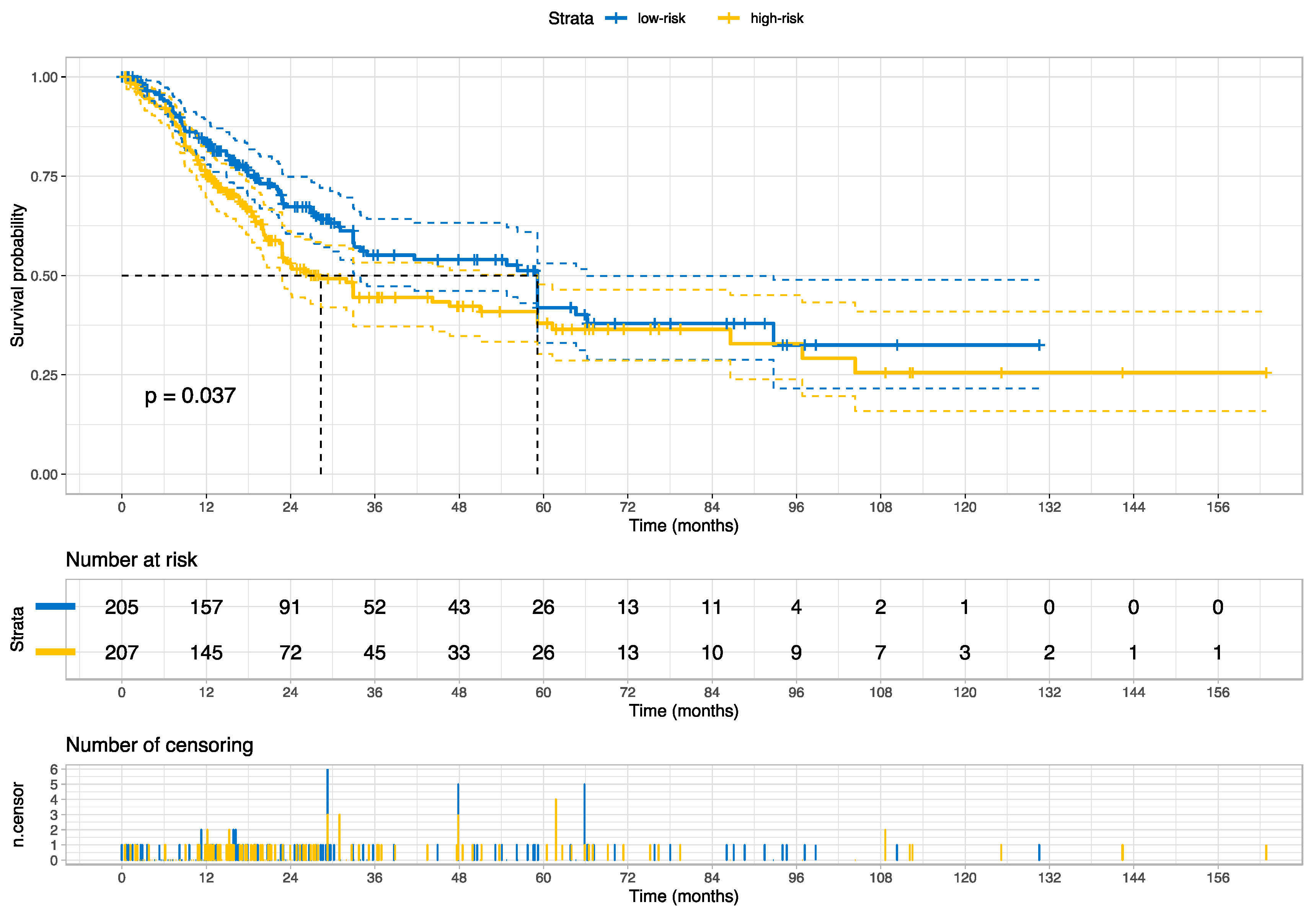
| High- vs. Low-risk group | 1.35 | (1.01–1.80) | 1.99 | 0.0462 |
| Age at diagnosis | 1.02 | (1.00–1.03) | 2.32 | 0.0201 |
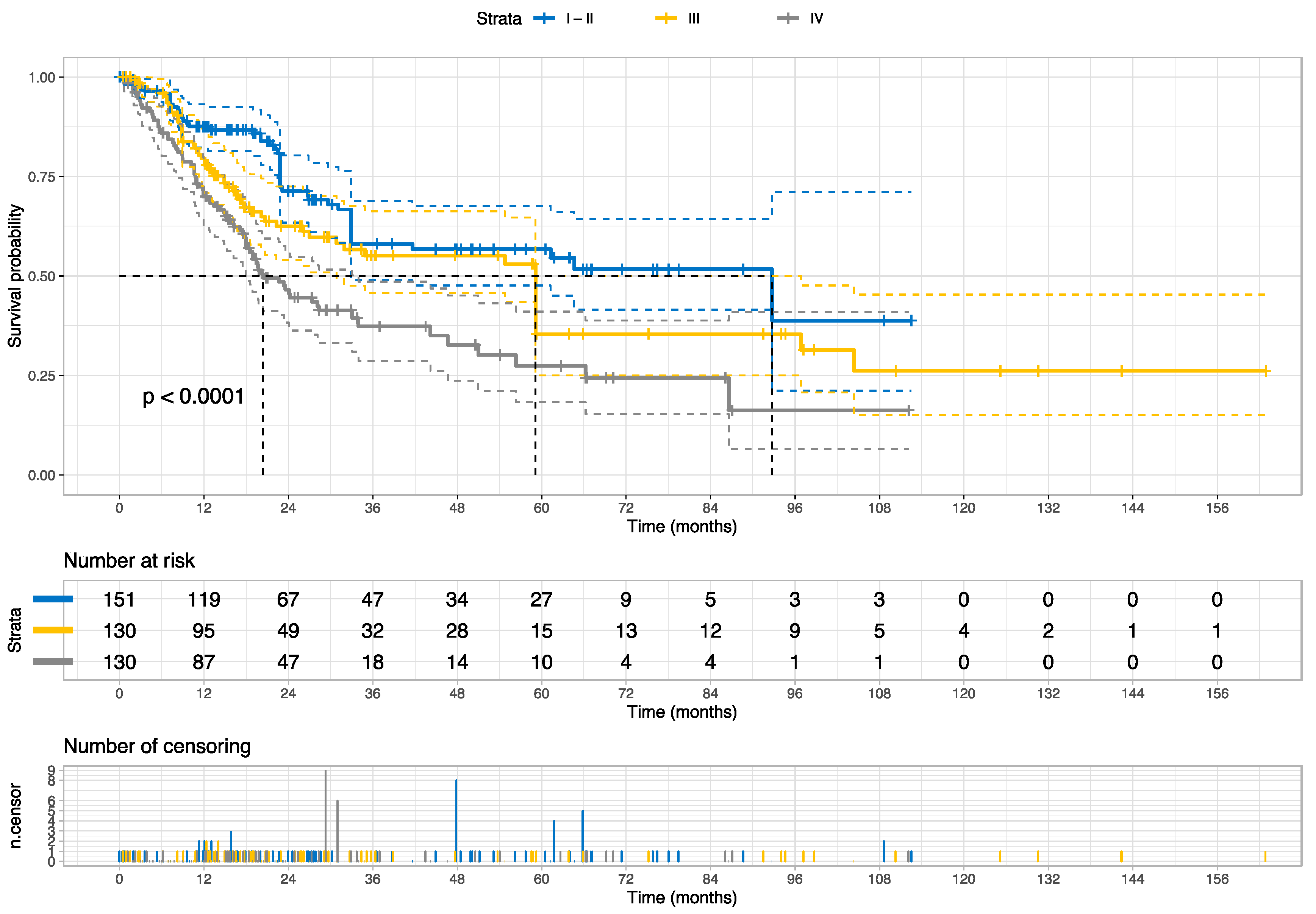
| AJCC pathologic tumor stage | ||||
| I/II (ref.) | – | – | – | – |
| III | 1.51 | (1.03–2.21) | 2.10 | 0.0357 |
| IV | 2.21 | (1.54–3.18) | 4.30 | <0.0001 |
| Signature | p Value | Features Associated with  Low-Risk or Low-Risk or  High-Risk Group High-Risk Group |
|---|---|---|
| microRNA cluster | 0.003998001 | Cluster 3 Cluster 1 |
| mutation in TSC1 | 0.006496752 | TSC1 mutation |
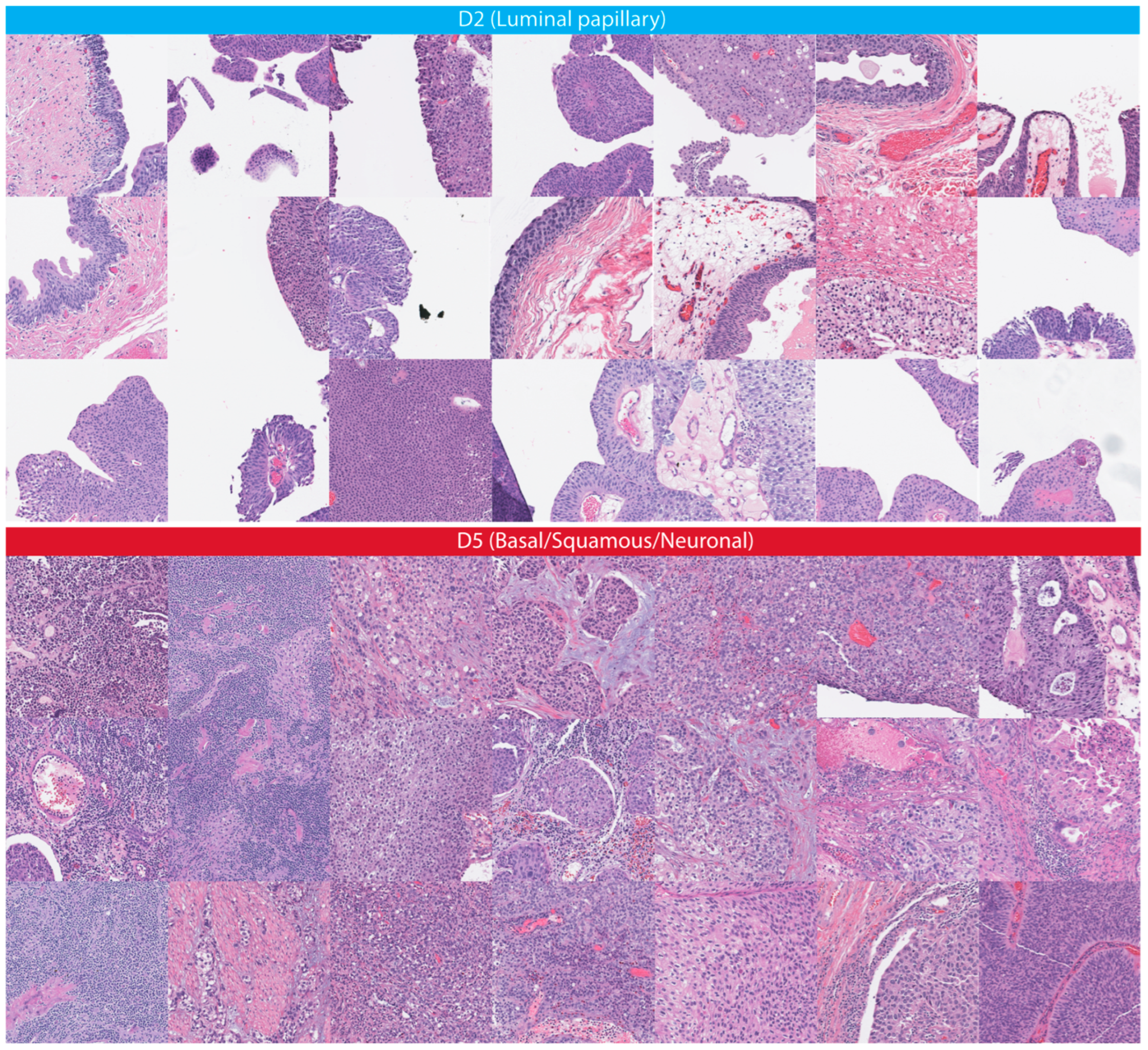
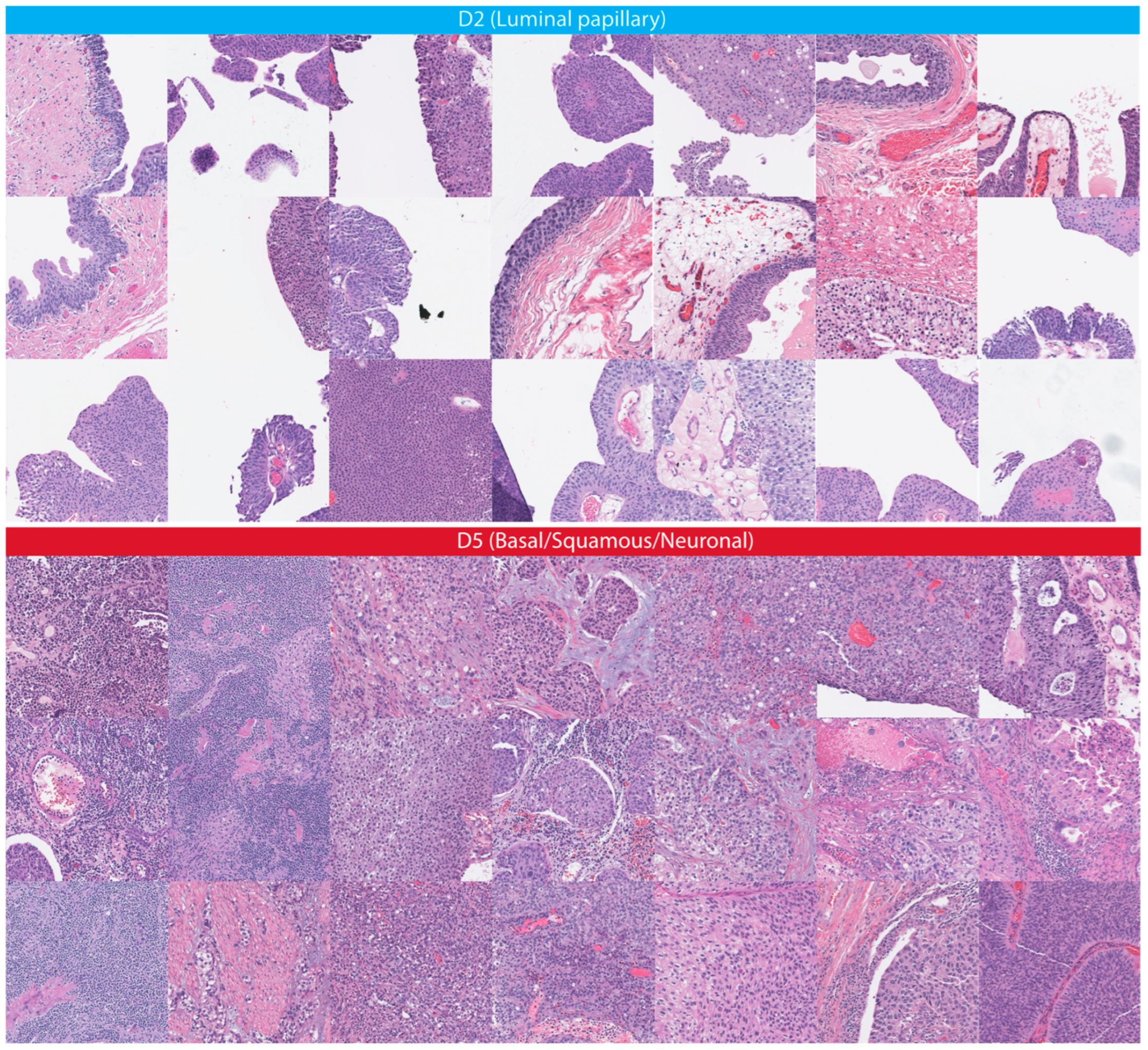
| mRNA cluster | 0.009995002 | Luminal papillary Basal/Squamous Neuronal |
| mutation in FGFR3 | 0.010994503 | FGFR3 mutation |
| lncRNA cluster | 0.012493753 | Cluster 3 Cluster 4 |
| mutation in ERBB3 | 0.016991504 | ERBB3 mutation |
| mutation in FAT1 | 0.023488256 | FAT1 mutation |
| mutation in PIK3CA | 0.028485757 | PIK3CA mutation |
| mutation in KANSL1 | 0.033983008 | KANSL1 mutation |
| mutation in TMCO4 | 0.038480760 | TMCO4 mutation |
| mutation in KDM6A | 0.044977511 | KDM6A mutation |
| mutation in METTL3 | 0.057971014 | METL3 mutation |
| Squamous pathology | 0.066466767 | Squamous histopathology |
| mutation in PSIP1 | 0.075462269 | PSIP1 mutation |
| mutation in ZNF773 | 0.092453773 | ZNF773 mutation |
| Hypomethylation cluster | 0.092953523 | Cluster 4 Cluster 2 |
| mutation in GNA13 | 0.093953023 | GNA13 mutation |
| Risk Groups | Molecular Signatures | |
|---|---|---|
| mRNA | ||
| Luminal papillary | Basal/Squamous/Neuronal | |
| Low-risk | 85 (59%) | 56 (36%) |
| High-risk | 58 (41%) | 101 (64%) |
| lncRNA | ||
| Cluster 3 | Cluster 4 | |
| Low-risk | 47 (64%) | 61 (41%) |
| High-risk | 26 (36%) | 87 (59%) |
| miRNA | ||
| Cluster 3 | Cluster 1 | |
| Low-risk | 77 (62%) | 30 (39%) |
| High-risk | 47 (38%) | 47 (61%) |
| DNA hypomethylation | ||
| Cluster 4 | Cluster 2 | |
| Low-risk | 23 (68%) | 27 (39%) |
| High-risk | 11 (32%) | 42 (61%) |
| TSC1 Gene | ||
|---|---|---|
| Risk groups | wild-type | mutated |
| Low-risk | 177 (47%) | 28 (72%) |
| High-risk | 196 (53%) | 11 (28%) |
| ERBB3 Gene | ||
|---|---|---|
| Risk groups | wild-type | mutated |
| Low-risk | 175 (48%) | 30 (67%) |
| High-risk | 192 (52%) | 15 (33%) |
| FGFR3 Gene | ||
|---|---|---|
| Risk groups | wild-type | mutated |
| Low-risk | 163 (47%) | 42 (65%) |
| High-risk | 184 (53%) | 23 (35%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eminaga, O.; Leyh-Bannurah, S.-R.; Shariat, S.F.; Krabbe, L.-M.; Lau, H.; Xing, L.; Abbas, M. Artificial Intelligence Reveals Distinct Prognostic Subgroups of Muscle-Invasive Bladder Cancer on Histology Images. Cancers 2023, 15, 4998. https://doi.org/10.3390/cancers15204998
Eminaga O, Leyh-Bannurah S-R, Shariat SF, Krabbe L-M, Lau H, Xing L, Abbas M. Artificial Intelligence Reveals Distinct Prognostic Subgroups of Muscle-Invasive Bladder Cancer on Histology Images. Cancers. 2023; 15(20):4998. https://doi.org/10.3390/cancers15204998
Chicago/Turabian StyleEminaga, Okyaz, Sami-Ramzi Leyh-Bannurah, Shahrokh F. Shariat, Laura-Maria Krabbe, Hubert Lau, Lei Xing, and Mahmoud Abbas. 2023. "Artificial Intelligence Reveals Distinct Prognostic Subgroups of Muscle-Invasive Bladder Cancer on Histology Images" Cancers 15, no. 20: 4998. https://doi.org/10.3390/cancers15204998
APA StyleEminaga, O., Leyh-Bannurah, S.-R., Shariat, S. F., Krabbe, L.-M., Lau, H., Xing, L., & Abbas, M. (2023). Artificial Intelligence Reveals Distinct Prognostic Subgroups of Muscle-Invasive Bladder Cancer on Histology Images. Cancers, 15(20), 4998. https://doi.org/10.3390/cancers15204998