Simple Summary
Many aspects of the regulatory mechanisms of somatic cell reprogramming—the conversion of any cell type into pluripotent stem cells—still remain elusive. The tumor suppressor CYLD regulates several signaling pathways involved in this process. However, its potential role in reprogramming has not been investigated. In this work, we present evidence that CYLD exerts important regulatory control at the early stages of reprogramming. Loss of CYLD catalytic activity leads to the reduced reprogramming efficiency of mouse embryonic fibroblasts. Whole proteome analysis during early reprogramming stages revealed that CYLD DUB deficiency impedes a vital early reprogramming step known as the mesenchymal-to-epithelial transition (MET). Our findings expand our knowledge of early reprogramming mechanics and reveal a novel role for CYLD as an extracellular matrix regulator.
Abstract
CYLD is a tumor suppressor gene coding for a deubiquitinating enzyme that has a critical regulatory function in a variety of signaling pathways and biological processes involved in cancer development and progression, many of which are also key modulators of somatic cell reprogramming. Nevertheless, the potential role of CYLD in this process has not been studied. With the dual aim of investigating the involvement of CYLD in reprogramming and developing a better understanding of the intricate regulatory system governing this process, we reprogrammed control (CYLDWT/WT) and CYLD DUB-deficient (CYLDΔ9/Δ9) mouse embryonic fibroblasts (MEFs) into induced pluripotent stem cells (iPSCs) through ectopic overexpression of the Yamanaka factors (Oct3/4, Sox2, Klf4, c-myc). CYLD DUB deficiency led to significantly reduced reprogramming efficiency and slower early reprogramming kinetics. The introduction of WT CYLD to CYLDΔ9/Δ9 MEFs rescued the phenotype. Nevertheless, CYLD DUB-deficient cells were capable of establishing induced pluripotent colonies with full spontaneous differentiation potential of the three germ layers. Whole proteome analysis (Data are available via ProteomeXchange with identifier PXD044220) revealed that the mesenchymal-to-epithelial transition (MET) during the early reprogramming stages was disrupted in CYLDΔ9/Δ9 MEFs. Interestingly, differentially enriched pathways revealed that the primary processes affected by CYLD DUB deficiency were associated with the organization of the extracellular matrix and several metabolic pathways. Our findings not only establish for the first time CYLD’s significance as a regulatory component of early reprogramming but also highlight its role as an extracellular matrix regulator, which has profound implications in cancer research.
Keywords:
CYLD; somatic cell reprogramming; MET; EMT; ECM organization; TGF-beta; pluripotency; dedifferentiation 1. Introduction
Somatic cell reprogramming is a process by which somatic cells can be transformed into pluripotent stem cells [1]. Although many methodologies have been developed to achieve this goal, such as chemical reprogramming [2,3,4], plasmid-based [5], mRNA-based [6], direct delivery [7] and piggybac transposition [8], the most widespread and well-studied is the virus-delivered, transgenic overexpression of OCT3/4, SOX2, KLF4 and c-myc, also known as Yamanaka factors, which can be applied in numerous cutting-edge fields [9], such as disease modeling [10,11,12], rejuvenation [13] and tissue regeneration [14,15,16]. Despite the significant strides made in understanding the mechanisms involved in this process, reprogramming remains a remarkably complex phenomenon, with a large number of heterogeneous factors regulating both reprogramming progression and efficiency [17,18,19,20]. Just a few examples include the implication of numerous independently acting genes [21,22,23,24,25], small molecules [26,27,28,29], most studied signaling pathways [30,31,32,33,34,35,36,37,38], epigenetic modifications [39,40,41,42,43,44] and micro-RNAs [45,46,47,48]. Understanding the involvement of each of these heterogeneous mechanisms and then combining them is imperative to fully decipher the regulatory network of reprogramming.
Several pieces of evidence indicate that somatic cell reprogramming resembles the process of carcinogenesis. The Yamanaka factors are known oncogenes implicated in various forms of cancer and also associated with poor prognosis [49,50,51,52,53,54,55,56,57,58,59]. In addition, tumor suppressor genes, such as p53 and p21, are reprogramming barriers, and loss of their activity can dramatically increase reprogramming efficiency [60,61,62]. Moreover, many changes observed during reprogramming, such as the inhibition of apoptosis and senescence, increased proliferation rates, epithelial-to-mesenchymal transition and the Warburg effect, are well-established and common processes in tumor development [20,63,64]. Furthermore, during early reprogramming, cells undergo drastic morphological changes [65], such as the loss of somatic cell identity and the establishment of new intercellular contacts [66,67], phenomena also observed in tumorigenesis [68,69]. Based on the above, it has been suggested that reprogramming can be used not only as a model for the study of numerous cancer-related processes, including the EMT [64], development and differentiation of cancer stem cells [70,71,72] and oncogenic transformation [73,74,75,76,77], but also for the discovery of novel diagnostic and therapeutic targets [72,78,79].
CYLD is a 60 kb long gene encoding a 956-amino-acid protein (CYLD) that functions primarily as a deubiquitinating enzyme [80]. The contribution of CYLD to the regulation of NF-κB activity has been extensively studied [80,81], where it appears to act as both a K-63 and M-1 deubiquitinase [82,83,84]. Furthermore, CYLD has been shown to have a regulatory role in other signaling pathways, including TGF-beta [85,86,87], Wnt-beta catenin [88,89], Hippo [85,90], JNK [91,92,93], ERK [94] and Akt [95,96,97]. Regarding its biological role, CYLD has been studied mainly in terms of its role in carcinogenesis. It has been shown to be a tumor suppressor gene, as both a decrease in its expression and loss of function have been associated with adverse effects in many cancers [85,87,88,91,92,93,98,99,100,101]. Despite that in most cases the molecular basis of CYLD involvement remains elusive, in some types of cancer a relevant molecular mechanism has been identified. In breast cancer and head and neck squamous cell carcinoma (HNSCC), CYLD DUB deficiency leads to TGF-beta pathway activation and the promotion of epithelial-to-mesenchymal transition (EMT) [85,87]. On the other hand, the role of CYLD in multiple myeloma depends mainly on the regulation of WNT, as the loss of CYLD leads to pathway hyperactivation and cancer aggressiveness [88]. The regulation of NF-κB by CYLD plays a critical role in lung cancer [101,102], B-cell receptor-dependent lymphomas [103], glioblastoma [104] and pulmonary adenocarcinoma [105], among others.
CYLD may also have a regulatory role in cellular reprogramming. As already mentioned, CYLD is a tumor suppressor gene and modulates several pathways crucial in reprogramming progression, most notably TGF-beta, Hippo and Wnt. Moreover, in some cases, CYLD DUB deficiency has been associated with cancer stemness [104,106,107]. In this study, we reveal for the first time the regulatory role of CYLD in somatic cell reprogramming. CYLD DUB deficiency leads to a decrease in reprogramming efficiency, without affecting the pluripotency potential of induced stem cells and the in vitro spontaneous differentiation ability. CYLD DUB-deficient cells are characterized by the slower induction of pluripotency and expression of epithelial markers and deactivation of mesenchymal genes, particularly those related to the organization of the extracellular matrix (ECM). Taken together, we propose that CYLD is a key regulator of successful early reprogramming development.
2. Materials and Methods
2.1. Generation of Lentiviral Particles for Somatic Cell Reprogramming
The reprogramming protocol used is based on the co-expression of the OSKM reprogramming cassette using the polycistronic vector TetO-FUW-OSKM (TetO-FUW-OSKM was a gift from Rudolf Jaenisch (Addgene plasmid # 20321; https://www.addgene.org/20321/, accessed on 14 October 2023)). The reprogramming cassette is doxycycline-inducible, based on the tet-on system, and its expression is controlled by the M2rtTA protein. As an M2rtTA vector, we used FUW-M2rtTA (FUW-M2rtTA was a gift from Rudolf Jaenisch (Addgene plasmid #20342; https://www.addgene.org/20342/, accessed on 14 October 2023)). Standard HBS calcium phosphate transfection was used for the production of lentiviral particles in human embryonic kidney cells 293T (HEK293T). The plasmids pMD2.G (pMD2.G was a gift from Didier Trono (Addgene plasmid # 12259; https://www.addgene.org/12259/, accessed on 14 October 2023)) and psPAX2 (psPAX2 was a gift from Didier Trono (Addgene plasmid # 12260; https://www.addgene.org/12260/, accessed on 14 October 2023)) were utilized as envelope and packaging vectors, respectively. Briefly, the culture medium was changed 4 h before transfection. The transfection cocktail consisted of 50% HBS 2X, 5% CaCl2 2 M, 10 μg of packaging and envelope plasmids and 20 μg of transfer plasmids. Three days after the medium change in transfected HEK293T, lentivirus-containing supernatants were collected for further use.
2.2. Isolation of Mouse Embryonic Fibroblasts (MEFs)
MEFs were generated as previously described [108]. CYLDWT/WT (WT) and CYLDΔ9/Δ9 (Δ9) MEFs were used for somatic cell reprogramming experiments, whereas CYLDWT/Δ9 MEFs were utilized for feeder cell preparation. Initial experiments were optimized with matching genetic background feeders and CYLDWT/Δ9 cells. No difference in any experimental outcome was observed, so the scaled-up experiments were performed using only heterozygote MEF feeder cells, as we could reduce the amount of animal use also. Three independent MEF cell lines were established, two of them derived from littermate embryos of the same crossing (crossing b2, cell lines b2_1 and b2_2) and the third one from littermate embryos of a different crossing (crossing b3, cell lines b3). All subsequent experiments (reprogramming efficiency/kinetics/whole proteome analysis, pluripotency maintenance, in vitro spontaneous differentiation) were conducted using the three biological replicates described above. Finally, each biological replicate was analyzed in at least three technical replicates for each experiment presented.
2.3. Genotyping
Genotyping of animals and cells was conducted according to the protocol described by Trompouki et al. [108] and Karatzas et al. [109]. Briefly, genomic DNA from selected samples was isolated by using the PureLink™ Genomic DNA Mini Kit (Invitrogen™, Waltham, MA, USA). The following PCR primers were used to characterize the CYLD locus: FWD1: 5 -GATGGCTCTTGTCACCACTT-3′, F6: 5′-CGTGAACAGATGTGATGAAGGC-3′ and R6: 5′-CTACCATCCCTGCTAACCAC-3′.
2.4. Production of Pinducer20 C601S and Pinducer21 FCYLD
Construction of the lentiviral expression vectors pINDUCER20 FLAGCYLDC601S and pINDUCER21 FLAGCYLDWT was performed using the Gateway protocol. In more detail, pENTR1A FLAGCYLDWT and pENTR1A FLAGCYLDC601S inserts flanked by the attL1 and attL2 regions (previously constructed in our lab) were transferred between the attR1 and attR2 regions of each of the two target vectors pINDUCER20 (Addgene plasmid #44012, https://www.addgene.org/44012/, accessed on 14 October 2023) and pINDUCER21 (Addgene plasmid #46948, https://www.addgene.org/46948/, accessed on 14 October 2023). This transfer was achieved by the LR recombination reaction using the LR Clonase II enzyme mixture (Invitrogen™, Waltham, MA, USA).
For the construction of pENTR1A FCYLDWT and pENTR1A FCYLD C601S, Gateway™ cloning (Invitrogen™, Waltham, MA, USA) was utilized. More specifically, the pENTR™ 1A Dual Selection Vector (Invitrogen, A10462) was cleaved with the enzymes KpnI (Fermentas, Waltham, MA, USA) and XhoI (Fermentas, Waltham, MA, USA). The enzyme reaction products were dephosphorylated using SAP (Fermentas, Waltham, MA, USA). Subsequently, a ligation reaction between FLAGCYLDWT, FLAGCYLDC601S and the empty pENTR1A vector was performed using T4 Ligase (Fermentas, Waltham, MA, USA) with a molar ratio of linear vector to linear insert of 1:1.
2.5. Western Blot
Cell lysis was performed using NP-40 buffer (50 mM Tris-Cl, pH 8.0, 150 mM NaCl and 1% NP-40) supplemented with a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Protein denaturation was achieved after diluting (1:1) the samples with 2× Laemmli Sample Buffer (Bio-Rad Laboratories, Hercules, CA, USA), followed by heating at 95 °C for 5 min. The samples were analyzed with SDS-PAGE, and proteins were electrophoretically transferred to PVDF membrane for Western blot analysis. Membranes were treated with blocking solution, consisting of 5% non-fat dry milk in PBST, for 1 h at RT. For the detection of CYLD, we used the cylindromatosis 1 antibody (E-4, sc-74434, Santa Cruz Biotechnology, Dallas, TX, USA), and for b-actin, we used the beta actin antibody (C-4, Santa-Cruz). As a secondary antibody, we used m-IgGκ BP-HRP (sc-516102, Santa Cruz Biotechnology, Dallas, TX, USA) for both CYLD and b-actin. In all cases, the antibodies were diluted in blocking solution, and the membrane was treated for 1 h at RT. Membrane-bound antibodies were detected with an enhanced chemiluminescence detection kit (Pierce, Waltham, MA, USA) using a Typhoon FLA 7000 imaging system (GE Healthcare Life Sciences, Chicago, IL, USA). The E-4 CYLD antibody interacts with the C-terminal 419 amino acids of human CYLD.
2.6. Cell Culture
MEFs were cultured on plates treated with a 0.1% gelatin solution (20 min on 37 °C). MEF culturing medium consisted of high-glucose DMEM, 10% FBS, 1X NEAA, 1X penicillin–streptomycin, 1X GlutaMAXTM supplement (Gibco-Invitrogen, Waltham, MA, USA) and 0.1 mM β-mercaptoethanol. Induced pluripotent stem cells (iPSCs) were cultured on feeder cells plated onto gelatin-coated plates. iPSC medium consisted of high-glucose DMEM (Gibco-Invitrogen, Waltham, MA, USA), 15% KnockOut™ Serum Replacement (Gibco-Invitrogen, Waltham, MA, USA), 1X NEAA (Gibco-Invitrogen, Waltham, MA, USA), 1X penicillin–streptomycin (Gibco-Invitrogen, Waltham, MA, USA), 1X GlutaMAXTM supplement (Gibco-Invitrogen, Waltham, MA, USA), 0.1 mM β-mercaptoethanol (Gibco-Invitrogen, Waltham, MA, USA) and 10 ng/mL mouse leukemia inhibitory factor (LIF Recombinant Mouse Protein, embryonic stem cell-qualified, Gibco-Invitrogen, Waltham, MA, USA). All cell types used were tested for mycoplasma.
2.7. Feeder Cell Preparation
CYLDWT/Δ9 MEFs were treated with a mitomycin C (Sigma-Aldrich, St. Louis, MO, USA) solution (10 μg/mL in DMEM) for 2 h at 37 °C. Cells were subsequently washed with PBS and immediately trypsinized and cryopreserved for future use.
2.8. Somatic Cell Reprogramming
Reprogramming was conducted according to Papathanasiou et al. [110]. The day before infection, MEFs were passaged at a ratio of 5 × 105 cells/p100 plate. The infection cocktail was prepared with a ratio of 3 mL OSKM virus + 3 mL M2rtTA virus + 2 mL MEF medium/p100 plate. Cells were cultured for 24 h with the infection cocktail, which was then replaced with MEF medium. The next day, a DOX-containing medium (2 μg/mL) was added to the cells, which initiated the reprogramming process (D0). On day 6, early iPSC colonies were dissociated with accutase and plated on wells with a ratio of 105 cells/p100 plate and cultured with DOX-containing (2 μg/mL) iPSC medium. Cells were cultured until day 18, when reprogramming efficiency was calculated via ALP staining.
Apart from the three MEF cell lines (mentioned in Section 2.2), two additional derivative cell lines were established using the three separate Δ9 MEF isolates. For the first one, we inserted into Δ9 MEFs a plasmid expressing the WT CYLD (from now on termed Δ9 FCYLD), and for the second one, we inserted a plasmid expressing another catalytically inactive form of CYLD known as C601S (from now on termed Δ9 C601S). The difference between C601S and Δ9 is that although both are catalytically inactive, the former is a full-length protein, rendered inactive due to the presence of a single point mutation in the catalytic center of the protein, whereas the latter is a truncated CYLD variant lacking its carboxyl terminus, including the catalytic center. In both Δ9 FCYLD and Δ9 C601S lines, the expression of the CYLD transgene is doxycycline-inducible. At the start of reprogramming, every cell line used was of similar passage. Moreover, we ensured the Yamanaka reprogramming cassette was expressed in comparable levels in each sample (Figure S1).
2.9. ALP Staining
On day 18 of reprogramming, the medium was removed, and cells were fixed with a 4% PFA solution for 10 min at 4 °C. After washing with PBS (1X), cells were treated with the ALP (Alkaline Phosphatase) staining solution, containing 200 μL NBT (nitro-blue tetrazolium chloride)/BCIP (5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt) stock solution (Sigma-Aldrich, St. Louis, MO, USA) in 10 mL of NTMT (NaCl, Tris-Cl, MgCl2, Tween-20) buffer in the dark for 10 min. Reprogramming efficiency was calculated as the number of ALP-stained colonies/number of cells plated onto feeders × 100.
2.10. Colony Harvesting
Using a sharp, glass Pasteur pipette, an incision was made along the border of the iPSC colony in order to separate it from the surrounding cells. The colony was then collected with a Gilson PIPETMAN (Gilson, Madison, WI, USA) and placed in medium-containing collection tubes on ice. Only colonies from the same plate were pooled together and chosen based on their phenotypic characteristics. Once enough colonies were collected, the total sample was flash frozen and stored for future use (−80 °C). A representative example can be found in Figure S2.
2.11. RNA Extraction, cDNA Synthesis and Quantitative Real-Time PCR (qPCR)
RNA extraction was conducted using the NucleoSpin RNA Plus RNA purification kit (Macherey-Nagel, Düren, Germany). RNA samples were stored at −80 °C, and RNA integrity was tested with bleach agarose gel [111]. cDNA was synthesized using the PrimeScript™ 1st strand cDNA Synthesis Kit (Takara Bio USA, San Jose, CA, USA). Real-time quantitative PCRs (qPCRs) were performed in duplicates using 5 ng of cDNA per reaction amplified by SYBR FAST qPCR Master Mix (KK4602, Kapa Biosystems, Wilmington, MA, USA) in a StepOne™ Real-Time PCR System (Applied Biosystems™, Warrington, UK) and a 7500 Fast Dx Real-Time PCR Instrument (Applied Biosystems™, Warrington, UK). CYLD qPCR primers recognize a region from exon 2 to exon 3 (spanning from 645 to 753, transcript NM_173369.3). The complete list of primer sequences can be found in Table S1.
2.12. Immunofluorescence
Cells were fixed with a 4% PFA solution for 10 min at room temperature. Samples were subsequently treated with a permeabilization solution consisting of 0.25% PBS/Triton-X 100 for 10 min and a blocking solution consisting of 1% BSA in PBST (PBS + 0.1% Tween 20, Sigma-Aldrich, St. Louis, MO, USA) for 30 min to block unspecific binding of the antibodies. Samples were incubated with the primary antibody in blocking solution for 1 h at RT and the secondary antibody in blocking solution for 1 h at RT in the dark. Finally, cells were incubated with DAPI solution (1 μg/mL) for 1 min and visualized using a Zeiss LSM 780 confocal microscope (Zeiss, Oberkochen, Germany). The following primary antibodies were used: OCT3/4 (H-134, sc-9081, Santa Cruz Biotechnology, Dallas, TX, USA), NANOG (C-4, sc-376915, Santa Cruz Biotechnology, Dallas, TX, USA) and LIN28 (AF3757, R&D Systems, Minneapolis, MN, USA). As a secondary antibody, Alexa Fluor™ 555 Highly Cross-Adsorbed Secondary Antibody (Invitrogen™, Waltham, MA, USA) was used. More specifically, Donkey anti-Rabbit for OCT3/4, Donkey anti-Mouse for NANOG and Donkey anti-Goat for LIN28.
2.13. In Vitro Spontaneous Differentiation Assay
The differentiation potential of the produced WT and Δ9 iPSC cell lines was assessed via the formation of Embryoid Bodies (EBs) and their in vitro spontaneous differentiation, as described by Liao et al. [112]. Briefly, iPSCs were dissociated with accutase and cultured into low-adhesion tissue culture plates with LIF-free iPSC medium. Embryoid Bodies (EBs) were formed after 3 days of suspension culture conditions. EBs were then trypsinized, and a single-cell suspension was plated in gelatin-coated wells and cultured in MEF medium for 14 days. Samples were harvested on days 0, 1, 2, 3, 4, 7, 10 and 14 of the differentiation procedure.
2.14. Proteomics
Samples harvested as described in Section 2.10 were lysed with FASP buffer (0.1 M Tris-HCl pH 7.6, 4% SDS, 0.1 M DTT). Supernatants were collected and stored at −20 °C. The protein extracts were processed by tryptic digestion using the Sp3 protocol, including an alkylation step in 100 mM iodoacetamide (Acros Organics/Thermo Scientific Chemicals, Waltham, MA, USA). Then, 20 μg of beads (1:1 mixture of hydrophilic and hydrophobic SeraMag carboxylate-modified beads, Cytiva Life Sciences, Marlborough, MA, USA) was added to each sample in 50% ethanol. Protein clean-up was performed on a magnetic rack. The beads were washed two times with 80% ethanol and once with 100% acetonitrile (Fisher Scientific, Waltham, MA, USA). The proteins captured on beads were digested overnight at 37 °C under vigorous shaking (1200 rpm, Eppendorf Thermomixer, Eppendorf, Hamburg, Germany) with 1 μg Trypsin/LysC (MS grade, Promega) prepared in 25 mM ammonium bicarbonate. The next day, the peptides were purified using a modified Sp3 clean-up protocol, solubilized in the mobile phase A (0.1% Formic acid in water) and sonicated. The peptide concentration was determined through an absorbance at 280 nm measurement using NanoPhotometer® P330 (Implen, Westlake Village, CA, USA).
Samples were analyzed with a liquid chromatography–tandem mass spectrometry (LC-MS/MS) setup consisting of a Dionex Ultimate 3000 nanoRSLC (Thermo-Fisher Scientific, Waltham, MA, USA), coupled in line with a Thermo Q Exactive HF-X Orbitrap mass spectrometer (Thermo-Fisher Scientific, Waltham, MA, USA). Peptidic samples were directly injected and separated on a 25 cm long analytical C18 column (PepSep, 1.9 μm 3 beads, 75 µm ID) using a one-hour-long run, starting with a gradient of 7% Buffer B (0.1% Formic acid in 80% acetonitrile) to 35% for 40 min and followed by an increase to 45% in 5 min and a second increase to 99% in 0.5 min and then kept constant for equilibration for 14.5 min. A full MS was acquired in profile mode using a Q Exactive HF-X Hybrid Quadrupole-Orbitrap mass spectrometer, operating in the scan range of 375–1400 m/z using 120 K resolving power with an AGC of 3 × 106 and maximum IT of 60 ms followed by a data-independent acquisition method using 8 Th windows (a total of 39 loop counts), each with 15 K resolving power with an AGC of 3 × 105 and max IT of 22 ms and normalized collision energy (NCE) of 26.
Orbitrap raw data were analyzed in DIA-NN (Data-Independent Acquisition by Neural Networks) [113] against the Mouse Proteome (downloaded from Uniprot, 21,946 proteins entries, downloaded 11/2022) using the library free mode of the software (version 1.8.1) allowing up to two tryptic missed cleavages and a maximum of three variable modifications/peptide. A spectral library was created from the DIA runs and used to reanalyze them (double search mode). A DIA-NN search was used with oxidation of methionine residues, N-terminal methionine excision and acetylation of the protein N-termini set as variable modifications and carbamidomethylation of cysteine residues as a fixed modification. The match between runs feature was used for all analyses, and the output (precursor) was filtered at 0.01 FDR. The protein inference was performed on the level of genes using only proteotypic peptides.
The proteomics data were processed with the Perseus software (version 1.6.15.0, Max Planck Institute of Biochemistry) [114]. Values were log(2) transformed, a threshold of 70% of valid values in at least one group was applied, and the missing values were replaced from normal distribution. For statistical analysis, a Student’s t-test was performed, and permutation-based FDR was calculated. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [115] partner repository with the dataset identifier PXD044220 (https://www.ebi.ac.uk/pride/ (accessed on 14 October 2023).
2.15. Statistical Analysis
Graphs were plotted using GraphPad Prism Software 6 (GraphPad Software, La Jolla, CA, USA). All values are presented as mean ± SEM from at least three independent experiments with suitable number of technical replicates (at least three). A two-tailed Student’s t-test was used to estimate the statistical significance between two groups. Significance was set up at p ≤ 0.05 (unless stated otherwise).
3. Results
3.1. CYLD DUB Deficiency Affects Reprogramming Efficiency
Initially, it was tested whether CYLD plays any regulatory role in somatic cell reprogramming. For this purpose, WT, Δ9, Δ9FCYLD and Δ9C601S MEFs were reprogrammed into pluripotent stem cells. The expression of CYLD in all cell types used was measured with both qPCR and Western blot (Figure 1a,b) (the uncropped blot is presented in Figure S3). At the end of the reprogramming period (day 18), cells were stained for Alkaline Phosphatase (ALP), which is highly expressed in undifferentiated cells, thus it is used as a surface pluripotency marker [1]. A representative example of ALP staining is presented in Figure 1c, while the staining results of each sample can be found in Figure S4. Overall, CYLD DUB deficiency caused a ~75% reduction in reprogramming efficiency compared to WT MEFs (mean efficiency = 0.8% for MEFs carrying the WT CYLD and 0.21% for cells carrying a catalytically inactive CYLD). Importantly, the introduction of WT CYLD in Δ9 MEFs was able to rescue the reprogramming efficiency, which became comparable to that of WT MEFs. On the other hand, the C601S variant had no effect (Figure 1c,d). This indicates that the observed phenotype is caused solely by CYLD catalytic inactivation.
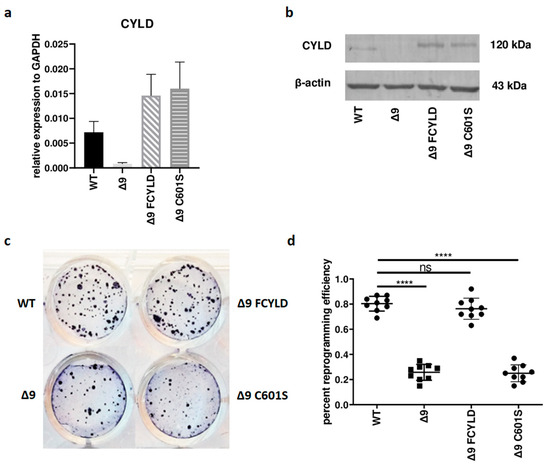
Figure 1.
CYLD DUB deficiency reduces reprogramming efficiency. (a,b) CYLD expression levels in the indicated MEFs (WT, Δ9, Δ9 FCYLD, Δ9 C601S) determined by qPCR (a) and immunoblotting (b). (c) Representative example of ALP-positive staining. CYLD catalytic inactivity leads to the formation of a smaller number of iPSC colonies. (d) Cumulative results of ALP staining in WT and Δ9 reprogrammed MEFs of the three established cell lines (b2_1, b2_2 and b3). CYLD DUB deficiency leads to a mean 75% reduction in reprogramming efficiency compared to MEFs expressing the WT CYLD (raw ALP staining results of the three MEF cell lines are presented in Figure S4). Data shown are the mean ± SEM from n = 3 independent experiments, two-tailed Student’s t-test (**** p ≤ 0.0001, Abbreviation: ns, not significant). The uncropped blot is shown in Supplementary Figure S3.
3.2. Δ9 CYLD Does Not Affect iPSC Pluripotency
Despite the reduction observed in cell reprogramming efficiency, Δ9 MEFs formed colonies of apparent reprogrammed cells. Consequently, it was tested whether Δ9 iPSCs are fully pluripotent. For this purpose, WT and iPSC colonies were harvested on day 18 and established stable, monoclonal iPSC cell lines from all WT and Δ9 MEF cell lines reprogrammed. iPSC cultures were maintained in DOX-free conditions. After 10 consecutive passages, Δ9 iPSC exhibited an indistinguishable morphology to control iPSCs (Figure 2a) and expressed comparable, high levels of core (OCT3/4, NANOG) and late (DPPA5A, REX1, UTF1) [17,116,117] pluripotency markers, as well as epithelial markers (CDH1, OCLN) [118,119] (Figure 2b). Additionally, in both iPSC types, mesenchymal (CDH2, SNAI1, ZEB2) [118,119] and fibroblast markers (ACTA2, CD44, THY1) [120,121] were expressed in comparable levels and presented a dramatic decrease compared to MEFs (Figure 2b). Furthermore, both iPSC types expressed comparable, low levels of T and EOMES, which are markers of primed pluripotency and incomplete reprogramming [122] (Figure S5a). The protein expression of pluripotency markers was further investigated with immunofluorescence. It was observed that both the expression and localization of pluripotency markers OCT3/4, NANOG and LIN28 were comparable in WT and Δ9 iPSC colonies. In conclusion, CYLD inactivation does not affect the pluripotency maintenance of iPSCs.
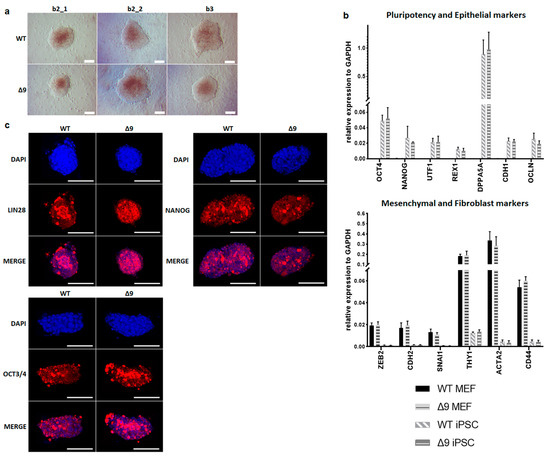
Figure 2.
CYLD is dispensable for pluripotency maintenance. (a) Bright-field microscopy images of representative iPSC monoclonal colonies after 10 consecutive passages (scale bar: 100 μm). Three WT and Δ9 monoclonal cell lines are presented, derived from the three WT and Δ9 MEF established cell lines (b2_1, b2_2 and b3). (b) Relative quantitation by qPCR of pluripotency (OCT3/4, NANOG, UTF1, REX1, DPPA5A), epithelial (CDH1, OCLN), mesenchymal (ZEB2, CDH2, SNAI1) and fibroblast (THY1, ACTA2, CD44) markers in the three WT and Δ9 monoclonal iPSC cell lines described in (a). (c) Representative immunofluorescence staining of the pluripotency markers OCT3/4, NANOG and LIN28 in WT and Δ9 iPSCs (scale bar: 100 μm). Data shown are the mean ± SEM from n = 3 independent experiments.
3.3. CYLD Is Dispensable for In Vitro Spontaneous Differentiation
A hallmark of pluripotency is the ability of stem cells to spontaneously differentiate into cells from the three embryonic germ layers. Therefore, it was tested whether CYLD DUB deficiency could potentially affect this process. For this purpose, WT and Δ9 iPSCs derived from the three independent MEF cell lines were cultured on suspension, under LIF-free conditions. After 3 days, both cell types formed Embryoid Bodies (EBs) of similar morphology (Figure S5b). Since the differentiation process starts with the formation of EBs, it was tested whether the downregulation of pluripotency genes followed a similar pattern in WT and Δ9 cells. It was observed that both cell types presented comparable downregulation rates (Figure S5c). On day 3, EBs were plated on gelatin-coated plates and cultured on FBS-containing medium. On the 14th day of culture, both cell types expressed comparable levels of endodermic (AFP, GATA4), mesodermic (ACTA2, BRACHYURY) and ectodermic (NESTIN, PAX6) markers [123,124] (Figure S5d). It was then investigated whether CYLD DUB deficiency could affect the kinetics of differentiation. To this end, cells were allowed to differentiate spontaneously and sampled from days 0-4, 7, 10 and 14 to assess the downregulation rate of pluripotency genes (OCT3/4, NANOG) and the upregulation of developmental genes (Endoderm: AFP, GATA4; Mesoderm: ACTA2, BRACHYURY; Ectoderm: NESTIN, PAX6; Trophoectoderm: EOMES). In both cases, the tested cell types exhibited comparable behavior. In conclusion, CYLD DUB deficiency does not affect the spontaneous differentiation potential of iPSCs (Figure S6).
3.4. CYLD Regulates Early Reprogramming
It has been firmly established that somatic cell reprogramming can be separated into three distinct phases: initiation (or early reprogramming), maturation (intermediate) and stabilization (late) [17,63]. Aiming to identify which phase (or phases) is mostly affected by CYLD DUB deficiency, early iPSC colonies derived from the three independent MEF cell lines were harvested from specific days, which are representative of each of the three phases—5 for early, d10 for intermediate and d16 for late reprogramming. Colonies were manually harvested and chosen based on their morphological characteristics (Figure S7a). More specifically, clusters of cells with cuboidal morphology (early iPSCs) [125] and compact colonies with distinct borders and well-defined edges (late iPSCs) [126] were chosen. The expression of markers specific to each phase was measured with qPCR. Both cell types exhibited similar expression patterns on days 10 and 16 (Figure S7b,c). Of particular importance for these phases are SOX2 and ESRRB since they are activated during the maturation phase [63,127]. In a similar manner, the expression of late pluripotency markers GBX2 [128], DPPA5A [117], UTF1 [17] and REX1 [116] follows the same trend in all cases. However, CYLD DUB-deficient cells presented lower expression levels of epithelial genes, such as CDH1 and EPCAM (Figure 3a), which are considered early reprogramming markers [17,63,118]. The downregulation of mesenchymal and somatic (fibroblast) genes is another key event of early reprogramming [17,63,118]; however, at day 5, no statistically significant difference in the expression of said markers (fibroblast: TGFB3, THY1 [120,127]; mesenchymal: SNAI1, ZEB2 [118,119]) between CYLD DUB-deficient and WT cells was detected (Figure S7b). On the other hand, CYLD DUB-deficient early iPSCs expressed lower levels of the core pluripotency genes OCT3/4 (endogenous) and NANOG compared to their WT counterparts (Figure 3b). This indicates that CYLD affects somatic cell reprogramming by regulating the initiation phase.
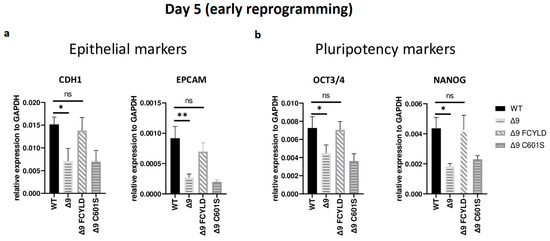
Figure 3.
CYLD regulates early reprogramming. qPCR-based relative quantitation of epithelial (CDH1, EPCAM) (a) and pluripotency (OCT3/4, NANOG) (b) genes on day 5, which corresponds to the early (initiation) phase of reprogramming. Tested mesenchymal markers for this day are presented in Figure S7b. Harvested day 5 early iPSC colonies were derived from reprogrammed WT and Δ9 MEFs of the three established MEF cell lines (b2_1, b2_2 and b3) and MEFs from the three independent Δ9 FCYLD and Δ9 C601S derivative cell lines. Data shown are the mean ± SEM from n = 3 independent experiments for each cell line, two-tailed Student’s t-test (* p ≤ 0.05, ** p ≤ 0.01. Abbreviation: ns, not significant **).
3.5. CYLD DUB Deficiency Delays Early Reprogramming Progression
Early reprogramming is a complex and multifaceted process in which many mechanisms are involved. Among the better-studied steps are the mesenchymal-to-epithelial transition (MET), the glycolytic shift, the loss of somatic cell identity and the increase in proliferation rates [20,63]. Interestingly, there are two distinct events observed during early reprogramming. The first days of reprogramming are characterized by elevated levels of mesenchymal genes, such as CDH2, SNAI1 and ZEB2, followed by the activation of epithelial and epidermal genes, like CDH1-OCLN and KRT14-KRT17, respectively, and the concomitant downregulation of mesenchymal and somatic genes [120,129,130]. This set of changes is considered a hallmark of MET [118,131]. Based on this, it has been suggested that the initiation phase can be split into two stages, a mesenchymal one, which takes place during the first days after the expression of the reprogramming cassette, followed by an epidermal stage [19]. Based on previous studies with the same reprogramming protocol [110], MET starts at day 3, when the first early iPSC colonies become observable, whereas the initiation phase lasts until day 6, when cells are plated onto feeders.
In order to better understand the way in which CYLD DUB deficiency affects these processes, early iPSC colonies from the three independent MEF cell lines were harvested during the initiation phase, and the relative expression of markers characteristic to each event was measured. High-quality and without fibroblast contamination, early iPSC colonies from all days considered to be part of the second stage of early reprogramming (D3–D6) were harvested (phenotypic characteristics of these colonies are described in Figure S8a). CYLD DUB-deficient cells express lower levels of core pluripotency and epithelial genes from the start and throughout the initiation phase. However, on day 6, the expression levels were on par with WT and Δ9 FCYLD early iPSCs. On the other hand, the mesenchymal and somatic genes tested exhibited similar expression patterns and were adequately downregulated by day 3 (Figure S8c).
These results indicate that CYLD DUB deficiency impedes early reprogramming progression. To test this hypothesis, we harvested samples from day 2, a transitional state before the initiation of MET, where cells alter their shape and are clustered together but have not yet formed pre-iPSC colonies (Figure S8a). Indeed, cells lacking catalytically active CYLD express higher levels of both mesenchymal (FN1, TGFB1) [119,132] and somatic markers (TGFB3, THY1) [120,127]. On par with that, the expression levels of epithelial genes (CDH1, OCLN) [119] are lower compared to WT and Δ9 FCYLD cells (Figure 4). Interestingly, even though CYLD DUB-deficient cells managed to successfully downregulate the mesenchymal and somatic markers tested by day 3, this was not the case with the epidermal and epithelial markers, which remained at notably lower levels compared to control cells. Taken together, CYLD DUB deficiency affects early reprogramming by hindering the progression of MET. All studied genes can be found in Figure S8d.
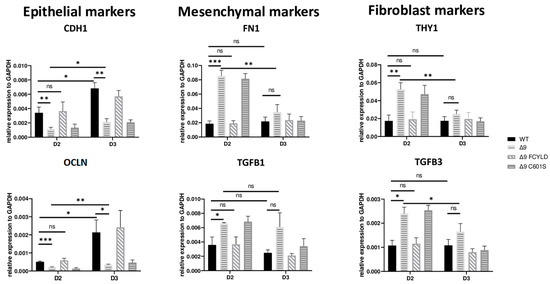
Figure 4.
CYLD is required for MET activation. qPCR-based relative quantitation of the indicated epithelial, mesenchymal and fibroblast markers in the indicated cell types on day 2 of the reprogramming process. Harvested D2 and D3 early iPSCs were derived from reprogrammed WT and Δ9 MEFs of the three established cell lines (b2_1, b2_2 and b3) and MEFs from the three independent Δ9 FCYLD and Δ9 C601S derivative cell lines. Data shown are the mean ± SEM from n = 3 independent experiments for each cell line, two-tailed Student’s t-test (* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, Abbreviation: ns, not significant).
3.6. CYLD Regulates MET and ECM Organization
Based on the results mentioned above, it was clear that CYLD DUB deficiency compromises the delicate balance that leads to MET activation. In order to gain insight into the way CYLD DUB deficiency disrupts the early reprogramming network, WΤ/Δ9 D0 MEFs of the three independent cell lines and the D3–D4 early iPSC colonies (of said cell lines) were harvested and subjected to whole proteome analysis through LC/MS. Day 3 proteome corresponds to the transcriptome of day 2 samples and day 4 proteome to that of day 3 samples. A total of 7163 proteins were identified (the z-score of identified proteins is given in Table S2). On D4, there were 5368 statistically significant Differentially Expressed Proteins (DEPs) in Δ9 early iPSCs compared to WT (2647 upregulated and 2721 downregulated). Similarly, on D3, there were 4910 DEPs in Δ9 early iPSCs (2347 upregulated and 2562 downregulated). After applying a threshold of |log2fc ≥ 1.5| and log2 (p-Value) ≥ 1.5, the number of DEPs dropped to 436 for D3 (199 upregulated and 237 downregulated) and 522 for D4 (142 upregulated and 380 downregulated) (Figure 5a). It was observed that the most overexpressed proteins in Δ9 early iPSCs are associated with the extracellular matrix, as identified by the use of the MatrisomeDB database and the literature research (Emilin1, Fbln1, Fbln2, Fn1, Igsf8, Pcolce2, Plg, Prss23, Vtn) [133,134,135]. On the other hand, markers of early reprogramming as well as epithelial and epidermal proteins associated with MET (Aldh3a1, Lgals7, Nccrp1, Sfn, Krt14, Krt17) [120,130,136,137,138] were downregulated (blue dots).
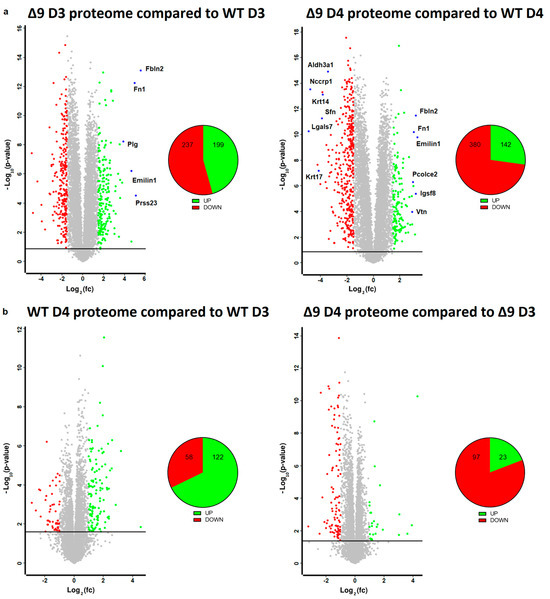
Figure 5.
Whole proteome profiling of D3 and D4 early iPSCs. (a) Volcano plots (FDR = 0.05, S0 = 0) comparing the Δ9 early iPSCs proteome to that of WT early iPSCs (D3: left plot, D4: right plot). A threshold of |log2fc ≥ 1.5| and log2 (p-value) ≥ 1.5 is applied to determine the statistically significant Differentially Expressed Proteins (DEPs). Proteins downregulated in Δ9 samples compared to WT samples are marked as red, whereas upregulated proteins are marked as green. Proteins of interest are highlighted in blue. CYLD DUB deficiency led to the upregulation of 199 and downregulation of 237 proteins compared to WT day 3 early iPSCs. Similarly, 142 proteins were upregulated and 380 downregulated compared with WT samples on day 4. At both time points, some of the most overexpressed proteins in Δ9 early iPSCs are associated with the extracellular matrix, whereas epithelial, epidermal and early reprogramming markers are underrepresented. (b) Volcano plots (FDR = 0.05, S0 = 0) comparing the proteome of D4 early iPSCs proteome to that of D3 early iPSCs (WT: left plot, Δ9: right plot). A threshold of |log2fc ≥ 1| and log2 (p-value) ≥ 1.5 is applied to determine the statistically significant DEPs. D3 and D4 harvested early iPSCs are derived from WT and Δ9 MEFs of the three established cell lines (b2_1, b2_2 and b3). Each sample was analyzed in at least two technical replicates.
Due to the low number of DEPs between the D3–D4 transition, a less strict threshold of |log2fc ≥ 1| was applied in this analysis. It was revealed that the majority of WT D4 DEPs are upregulated in comparison to D3 iPSCs (122 DEPs upregulated and 58 downregulated), whereas the opposite trend is observed in Δ9 early iPSCs (23 DEPs upregulated and 97 downregulated) (Figure 5b). Both WT and Δ9 MEFs followed a similar pattern during the transition from D0 to D3, with most DEPs being upregulated. However, this effect was more pronounced in Δ9 cells (Figure S9a). Finally, our analysis revealed that CYLD DUB deficiency also affected the proteome of MEFs. More specifically, 311 DEPs were downregulated and 82 upregulated in Δ9 MEFs compared to WT MEFs at day 0 (Figure S9b).
Pathway enrichment analysis with Metascape [139] revealed that DEPs upregulated in Δ9 cells are strongly correlated with extracellular matrix organization, cell–substrate adhesion and several developmental processes (for example, hemostasis, response to wounding and regulation of blood coagulation) (Figure 6a). Most of the processes overrepresented in Δ9 cells, notably the extracellular matrix organization, cell adhesion and the integrin pathway, are known to be downregulated during the MET, where structural reorganization occurs [130,140]. Indeed, iPSCs express a different set of ECM proteins compared to MEFs [48]. The remodeling of ECM (destruction of MEF-associated ECM and development of iPSC-associated ECM) during reprogramming is paramount for the establishment of the unique iPSC morphology and microenvironment [48,141]. Intriguingly, one of the enriched pathways associated with Δ9 D4 upregulated DEPs is the “Regulation of Epithelial Cell Proliferation Pathway”. However, most of the associated DEPs are associated with EMT progression, so they are expected to play a negative role in epithelial cell identity (Table S3). Conversely, downregulated DEPs are mostly associated with several metabolic processes and cytoskeletal organization. Interestingly, pathway analysis of the D3–D4 transition provided markedly different results for WT and Δ9 cells (Figure 6b). More specifically, upregulated DEPs in WT D4 early iPSCs are primarily associated with the formation of the cornified envelope and regulation of proteolysis. The number of downregulated DEPs was relatively small (58), and as a result, their association with the presented processes is weak (low −log10 (p-Value)). On the contrary, downregulated DEPs of Δ9 cells presented a strong correlation with extracellular matrix organization and the integrin pathway. The number of upregulated DEPs was very small, so the pathway analysis failed to construct any correlation. Regarding the D0-D3 transition, upregulated DEPs presented many similarities between WT and Δ9 MEFs, with most proteins being associated with the same biosynthetic and metabolic processes (for example, the biosynthesis of cofactors and tRNA, nucleoside monophosphate and amino acid metabolic process). However, the pattern was not the same with the downregulated proteins. In WT cells, downregulated DEPs presented a strong association with the extracellular matrix organization, a process that is completely absent from Δ9 cells. Similarly, processes highly enriched in Δ9 cells, such as vasculature development and the integrin 1 pathway, are not observed in WT cells (Figure S10a). Finally, comparison of D0 MEFs revealed that most downregulated DEPs in Δ9 cells are associated with several metabolic processes, whereas upregulated DEPs presented a weak association. Interestingly, the combined analysis was enriched in several processes associated with the immune response, such as the NF-κB pathway, response to viral infection and macroautophagy (Figure S10b).
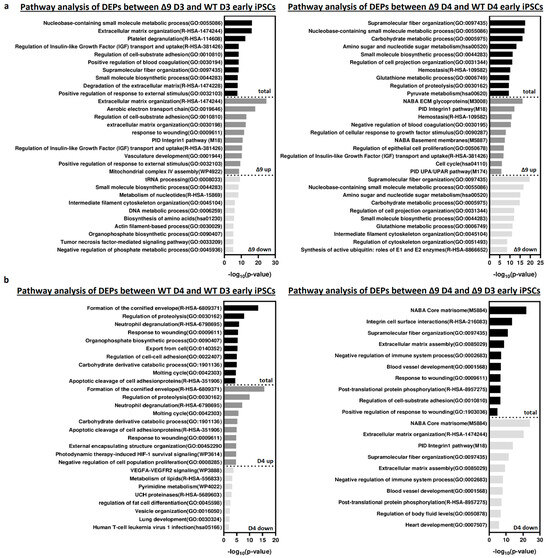
Figure 6.
Pathway enrichment analysis of DEPs from D3 and D4 early iPSCs. (a) Analysis of DEPs between Δ9 D3 (left) and D4 (right) compared to their WT counterparts. Upregulated DEPs significantly correlate with extracellular matrix organization, cell adhesion and several developmental processes. On the other hand, downregulated DEPs participate in several metabolic processes. (b) Analysis of D3–D4 transition DEPs in WΤ (left) and Δ9 (right) early iPSCs. Upregulated DEPs in WT cells are mostly associated with the formation of the cornified envelope and the regulation of proteolysis. Downregulated DEPs are fewer, and the enrichment analysis presented a weak association with some metabolic and developmental processes, the most notable of them being the VEGF signaling pathway. In the case of Δ9 early iPSCs, downregulated DEPs have a strong link with extracellular matrix organization and the integrin 1 pathway. The very small number (23) of upregulated proteins did not present a significant association with any process. Pathway analysis was conducted with Metascape [139].
Pathway analysis of D3 and D4 samples indicated that a set of 38 extracellular matrix-associated proteins are differentially expressed (Table 1). Notable members of this group are Fn1, Emilin1 and Fbln2, which were shown above to be the most overexpressed proteins in Δ9 cells. A heatmap presenting the expression patterns of the extracellular matrix proteins across the tested samples is available in Figure 7a. As it can be clearly observed, most of these proteins are significantly overexpressed in Δ9 cells on day 3 of reprogramming. Moreover, this trend is also present, although to a lesser extent, on day 4.

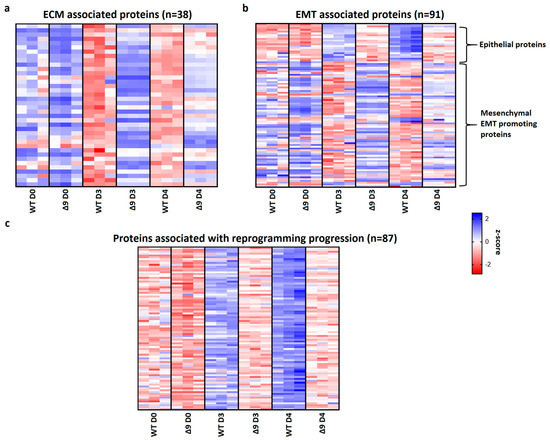
Figure 7.
CYLD regulates MET. (a) Expression pattern of 38 extracellular matrix-related proteins that were identified from pathway analysis. On both D3 and D4, Δ9 early iPSCs express higher levels of these proteins compared to WT cells, although this is more prominent on D3. (b) Expression pattern of 91 identified EMT−related proteins. Epithelial−related proteins are presented on top. CYLD DUB−deficient cells express lower levels of epithelial proteins and higher levels of mesenchymal proteins compared to WT cells. While mesenchymal proteins are slightly downregulated on D4 compared to D3, Δ9 cells were not able to increase the expression of epithelial proteins on D4. (c) Expression pattern of 87 identified reprogramming-associated proteins. Activation of these proteins is concomitant to MET initiation. CYLD DUB-deficient cells present vastly lower expression levels of all these proteins compared to WT cells. On the other hand, WT early iPSCs steadily increase most of them on D4 compared to D3. Three independent WT and Δ9 MEF cell lines were used (b2_1, b2_2 and b3, respectively). Harvested D3 and D4 early iPSC colonies are derived from these reprogrammed WT and Δ9 MEF cell lines. Each sample was analyzed in at least two technical replicates (z−scores of DEPs from every sample are presented in Table S2).
Since many of these proteins are also involved in the EMT, and taking under consideration that hallmark epithelial genes such as CDH1 and EPCAM are downregulated in Δ9 cells, the number of D3 and D4 DEPs associated with the EMT was investigated. Through the literature search, the use of the EMTome portal [142] and the Hallmark Epithelial Mesenchymal Transition Gene Set (Molecular Signature Database, MSigDB) [143], 91 EMT-associated DEPs were identified; 21 epithelial and 70 mesenchymal or EMT-promoting DEPs. A heatmap presenting the expression pattern of these proteins on days 0, 3 and 4 is available in Figure 7b.
The expression levels of epithelial proteins are substantially lower in both D3 and D4 compared to WT early iPSCs. At the same time, most mesenchymal and EMT-promoting proteins are upregulated in Δ9 early iPSCs, although it is worth noting that on D4 their expression has decreased. D0 MEFs show comparable expression levels of most proteins. Finally, we identified 87 proteins whose expression is associated with successful reprogramming (Figure 7c), based on previous research [17,19,120,129,130,137,138,140]. Regardless of their different expression patterns throughout reprogramming, these proteins are activated during MET. While all these proteins have been successfully upregulated in Δ9 early iPSCs compared to MEFs, their expression is considerably lower compared to WT early iPSCs, suggesting a lower rate of reprogramming kinetics. To summarize, our proteomic results indicate that Δ9 early iPSCs express higher levels of EMT-associated markers and extracellular matrix proteins, while failing to adequately activate epithelial and MET-associated proteins (the complete list of genes included in Figure 7 heatmaps is available in Table S2).
4. Discussion
The current study revealed for the first time the regulatory role of CYLD in somatic cell reprogramming. MEFs lacking a catalytically active protein presented lower reprogramming efficiency and slower early reprogramming kinetics compared to WT MEFs, with the transition from the first to the second stage of the initiation phase being most affected (Figure 8). However, it is worth noting that CYLD DUB-deficient cells were able to be fully reprogrammed. Δ9 iPSCs were stable after 10 consecutive passages and presented similar phenotypic and molecular characteristics to WT iPSCs. Moreover, they could spontaneously differentiate into cells of the three germ layers and followed the same differentiation kinetics pattern as WT cells. It would be very interesting to investigate whether this is the case with directed differentiation also, especially towards cell types that are known to be negatively affected by CYLD inactivation, such as hepatocytes [91,144,145,146], cardiomyocytes [147,148,149,150] and neurons [151,152,153,154].
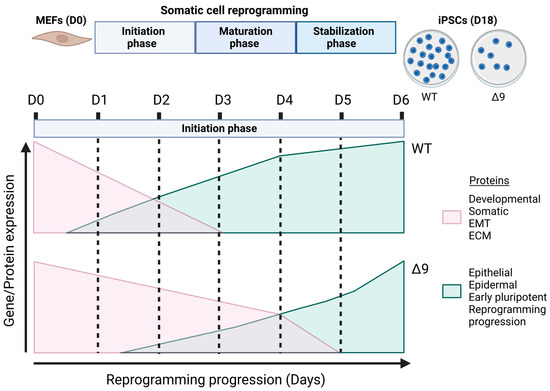
Figure 8.
Proposed role of CYLD in somatic cell reprogramming. CYLD DUB-deficient MEFs (CYLDΔ9/Δ9) present a 75% reduction in reprogramming efficiency compared to WT MEFs (CYLDWT/WT), as determined through ALP staining. Gene profiling and whole proteome analysis identified the initiation phase of reprogramming as mostly affected by CYLD DUB deficiency, hindering early reprogramming progression. Δ9 early iPSCs are characterized by higher expression levels of developmental, somatic (fibroblast), EMT- and ECM-associated proteins (which act as reprogramming barriers) and lower expression levels of epithelial, early pluripotency and reprogramming-progression-associated proteins. As a result, the process of MET is severely affected (this figure was generated with BioRender, https://www.biorender.com/). Abbreviations: D= day of reprogramming.
The mechanism by which CYLD DUB deficiency slows down the transition to the second stage of initiation phase is likely the obstruction of MET through the promotion of the opposite process, EMT. As our proteomics results demonstrated, CYLD DUB-deficient cells are characterized by the aberrant upregulation of mesenchymal and EMT-promoting proteins and low expression of epithelial proteins, in contrast to WT early iPSCs. This correlates with the inadequate activation of proteins associated with successful reprogramming. As previously stated, these proteins are also markers of the MET since they are activated during that process [120,130,140]. These results strengthen our early reprogramming kinetic experiments, where we demonstrated that CYLD DUB-deficient cells activate epithelial and pluripotency genes at a lower rate. Indeed, analysis of the D3-D4 transition supports this observation. In more detail, the main difference between WT D3 and D4 early iPSCs is the upregulation of MET- and reprogramming-associated proteins. On the other hand, Δ9 D4 early iPSCs are still in the process of downregulating mesenchymal and ECM-associated genes, as the pathway analysis of D3–D4 transition revealed.
The reason behind the EMT-promoting phenotype of Δ9 cells is not clear yet. As previously mentioned, the first transcriptional wave observed during somatic cell reprogramming is characterized by the downregulation of somatic and mesenchymal genes, along with genes associated with several developmental processes, such as vasculature, bone and heart development [120,129,130,140]. The major event of this stage (which is also called the mesenchymal stage [19]) is the downregulation of Thy1 [120,129] and roughly corresponds to the first day of our reprogramming model. The absence of Thy1 from the cell surface signifies the end of this stage [120,129]. Whether EMT- and ECM-associated genes are hyperactivated in Δ9 cells at this stage or their downregulation follows a slower rate is not known. Since cells at D1 cannot be distinguished from their surroundings nor can they be individually harvested with our protocol, we were unable to isolate pure samples from this stage. However, it is worth mentioning that pathway analysis of the Δ9 D0-D3 transition did not include the process of extracellular matrix organization, which was the primary downregulated event of the WT D0-D3 transition. Furthermore, the proteomic results suggest that a number of key mesenchymal stage players, such as Tgf-β1 and Fn1, presented a small but statistically significant upregulation in Δ9 D3 early iPSCs compared to MEFs. While these observations likely point out to an aberrant activation of EMT during D1, other proteins, including Thy1 and Thbs1, are significantly downregulated in Δ9 D3 iPSCs compared to Δ9 MEFs. As a result, no safe conclusion can be made based on the current results, and further research on D1 cells is required. Regardless, the overexpression of EMT genes/proteins and several components of the TGF-beta pathway (such as TGFΒ1, TGFBi, LTBP1 and THBS1) is consistent with previous observations that associate CYLD DUB deficiency with TGF-beta activation and EMT promotion [85,86,87,108,155], although it is not yet known whether the underlying mechanism is the same in the case of somatic cell reprogramming.
Importantly, the negative effect of CYLD DUB deficiency on epithelial and epidermal gene activation is further supported by the transcriptomic analysis of WT and Δ9 D3 early iPSCs (data not shown). Importantly, 17 out of the 21 identified epithelial DEPs and several proteins associated with successful reprogramming progression showcased high correlation with the transcriptomic results, further supporting the findings of the present study.
A finding of significant importance is the ability of CYLD to regulate the processes of extracellular matrix organization, which is proposed for the first time in this study. This can have a considerable implication in cancer research, since this process is known to play a critical role in cancer development, growth and invasion [156,157,158,159]. Regarding its role in somatic cell reprogramming, several studies [48,120,130,140] have demonstrated that the downregulation of ECM genes is a hallmark of the MET and failure to do so is associated with poor reprogramming efficiency. Indeed, the ECM is a driver of the EMT [160], so the hyperactivation of ECM-related proteins in Δ9 cells may contribute independently to MET obstruction.
Another point worthy of discussion is the potential involvement of Irf6. Our results revealed that CYLD DUB-deficient cells failed to activate Irf6 during the first days of reprogramming (Table S2). It has been recently shown that irf6 is a member of a nine-transcription-factor (TF) complex that plays a key regulatory role in early reprogramming, facilitating the transition to the next phase [110]. More specifically, early iPSC colonies that express all nine transcription factors simultaneously have significantly higher chances of becoming fully reprogrammed. Interestingly, RNAi knockdown of Irf6 reduced reprogramming efficiency by 60%. It was found that the expression of Irf6 (along with Gli2 and Ovol1, two other components of the 9TF assembly) is induced between days 2 and 3 by mostly stochastic events related to the OSKM reprogramming cassette. However, it is highly likely that the processes triggered by CYLD DUB deficiency might interfere with these mechanisms.
Besides EMT and ECM regulation, our pathway analysis revealed that CYLD DUB deficiency negatively affects various metabolic processes (such as glutathione, nucleotide and carbohydrate metabolism) not only during reprogramming but also in MEFs. While it is known that metabolic changes play a crucial part in reprogramming, especially the metabolic shift from oxidative phosphorylation to glycolysis [161,162], this process was not observed in our pathway analysis. While further research is needed to identify the effect of CYLD DUB deficiency in these metabolic processes, one valid hypothesis is that these changes correlate with the promotion of EMT, as it has been demonstrated in cancer development [64,163]. One potential way to uncover the role of the CYLD-affected metabolic processes in somatic cell reprogramming is the study of the differential metabolites between WT and Δ9 early iPSCs [164].
5. Conclusions
The present study uncovered for the first time the effect of CYLD in somatic cell reprogramming. CYLD is part of the mechanism overseeing a successful mesenchymal-to-epithelial transition during early reprogramming and the successful establishment of pluripotency. Indeed, loss of CYLD promotes the EMT and slows down the progression of early reprogramming, leading to efficiency reduction. Nevertheless, CYLD DUB-deficient cells present self-renewal and differentiation abilities comparable to those of control iPSCs.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/cancers15204997/s1, Figure S1: Transduction efficiency test, Figure S2: Colony harvesting example, Figure S3: Uncropped Western blot from Figure 1b, Figure S4: Total ALP staining results, Figure S5: CYLD DUB deficiency does not affect iPSC pluripotency, Figure S6: Kinetics of differentiation, Figure S7: Kinetics of reprogramming, Figure S8: Kinetics of early reprogramming, Figure S9: Whole proteome profiling of D0 MEFs and D0-D3 transition, Figure S10: Pathway analysis of D0 MEFs and D0-D3 transitions, Table S1: Primer sequences used for qPCR, Table S2: Heatmap gene expression analysis, Table S3: Genes involved in pathway analysis described in Figure 6, Table S4: Genes involved in pathway analysis described in Figure S10.
Author Contributions
Conceptualization, D.D. and G.M.; methodology, N.B., M.S., M.P., P.M., A.P., K.X., G.M. and D.D.; formal analysis, N.B., M.S., G.M. and D.D.; resources, D.T., G.M. and D.D.; writing—original draft preparation, N.B., M.S., G.M. and D.D.; writing—review and editing, N.B., M.S., M.P, P.M., K.X., G.M. and D.D.; supervision, G.M. and D.D.; funding acquisition, G.M. and D.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the State Scholarships Foundation, grant number OΠΣ 5000432.
This work was supported by grants to DT from the Greek General Secretariat for Research and Technology (GSRT) (Cooperative Grants Synergasia I #969),
from the European Economic Area (EL0084) and
from the KMW offsets program.
We acknowledge the support of this work by the project “The Greek Research Infrastructure for Personalised Medicine (pMED-GR)” (MIS 5002802) which is implemented under the Action “Rein-forcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund).
Institutional Review Board Statement
Not applicable in this study.
Informed Consent Statement
Not applicable in this study.
Data Availability Statement
The data presented in this study (and Supplementary Materials) and available upon request. Project Name: Inactivation of tumor suppressor CYLD inhibits fibroblast re-programming to pluripotency. Project accession: PXD044220. The raw proteomic data can be found in the proteomics identification database (PRIDE): https://www.ebi.ac.uk/pride/ (accessed on 14 October 2023) [115]. Project Name: Inactivation of tumor suppressor CYLD inhibits fibroblast re-programming to pluripotency. Project accession: PXD044220.
Acknowledgments
We acknowledge the support of this work by the project “The Greek Research Infrastructure for Personalised Medicine (pMED-GR)” (MIS 5002802) which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Guan, J.; Wang, G.; Wang, J.; Zhang, Z.; Fu, Y.; Cheng, L.; Meng, G.; Lyu, Y.; Zhu, J.; Li, Y.; et al. Chemical Reprogramming of Human Somatic Cells to Pluripotent Stem Cells. Nature 2022, 605, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Fu, Y.; Zhu, J.; Liu, Y.; Zhang, Q.; Yi, Z.; Chen, S.; Jiao, Z.; Xu, X.; Xu, J.; et al. Single-Cell RNA-Seq Reveals Dynamic Early Embryonic-like Programs during Chemical Reprogramming. Cell Stem Cell 2018, 23, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Wang, A.Y.L.; Loh, C.Y.Y. Episomal Induced Pluripotent Stem Cells: Functional and Potential Therapeutic Applications. Cell Transplant. 2019, 28, 112S–131S. [Google Scholar] [CrossRef]
- Wang, A.Y.L. Application of Modified mRNA in Somatic Reprogramming to Pluripotency and Directed Conversion of Cell Fate. Int. J. Mol. Sci. 2021, 22, 8148. [Google Scholar] [CrossRef]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac Transposition Reprograms Fibroblasts to Induced Pluripotent Stem Cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Aboul-Soud, M.A.M.; Alzahrani, A.J.; Mahmoud, A. Induced Pluripotent Stem Cells (iPSCs)—Roles in Regenerative Therapies, Disease Modelling and Drug Screening. Cells 2021, 10, 2319. [Google Scholar] [CrossRef] [PubMed]
- Kouroupi, G.; Taoufik, E.; Vlachos, I.S.; Tsioras, K.; Antoniou, N.; Papastefanaki, F.; Chroni-Tzartou, D.; Wrasidlo, W.; Bohl, D.; Stellas, D.; et al. Defective Synaptic Connectivity and Axonal Neuropathology in a Human iPSC-Based Model of Familial Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2017, 114, E3679–E3688. [Google Scholar] [CrossRef]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling Inherited Metabolic Disorders of the Liver Using Human Induced Pluripotent Stem Cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing Sporadic and Familial Alzheimer’s Disease Using Induced Pluripotent Stem Cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Browder, K.C.; Reddy, P.; Yamamoto, M.; Haghani, A.; Guillen, I.G.; Sahu, S.; Wang, C.; Luque, Y.; Prieto, J.; Shi, L.; et al. In Vivo Partial Reprogramming Alters Age-Associated Molecular Changes during Physiological Aging in Mice. Nat. Aging 2022, 2, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Yamamoto, M.; Hishida-Nozaki, Y.; Shao, C.; Huang, L.; Wang, C.; Shojima, K.; Xue, Y.; Hang, Y.; Shokhirev, M.; et al. In Vivo Partial Cellular Reprogramming Enhances Liver Plasticity and Regeneration. Cell Rep. 2022, 39, 110730. [Google Scholar] [CrossRef]
- Wang, C.; Rabadan Ros, R.; Martinez-Redondo, P.; Ma, Z.; Shi, L.; Xue, Y.; Guillen-Guillen, I.; Huang, L.; Hishida, T.; Liao, H.-K.; et al. In Vivo Partial Reprogramming of Myofibers Promotes Muscle Regeneration by Remodeling the Stem Cell Niche. Nat. Commun. 2021, 12, 3094. [Google Scholar] [CrossRef]
- Chen, Y.; Lüttmann, F.F.; Schoger, E.; Schöler, H.R.; Zelarayán, L.C.; Kim, K.-P.; Haigh, J.J.; Kim, J.; Braun, T. Reversible Reprogramming of Cardiomyocytes to a Fetal State Drives Heart Regeneration in Mice. Science 2021, 373, 1537–1540. [Google Scholar] [CrossRef]
- Buganim, Y.; Faddah, D.A.; Jaenisch, R. Mechanisms and Models of Somatic Cell Reprogramming. Nat. Rev. Genet. 2013, 14, 427–439. [Google Scholar] [CrossRef]
- González, F.; Huangfu, D. Mechanisms Underlying the Formation of Induced Pluripotent Stem Cells. WIREs Dev. Biol. 2016, 5, 39–65. [Google Scholar] [CrossRef]
- Liu, K.; Song, Y.; Yu, H.; Zhao, T. Understanding the Roadmaps to Induced Pluripotency. Cell Death Dis. 2014, 5, e1232. [Google Scholar] [CrossRef]
- Meir, Y.-J.J.; Li, G. Somatic Reprogramming—Above and Beyond Pluripotency. Cells 2021, 10, 2888. [Google Scholar] [CrossRef]
- Fidalgo, M.; Faiola, F.; Pereira, C.-F.; Ding, J.; Saunders, A.; Gingold, J.; Schaniel, C.; Lemischka, I.R.; Silva, J.C.R.; Wang, J. Zfp281 Mediates Nanog Autorepression through Recruitment of the NuRD Complex and Inhibits Somatic Cell Reprogramming. Proc. Natl. Acad. Sci. USA 2012, 109, 16202–16207. [Google Scholar] [CrossRef]
- Lluis, F.; Ombrato, L.; Pedone, E.; Pepe, S.; Merrill, B.J.; Cosma, M.P. T-Cell Factor 3 (Tcf3) Deletion Increases Somatic Cell Reprogramming by Inducing Epigenome Modifications. Proc. Natl. Acad. Sci. USA 2011, 108, 11912–11917. [Google Scholar] [CrossRef]
- Popowski, M.; Templeton, T.D.; Lee, B.-K.; Rhee, C.; Li, H.; Miner, C.; Dekker, J.D.; Orlanski, S.; Bergman, Y.; Iyer, V.R.; et al. Bright/Arid3A Acts as a Barrier to Somatic Cell Reprogramming through Direct Regulation of Oct4, Sox2, and Nanog. Stem Cell Rep. 2014, 2, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Serrano, F.; Calatayud, C.F.; Blazquez, M.; Torres, J.; Castell, J.V.; Bort, R. Gata4 Blocks Somatic Cell Reprogramming By Directly Repressing Nanog. Stem Cells 2013, 31, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Han, Q.; Peng, T.; Peng, M.; Wei, B.; Li, D.; Wang, X.; Yu, S.; Yang, J.; Cao, S.; et al. The Oncogene C-Jun Impedes Somatic Cell Reprogramming. Nat. Cell Biol. 2015, 17, 856–867. [Google Scholar] [CrossRef]
- Yang, C.-S.; Lopez, C.G.; Rana, T.M. Discovery of Nonsteroidal Anti-Inflammatory Drug and Anticancer Drug Enhancing Reprogramming and Induced Pluripotent Stem Cell Generation. Stem Cells 2011, 29, 1528–1536. [Google Scholar] [CrossRef]
- Mali, P.; Chou, B.-K.; Yen, J.; Ye, Z.; Zou, J.; Dowey, S.; Brodsky, R.A.; Ohm, J.E.; Yu, W.; Baylin, S.B.; et al. Butyrate Greatly Enhances Derivation of Human Induced Pluripotent Stem Cells by Promoting Epigenetic Remodeling and the Expression of Pluripotency-Associated Genes. Stem Cells 2010, 28, 713–720. [Google Scholar] [CrossRef]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C Enhances the Generation of Mouse and Human Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of Pluripotent Stem Cells by Defined Factors Is Greatly Improved by Small-Molecule Compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef]
- Neganova, I.; Shmeleva, E.; Munkley, J.; Chichagova, V.; Anyfantis, G.; Anderson, R.; Passos, J.; Elliott, D.J.; Armstrong, L.; Lako, M. JNK/SAPK Signaling Is Essential for Efficient Reprogramming of Human Fibroblasts to Induced Pluripotent Stem Cells. Stem Cells 2016, 34, 1198–1212. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.A.; Scalf, S.M.; Zhang, J.; Hu, X.; Chen, X.; Eastman, A.E.; Yang, C.; Guo, S. YAP Non-Cell-Autonomously Promotes Pluripotency Induction in Mouse Cells. Stem Cell Rep. 2020, 14, 730–743. [Google Scholar] [CrossRef]
- Marson, A.; Foreman, R.; Chevalier, B.; Bilodeau, S.; Kahn, M.; Young, R.A.; Jaenisch, R. Wnt Signaling Promotes Reprogramming of Somatic Cells to Pluripotency. Cell Stem Cell 2008, 3, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Feng, G.; Wang, L.; Teng, F.; Wang, L.; Li, W.; Zhang, Y.; Zhou, Q. MeCP2 Deficiency Promotes Cell Reprogramming by Stimulating IGF1/AKT/mTOR Signaling and Activating Ribosomal Protein-Mediated Cell Cycle Gene Translation. J. Mol. Cell Biol. 2018, 10, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Barrandon, O.; Nichols, J.; Kawaguchi, J.; Theunissen, T.W.; Smith, A. Promotion of Reprogramming to Ground State Pluripotency by Signal Inhibition. PLoS Biol. 2008, 6, e253. [Google Scholar] [CrossRef]
- Maherali, N.; Hochedlinger, K. Tgfβ Signal Inhibition Cooperates in the Induction of iPSCs and Replaces Sox2 and cMyc. Curr. Biol. 2009, 19, 1718–1723. [Google Scholar] [CrossRef]
- Ichida, J.K.; Blanchard, J.; Lam, K.; Son, E.Y.; Chung, J.E.; Egli, D.; Loh, K.M.; Carter, A.C.; Di Giorgio, F.P.; Koszka, K.; et al. A Small-Molecule Inhibitor of Tgf-β Signaling Replaces Sox2 in Reprogramming by Inducing Nanog. Cell Stem Cell 2009, 5, 491–503. [Google Scholar] [CrossRef]
- Ho, R.; Papp, B.; Hoffman, J.A.; Merrill, B.J.; Plath, K. Stage-Specific Regulation of Reprogramming to Induced Pluripotent Stem Cells by Wnt Signaling and T Cell Factor Proteins. Cell Rep. 2013, 3, 2113–2126. [Google Scholar] [CrossRef]
- Tan, F.; Qian, C.; Tang, K.; Abd-Allah, S.M.; Jing, N. Inhibition of Transforming Growth Factor β (TGF-β) Signaling Can Substitute for Oct4 Protein in Reprogramming and Maintain Pluripotency. J. Biol. Chem. 2015, 290, 4500–4511. [Google Scholar] [CrossRef]
- Doege, C.A.; Inoue, K.; Yamashita, T.; Rhee, D.B.; Travis, S.; Fujita, R.; Guarnieri, P.; Bhagat, G.; Vanti, W.B.; Shih, A.; et al. Early-Stage Epigenetic Modification during Somatic Cell Reprogramming by Parp1 and Tet2. Nature 2012, 488, 652–655. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Chen, J.; Wu, Y.; et al. H3K9 Methylation Is a Barrier during Somatic Cell Reprogramming into iPSCs. Nat. Genet. 2013, 45, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Brady, J.J.; Damian, M.; Sacco, A.; Corbel, S.Y.; Blau, H.M. Reprogramming towards Pluripotency Requires AID-Dependent DNA Demethylation. Nature 2010, 463, 1042–1047. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, L.; Mao, S.-Q.; Li, Z.; Chen, J.; Zhang, R.-R.; Wu, H.-P.; Gao, J.; Guo, F.; Liu, W.; et al. Tet and TDG Mediate DNA Demethylation Essential for Mesenchymal-to-Epithelial Transition in Somatic Cell Reprogramming. Cell Stem Cell 2014, 14, 512–522. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, J.; Li, K.; Wu, T.; Huang, B.; Liu, W.; Kou, X.; Zhang, Y.; Huang, H.; Jiang, Y.; et al. Replacement of Oct4 by Tet1 during iPSC Induction Reveals an Important Role of DNA Methylation and Hydroxymethylation in Reprogramming. Cell Stem Cell 2013, 12, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Kretsovali, A.; Hadjimichael, C.; Charmpilas, N. Histone Deacetylase Inhibitors in Cell Pluripotency, Differentiation, and Reprogramming. Stem Cells Int. 2012, 2012, 184154. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Bao, X.; Liu, L.; Feng, S.; Zovoilis, A.; Liu, W.; Xue, Y.; Cai, J.; Guo, X.; Qin, B.; et al. MicroRNA Cluster 302–367 Enhances Somatic Cell Reprogramming by Accelerating a Mesenchymal-to-Epithelial Transition. J. Biol. Chem. 2011, 286, 17359–17364. [Google Scholar] [CrossRef]
- Xu, N.; Papagiannakopoulos, T.; Pan, G.; Thomson, J.A.; Kosik, K.S. MicroRNA-145 Regulates OCT4, SOX2, and KLF4 and Represses Pluripotency in Human Embryonic Stem Cells. Cell 2009, 137, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-S.; Li, Z.; Rana, T.M. microRNAs Modulate iPS Cell Generation. RNA 2011, 17, 1451–1460. [Google Scholar] [CrossRef]
- Li, Z.; Dang, J.; Chang, K.-Y.; Rana, T.M. MicroRNA-Mediated Regulation of Extracellular Matrix Formation Modulates Somatic Cell Reprogramming. RNA 2014, 20, 1900–1915. [Google Scholar] [CrossRef]
- Zhao, F.-Q.; Misra, Y.; Li, D.-B.; Wadsworth, M.; Krag, D.; Weaver, D.; Tessitore, J.; Li, D.-W.; Zhang, G.; Tian, Q.; et al. Differential Expression of Oct3/4 in Human Breast Cancer and Normal Tissues. Int. J. Oncol. 2018, 52, 2069–2078. [Google Scholar] [CrossRef]
- Clemente-Periván, S.I.; Gómez-Gómez, Y.; Leyva-Vázquez, M.A.; Lagunas-Martínez, A.; Organista-Nava, J.; Illades-Aguiar, B. Role of Oct3/4 in Cervical Cancer Tumorigenesis. Front. Oncol. 2020, 10, 247. [Google Scholar] [CrossRef]
- Yu, S.S.; Cirillo, N. The Molecular Markers of Cancer Stem Cells in Head and Neck Tumors. J. Cell. Physiol. 2020, 235, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Novak, D.; Hüser, L.; Elton, J.J.; Umansky, V.; Altevogt, P.; Utikal, J. SOX2 in Development and Cancer Biology. Semin. Cancer Biol. 2020, 67, 74–82. [Google Scholar] [CrossRef]
- Chaudhary, S.; Islam, Z.; Mishra, V.; Rawat, S.; Ashraf, G.M.; Kolatkar, P.R. Sox2: A Regulatory Factor in Tumorigenesis and Metastasis. Curr. Protein Pept. Sci. 2019, 20, 495–504. [Google Scholar] [CrossRef]
- Deng, X.; Kong, F.; Li, S.; Jiang, H.; Dong, L.; Xu, X.; Zhang, X.; Yuan, H.; Xu, Y.; Chu, Y.; et al. A KLF4/PiHL/EZH2/HMGA2 Regulatory Axis and Its Function in Promoting Oxaliplatin-Resistance of Colorectal Cancer. Cell Death Dis. 2021, 12, 485. [Google Scholar] [CrossRef]
- Chen, J.; Li, H.; Zhang, B.; Xiong, Z.; Jin, Z.; Chen, J.; Zheng, Y.; Zhu, X.; Zhang, S. ABI2 -mediated MEOX2/KLF4-NANOG Axis Promotes Liver Cancer Stem Cell and Drives Tumour Recurrence. Liver Int. 2022, 42, 2562–2576. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, L.; Shi, G.; Sun, S. The Deubiquitinase OTUD1 Inhibits Non-small Cell Lung Cancer Progression by Deubiquitinating and Stabilizing KLF4. Thorac. Cancer 2022, 13, 761–770. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC Oncogene—The Grand Orchestrator of Cancer Growth and Immune Evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Fatma, H.; Maurya, S.K.; Siddique, H.R. Epigenetic Modifications of C-MYC: Role in Cancer Cell Reprogramming, Progression and Chemoresistance. Semin. Cancer Biol. 2022, 83, 166–176. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of Induced Pluripotent Stem Cell Generation by the P53–P21 Pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cañamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf Locus Is a Barrier for iPS Cell Reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Panopoulos, A.D.; Herrerías, A.; Bissig, K.-D.; Lutz, M.; Berggren, W.T.; Verma, I.M.; Izpisua Belmonte, J.C. A High Proliferation Rate Is Required for Cell Reprogramming and Maintenance of Human Embryonic Stem Cell Identity. Curr. Biol. 2011, 21, 45–52. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Polo, J.M. Phases of Reprogramming. Stem Cell Res. 2014, 12, 754–761. [Google Scholar] [CrossRef]
- Lai, X.; Li, Q.; Wu, F.; Lin, J.; Chen, J.; Zheng, H.; Guo, L. Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Front. Cell Dev. Biol. 2020, 8, 760. [Google Scholar] [CrossRef]
- Smith, Z.D.; Nachman, I.; Regev, A.; Meissner, A. Dynamic Single-Cell Imaging of Direct Reprogramming Reveals an Early Specifying Event. Nat. Biotechnol. 2010, 28, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Han, Q.; Pei, D. EMT and MET as Paradigms for Cell Fate Switching. J. Mol. Cell Biol. 2012, 4, 66–69. [Google Scholar] [CrossRef]
- Chen, T.; Yuan, D.; Wei, B.; Jiang, J.; Kang, J.; Ling, K.; Gu, Y.; Li, J.; Xiao, L.; Pei, G. E-Cadherin-Mediated Cell–Cell Contact Is Critical for Induced Pluripotent Stem Cell Generation. Stem Cells 2010, 28, 1315–1325. [Google Scholar] [CrossRef]
- Schwager, S.C.; Taufalele, P.V.; Reinhart-King, C.A. Cell–Cell Mechanical Communication in Cancer. Cel. Mol. Bioeng. 2019, 12, 1–14. [Google Scholar] [CrossRef]
- Roy, N.; Hebrok, M. Regulation of Cellular Identity in Cancer. Dev. Cell 2015, 35, 674–684. [Google Scholar] [CrossRef]
- Rasmussen, T.P. Parallels between Artificial Reprogramming and the Biogenesis of Cancer Stem Cells: Involvement of lncRNAs. Semin. Cancer Biol. 2019, 57, 36–44. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and Reprogramming: Origins of Cancer Stem Cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Saito, S.; Lin, Y.-C.; Nakamura, Y.; Eckner, R.; Wuputra, K.; Kuo, K.-K.; Lin, C.-S.; Yokoyama, K.K. Potential Application of Cell Reprogramming Techniques for Cancer Research. Cell. Mol. Life Sci. 2019, 76, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Granados, K.; Poelchen, J.; Novak, D.; Utikal, J. Cellular Reprogramming—A Model for Melanoma Cellular Plasticity. Int. J. Mol. Sci. 2020, 21, 8274. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature Termination of Reprogramming In Vivo Leads to Cancer Development through Altered Epigenetic Regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef]
- Semi, K.; Matsuda, Y.; Ohnishi, K.; Yamada, Y. Cellular Reprogramming and Cancer Development. Int. J. Cancer 2013, 132, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Zaret, K.S. Reprogramming of Human Cancer Cells to Pluripotency for Models of Cancer Progression. EMBO J. 2015, 34, 739–747. [Google Scholar] [CrossRef]
- Huyghe, A.; Furlan, G.; Schroeder, J.; Cascales, E.; Trajkova, A.; Ruel, M.; Stüder, F.; Larcombe, M.; Yang Sun, Y.B.; Mugnier, F.; et al. Comparative Roadmaps of Reprogramming and Oncogenic Transformation Identify Bcl11b and Atoh8 as Broad Regulators of Cellular Plasticity. Nat. Cell Biol. 2022, 24, 1350–1363. [Google Scholar] [CrossRef]
- Xiong, S.; Feng, Y.; Cheng, L. Cellular Reprogramming as a Therapeutic Target in Cancer. Trends Cell Biol. 2019, 29, 623–634. [Google Scholar] [CrossRef]
- Gong, L.; Yan, Q.; Zhang, Y.; Fang, X.; Liu, B.; Guan, X. Cancer Cell Reprogramming: A Promising Therapy Converting Malignancy to Benignity. Cancer Commun. 2019, 39, 48. [Google Scholar] [CrossRef]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN Deubiquitinases in NF-κB Signaling and Cell Death: So Similar, yet so Different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Harhaj, E.W.; Dixit, V.M. Regulation of NF-κB by Deubiquitinases: Deubiquitinases Regulating NF-κB Signaling. Immunol. Rev. 2012, 246, 107–124. [Google Scholar] [CrossRef]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD Is a Deubiquitinating Enzyme That Negatively Regulates NF-κB Activation by TNFR Family Members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Schlicher, L.; Wissler, M.; Preiss, F.; Brauns-Schubert, P.; Jakob, C.; Dumit, V.; Borner, C.; Dengjel, J.; Maurer, U. SPATA 2 Promotes CYLD Activity and Regulates TNF -induced NF -κB Signaling and Cell Death. EMBO Rep. 2016, 17, 1485–1497. [Google Scholar] [CrossRef]
- Kupka, S.; De Miguel, D.; Draber, P.; Martino, L.; Surinova, S.; Rittinger, K.; Walczak, H. SPATA2-Mediated Binding of CYLD to HOIP Enables CYLD Recruitment to Signaling Complexes. Cell Rep. 2016, 16, 2271–2280. [Google Scholar] [CrossRef]
- Pseftogas, A.; Xanthopoulos, K.; Poutahidis, T.; Ainali, C.; Dafou, D.; Panteris, E.; Kern, J.G.; Varelas, X.; Hardas, A.; Gonidas, C.; et al. The Tumor Suppressor CYLD Inhibits Mammary Epithelial to Mesenchymal Transition by the Coordinated Inhibition of YAP/TAZ and TGFβ Signaling. Cancers 2020, 12, 2047. [Google Scholar] [CrossRef]
- Zhao, Y.; Thornton, A.M.; Kinney, M.C.; Ma, C.A.; Spinner, J.J.; Fuss, I.J.; Shevach, E.M.; Jain, A. The Deubiquitinase CYLD Targets Smad7 Protein to Regulate Transforming Growth Factor β (TGF-β) Signaling and the Development of Regulatory T Cells. J. Biol. Chem. 2011, 286, 40520–40530. [Google Scholar] [CrossRef]
- Shinriki, S.; Jono, H.; Maeshiro, M.; Nakamura, T.; Guo, J.; Li, J.-D.; Ueda, M.; Yoshida, R.; Shinohara, M.; Nakayama, H.; et al. Loss of CYLD Promotes Cell Invasion via ALK5 Stabilization in Oral Squamous Cell Carcinoma: Association of CYLD with OSCC-Related Invasion. J. Pathol. 2018, 244, 367–379. [Google Scholar] [CrossRef]
- Van Andel, H.; Kocemba, K.A.; De Haan-Kramer, A.; Mellink, C.H.; Piwowar, M.; Broijl, A.; Van Duin, M.; Sonneveld, P.; Maurice, M.M.; Kersten, M.J.; et al. Loss of CYLD Expression Unleashes Wnt Signaling in Multiple Myeloma and Is Associated with Aggressive Disease. Oncogene 2017, 36, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Haegebarth, A.; Kuper, I.; Edelmann, M.J.; Henraat, M.; Canninga-van Dijk, M.R.; Kessler, B.M.; Clevers, H.; Maurice, M.M. Loss of the Tumor Suppressor CYLD Enhances Wnt/β-Catenin Signaling through K63-Linked Ubiquitination of Dvl. Mol. Cell 2010, 37, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Z.; Wang, P.; Li, D.; Zhou, J.; Wu, S. CYLD Negatively Regulates Hippo Signaling by Limiting Hpo Phosphorylation in Drosophila. Biochem. Biophys. Res. Commun. 2014, 452, 808–812. [Google Scholar] [CrossRef]
- Nikolaou, K.; Tsagaratou, A.; Eftychi, C.; Kollias, G.; Mosialos, G.; Talianidis, I. Inactivation of the Deubiquitinase CYLD in Hepatocytes Causes Apoptosis, Inflammation, Fibrosis, and Cancer. Cancer Cell 2012, 21, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Patai, Á.V.; Hogenson, T.L.; Fernandez-Zapico, M.E.; Qin, B.; Sinicrope, F.A. Irreversible JNK Blockade Overcomes PD-L1-Mediated Resistance to Chemotherapy in Colorectal Cancer. Oncogene 2021, 40, 5105–5115. [Google Scholar] [CrossRef]
- Pannem, R.R.; Dorn, C.; Ahlqvist, K.; Bosserhoff, A.K.; Hellerbrand, C.; Massoumi, R. CYLD Controls C-MYC Expression through the JNK-Dependent Signaling Pathway in Hepatocellular Carcinoma. Carcinogenesis 2014, 35, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Herlyn, M.; Yang, X. TRIM15 and CYLD Regulate ERK Activation via Lysine-63-Linked Polyubiquitination. Nat. Cell Biol. 2021, 23, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Liu, K.; Zhang, S.; Gao, S.; Chen, W.; Li, D.; Huang, Y. Bisdemethoxycurcumin Inhibits Hepatocellular Carcinoma Proliferation Through Akt Inactivation via CYLD-Mediated Deubiquitination. Drug Des. Dev. Ther. 2020, 14, 993–1001. [Google Scholar] [CrossRef]
- Yang, W.-L.; Jin, G.; Li, C.-F.; Jeong, Y.S.; Moten, A.; Xu, D.; Feng, Z.; Chen, W.; Cai, Z.; Darnay, B.; et al. Cycles of Ubiquitination and Deubiquitination Critically Regulate Growth Factor–Mediated Activation of Akt Signaling. Sci. Signal. 2013, 6, ra3. [Google Scholar] [CrossRef]
- Lim, J.H.; Jono, H.; Komatsu, K.; Woo, C.-H.; Lee, J.; Miyata, M.; Matsuno, T.; Xu, X.; Huang, Y.; Zhang, W.; et al. CYLD Negatively Regulates Transforming Growth Factor-β-Signalling via Deubiquitinating Akt. Nat. Commun. 2012, 3, 771. [Google Scholar] [CrossRef]
- Haq, S.; Sarodaya, N.; Karapurkar, J.K.; Suresh, B.; Jo, J.K.; Singh, V.; Bae, Y.S.; Kim, K.-S.; Ramakrishna, S. CYLD Destabilizes NoxO1 Protein by Promoting Ubiquitination and Regulates Prostate Cancer Progression. Cancer Lett. 2022, 525, 146–157. [Google Scholar] [CrossRef]
- Wang, L.; Lin, Y.; Zhou, X.; Chen, Y.; Li, X.; Luo, W.; Zhou, Y.; Cai, L. CYLD Deficiency Enhances Metabolic Reprogramming and Tumor Progression in Nasopharyngeal Carcinoma via PFKFB3. Cancer Lett. 2022, 532, 215586. [Google Scholar] [CrossRef]
- Cui, Z.; Kang, H.; Grandis, J.R.; Johnson, D.E. CYLD Alterations in the Tumorigenesis and Progression of Human Papillomavirus–Associated Head and Neck Cancers. Mol. Cancer Res. 2021, 19, 14–24. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Mi, Z.; Jiang, X.; Sun, L.; Zheng, B.; Wang, J.; Meng, M.; Zhang, L.; Wang, Z.; et al. STAT3/miR-135b/NF-κB Axis Confers Aggressiveness and Unfavorable Prognosis in Non-Small-Cell Lung Cancer. Cell Death Dis. 2021, 12, 493. [Google Scholar] [CrossRef]
- Wang, J.; Tan, L.; Jia, B.; Yu, X.; Yao, R.; OUYang, N.; Yu, X.; Cao, X.; Tong, J.; Chen, T.; et al. Downregulation of m6A Reader YTHDC2 Promotes the Proliferation and Migration of Malignant Lung Cells via CYLD/NF-κB Pathway. Int. J. Biol. Sci. 2021, 17, 2633–2651. [Google Scholar] [CrossRef] [PubMed]
- Minderman, M.; Lantermans, H.C.; Grüneberg, L.J.; Cillessen, S.A.G.M.; Bende, R.J.; Van Noesel, C.J.M.; Kersten, M.J.; Pals, S.T.; Spaargaren, M. MALT1-Dependent Cleavage of CYLD Promotes NF-κB Signaling and Growth of Aggressive B-Cell Receptor-Dependent Lymphomas. Blood Cancer J. 2023, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, S.; Li, H.-L.; Luo, H.; Wu, X.; Lu, J.; Wang, H.-W.; Chen, Y.; Chen, D.; Wu, W.-T.; et al. FOSL1 Promotes Proneural-to-Mesenchymal Transition of Glioblastoma Stem Cells via UBC9/CYLD/NF-κB Axis. Mol. Ther. 2022, 30, 2568–2583. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Lei, Y.; Huang, Y.; Zhang, J.; Chen, Y.; Chen, K.; Lin, J.; Sun, S.; Lin, X. CYLD Expression in Dendritic Cells Involved in the Immunoregulation of Pulmonary Adenocarcinoma via NF-κB Pathway. Artif. Cells Nanomed. Biotechnol. 2020, 48, 137–142. [Google Scholar] [CrossRef]
- Zhou, S.; Yin, D.; Hu, Z.; Luo, C.; Zhou, Z.; Xin, H.; Yang, X.; Shi, Y.; Wang, Z.; Huang, X.; et al. A Positive Feedback Loop Between Cancer Stem-Like Cells and Tumor-Associated Neutrophils Controls Hepatocellular Carcinoma Progression. Hepatology 2019, 70, 1214–1230. [Google Scholar] [CrossRef]
- Xu, D.; Zhou, P.; Wang, Y.; Zhang, L.; Fu, W.; Ruan, B.; Xu, H.; Hu, C.; Tian, L.; Qin, J.; et al. Reciprocal Activation between STAT3 and miR-181b Regulates the Proliferation of Esophageal Cancer Stem-like Cells via the CYLD Pathway. Mol. Cancer 2016, 15, 40. [Google Scholar] [CrossRef]
- Trompouki, E.; Tsagaratou, A.; Kosmidis, S.K.; Dollé, P.; Qian, J.; Kontoyiannis, D.L.; Cardoso, W.V.; Mosialos, G. Truncation of the Catalytic Domain of the Cylindromatosis Tumor Suppressor Impairs Lung Maturation. Neoplasia 2009, 11, 469–476. [Google Scholar] [CrossRef]
- Karatzas, D.N.; Xanthopoulos, K.; Kotantaki, P.; Pseftogas, A.; Teliousis, K.; Hatzivassiliou, E.G.; Kontoyiannis, D.L.; Poutahidis, T.; Mosialos, G. Inactivation of CYLD in Intestinal Epithelial Cells Exacerbates Colitis-Associated Colorectal Carcinogenesis—A Short Report. Cell. Oncol. 2016, 39, 287–293. [Google Scholar] [CrossRef]
- Papathanasiou, M.; Tsiftsoglou, S.A.; Polyzos, A.P.; Papadopoulou, D.; Valakos, D.; Klagkou, E.; Karagianni, P.; Pliatska, M.; Talianidis, I.; Agelopoulos, M.; et al. Identification of a Dynamic Gene Regulatory Network Required for Pluripotency Factor-induced Reprogramming of Mouse Fibroblasts and Hepatocytes. EMBO J. 2021, 40, e102236. [Google Scholar] [CrossRef]
- Aranda, P.S.; LaJoie, D.M.; Jorcyk, C.L. Bleach Gel: A Simple Agarose Gel for Analyzing RNA Quality. Electrophoresis 2012, 33, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Marumoto, T.; Yamaguchi, S.; Okano, S.; Takeda, N.; Sakamoto, C.; Kawano, H.; Nii, T.; Miyamoto, S.; Nagai, Y.; et al. Inhibition of PTEN Tumor Suppressor Promotes the Generation of Induced Pluripotent Stem Cells. Mol. Ther. 2013, 21, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Hewapathirana, S.; García-Seisdedos, D.; Kamatchinathan, S.; Kundu, D.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Wang, W.; Yang, J.; Liu, H.; Lu, D.; Chen, X.; Zenonos, Z.; Campos, L.S.; Rad, R.; Guo, G.; Zhang, S.; et al. Rapid and Efficient Reprogramming of Somatic Cells to Induced Pluripotent Stem Cells by Retinoic Acid Receptor Gamma and Liver Receptor Homolog 1. Proc. Natl. Acad. Sci. USA 2011, 108, 18283–18288. [Google Scholar] [CrossRef]
- Wang, J.; Yu, H.; Ma, Q.; Zeng, P.; Wu, D.; Hou, Y.; Liu, X.; Jia, L.; Sun, J.; Chen, Y.; et al. Phase Separation of OCT4 Controls TAD Reorganization to Promote Cell Fate Transitions. Cell Stem Cell 2021, 28, 1868–1883. [Google Scholar] [CrossRef] [PubMed]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional Genomics Reveals a BMP-Driven Mesenchymal-to-Epithelial Transition in the Initiation of Somatic Cell Reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A Mesenchymal-to-Epithelial Transition Initiates and Is Required for the Nuclear Reprogramming of Mouse Fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A Molecular Roadmap of Reprogramming Somatic Cells into iPS Cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-Cell Analysis Uncovers Fibroblast Heterogeneity and Criteria for Fibroblast and Mural Cell Identification and Discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Evans, T. Activation-Induced Cytidine Deaminase Regulates Fibroblast Growth Factor/Extracellular Signal-Regulated Kinases Signaling to Achieve the Naïve Pluripotent State During Reprogramming. Stem Cells 2019, 37, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Ngondo, R.P.; Cirera-Salinas, D.; Yu, J.; Wischnewski, H.; Bodak, M.; Vandormael-Pournin, S.; Geiselmann, A.; Wettstein, R.; Luitz, J.; Cohen-Tannoudji, M.; et al. Argonaute 2 Is Required for Extra-Embryonic Endoderm Differentiation of Mouse Embryonic Stem Cells. Stem Cell Rep. 2018, 10, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhao, W.; Huang, Y.; Tong, H.; Xia, Y.; Jiang, Q.; Qin, J. Loss of Polycomb Group Protein Pcgf1 Severely Compromises Proper Differentiation of Embryonic Stem Cells. Sci. Rep. 2017, 7, 46276. [Google Scholar] [CrossRef]
- David, L.; Samavarchi-Tehrani, P.; Golipour, A.; Wrana, J.L. Looking into the Black Box: Insights into the Mechanisms of Somatic Cell Reprogramming. Genes 2011, 2, 81–106. [Google Scholar] [CrossRef]
- Nagasaka, R.; Matsumoto, M.; Okada, M.; Sasaki, H.; Kanie, K.; Kii, H.; Uozumi, T.; Kiyota, Y.; Honda, H.; Kato, R. Visualization of Morphological Categories of Colonies for Monitoring of Effect on Induced Pluripotent Stem Cell Culture Status. Regen. Ther. 2017, 6, 41–51. [Google Scholar] [CrossRef]
- Jo, J.; Hwang, S.; Kim, H.J.; Hong, S.; Lee, J.E.; Lee, S.-G.; Baek, A.; Han, H.; Lee, J.I.; Lee, I.; et al. An Integrated Systems Biology Approach Identifies Positive Cofactor 4 as a Factor That Increases Reprogramming Efficiency. Nucleic Acids Res. 2016, 44, 1203–1215. [Google Scholar] [CrossRef]
- Tai, C.-I.; Ying, Q.-L. Gbx2, a LIF/Stat3 Target, Promotes Reprogramming to and Retention of the Pluripotent Ground State. J. Cell Sci. 2013, 126, 1093–1098. [Google Scholar] [CrossRef]
- Yang, C.-S.; Chang, K.-Y.; Rana, T.M. Genome-Wide Functional Analysis Reveals Factors Needed at the Transition Steps of Induced Reprogramming. Cell Rep. 2014, 8, 327–337. [Google Scholar] [CrossRef]
- O’Malley, J.; Skylaki, S.; Iwabuchi, K.A.; Chantzoura, E.; Ruetz, T.; Johnsson, A.; Tomlinson, S.R.; Linnarsson, S.; Kaji, K. High-Resolution Analysis with Novel Cell-Surface Markers Identifies Routes to iPS Cells. Nature 2013, 499, 88–91. [Google Scholar] [CrossRef]
- Kim, S.-I.; Oceguera-Yanez, F.; Hirohata, R.; Linker, S.; Okita, K.; Yamada, Y.; Yamamoto, T.; Yamanaka, S.; Woltjen, K. KLF4 N-Terminal Variance Modulates Induced Reprogramming to Pluripotency. Stem Cell Rep. 2015, 4, 727–743. [Google Scholar] [CrossRef]
- Li, X.; Pei, D.; Zheng, H. Transitions between Epithelial and Mesenchymal States during Cell Fate Conversions. Protein Cell 2014, 5, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Taha, I.N.; Clauser, K.R.; Gao, Y.; Naba, A. MatrisomeDB: The ECM-Protein Knowledge Database. Nucleic Acids Res. 2020, 48, D1136–D1144. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.H.; Costa, L.G.; Shaffer, S.A.; Goodlett, D.R.; Guizzetti, M. Shotgun Proteomics Implicates Extracellular Matrix Proteins and Protease Systems in Neuronal Development Induced by Astrocyte Cholinergic Stimulation. J. Neurochem. 2009, 108, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Apóstolo, N.; Smukowski, S.N.; Vanderlinden, J.; Condomitti, G.; Rybakin, V.; Ten Bos, J.; Trobiani, L.; Portegies, S.; Vennekens, K.M.; Gounko, N.V.; et al. Synapse Type-Specific Proteomic Dissection Identifies IgSF8 as a Hippocampal CA3 Microcircuit Organizer. Nat. Commun. 2020, 11, 5171. [Google Scholar] [CrossRef]
- Jaber, M.; Radwan, A.; Loyfer, N.; Abdeen, M.; Sebban, S.; Khatib, A.; Yassen, H.; Kolb, T.; Zapatka, M.; Makedonski, K.; et al. Comparative Parallel Multi-Omics Analysis during the Induction of Pluripotent and Trophectoderm States. Nat. Commun. 2022, 13, 3475. [Google Scholar] [CrossRef]
- Bansho, Y.; Lee, J.; Nishida, E.; Nakajima-Koyama, M. Identification and Characterization of Secreted Factors That Are Upregulated during Somatic Cell Reprogramming. FEBS Lett. 2017, 591, 1584–1600. [Google Scholar] [CrossRef]
- Chondronasiou, D.; Martínez De Villarreal, J.; Melendez, E.; Lynch, C.J.; Pozo, N.D.; Kovatcheva, M.; Aguilera, M.; Prats, N.; Real, F.X.; Serrano, M. Deciphering the Roadmap of in Vivo Reprogramming toward Pluripotency. Stem Cell Rep. 2022, 17, 2501–2517. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Hansson, J.; Rafiee, M.R.; Reiland, S.; Polo, J.M.; Gehring, J.; Okawa, S.; Huber, W.; Hochedlinger, K.; Krijgsveld, J. Highly Coordinated Proteome Dynamics during Reprogramming of Somatic Cells to Pluripotency. Cell Rep. 2012, 2, 1579–1592. [Google Scholar] [CrossRef]
- Jiao, J.; Dang, Y.; Yang, Y.; Gao, R.; Zhang, Y.; Kou, Z.; Sun, X.-F.; Gao, S. Promoting Reprogramming by FGF2 Reveals That the Extracellular Matrix Is a Barrier for Reprogramming Fibroblasts to Pluripotency. Stem Cells 2013, 31, 729–740. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; Den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A Resource for Pan-Cancer Analysis of Epithelial-Mesenchymal Transition Genes and Signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Urbanik, T. CYLD Deletion Triggers Nuclear Factor-κB-Signaling and Increases Cell Death Resistance in Murine Hepatocytes. World J. Gastroenterol. 2014, 20, 17049. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.-X.; Huang, Z.; Yang, X.; Wang, X.; Zhao, L.-P.; Wang, P.-X.; Zhang, X.-J.; Alves-Bezerra, M.; Cai, L.; Zhang, P.; et al. The Deubiquitinating Enzyme Cylindromatosis Mitigates Nonalcoholic Steatohepatitis. Nat. Med. 2018, 24, 213–223. [Google Scholar] [CrossRef]
- Hellerbrand, C.; Massoumi, R. Cylindromatosis-A Protective Molecule against Liver Diseases: CYLD IN LIVER DISEASE. Med. Res. Rev. 2016, 36, 342–359. [Google Scholar] [CrossRef]
- Qi, L.; Zang, H.; Wu, W.; Nagarkatti, P.; Nagarkatti, M.; Liu, Q.; Robbins, J.; Wang, X.; Cui, T. CYLD Exaggerates Pressure Overload-Induced Cardiomyopathy via Suppressing Autolysosome Efflux in Cardiomyocytes. J. Mol. Cell. Cardiol. 2020, 145, 59–73. [Google Scholar] [CrossRef]
- Wang, H.; Lai, Y.; Mathis, B.J.; Wang, W.; Li, S.; Qu, C.; Li, B.; Shao, L.; Song, H.; Janicki, J.S.; et al. Deubiquitinating Enzyme CYLD Mediates Pressure Overload-Induced Cardiac Maladaptive Remodeling and Dysfunction via Downregulating Nrf2. J. Mol. Cell. Cardiol. 2015, 84, 143–153. [Google Scholar] [CrossRef]
- Zhou, Y.-J.; Duan, D.-Q.; Lu, L.-Q.; Tang, L.-J.; Zhang, X.-J.; Luo, X.-J.; Peng, J. The SPATA2/CYLD Pathway Contributes to Doxorubicin-Induced Cardiomyocyte Ferroptosis via Enhancing Ferritinophagy. Chem.-Biol. Interact. 2022, 368, 110205. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Gao, S.; Yang, Y.; Li, H.; Zhou, J.; Chen, M.; Yang, S.; Zhang, Y.; Zhang, L.; Meng, X.; et al. CYLD Deubiquitinates Plakoglobin to Promote Cx43 Membrane Targeting and Gap Junction Assembly in the Heart. Cell Rep. 2022, 41, 111864. [Google Scholar] [CrossRef] [PubMed]
- Zajicek, A.S.; Ruan, H.; Dai, H.; Skolfield, M.C.; Phillips, H.L.; Burnette, W.J.; Javidfar, B.; Sun, S.-C.; Akbarian, S.; Yao, W.-D. Cylindromatosis Drives Synapse Pruning and Weakening by Promoting Macroautophagy through Akt-mTOR Signaling. Mol. Psychiatry 2022, 27, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, D.; Yang, L.; Long, C. Deficiency of the CYLD Impairs Fear Memory of Mice and Disrupts Neuronal Activity and Synaptic Transmission in the Basolateral Amygdala. Front. Cell. Neurosci. 2021, 15, 740165. [Google Scholar] [CrossRef]
- Pirooznia, S.K.; Wang, H.; Panicker, N.; Kumar, M.; Neifert, S.; Dar, M.A.; Lau, E.; Kang, B.G.; Redding-Ochoa, J.; Troncoso, J.C.; et al. Deubiquitinase CYLD Acts as a Negative Regulator of Dopamine Neuron Survival in Parkinson’s Disease. Sci. Adv. 2022, 8, eabh1824. [Google Scholar] [CrossRef] [PubMed]
- Ganjam, G.K.; Terpolilli, N.A.; Diemert, S.; Eisenbach, I.; Hoffmann, L.; Reuther, C.; Herden, C.; Roth, J.; Plesnila, N.; Culmsee, C. Cylindromatosis Mediates Neuronal Cell Death in Vitro and in Vivo. Cell Death Differ. 2018, 25, 1394–1407. [Google Scholar] [CrossRef]
- Nguyen, J.; Massoumi, R.; Alliston, T. CYLD, a Mechanosensitive Deubiquitinase, Regulates TGFβ Signaling in Load-Induced Bone Formation. Bone 2020, 131, 115148. [Google Scholar] [CrossRef] [PubMed]
- Läubli, H.; Borsig, L. Altered Cell Adhesion and Glycosylation Promote Cancer Immune Suppression and Metastasis. Front. Immunol. 2019, 10, 2120. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the Extracellular Matrix in Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Cox, T.R. The Matrix in Cancer. Nat. Rev. Cancer 2021, 21, 217–238. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2019, 7, 135. [Google Scholar] [CrossRef]
- Ishida, T.; Nakao, S.; Ueyama, T.; Harada, Y.; Kawamura, T. Metabolic Remodeling during Somatic Cell Reprogramming to Induced Pluripotent Stem Cells: Involvement of Hypoxia-Inducible Factor 1. Inflamm. Regen. 2020, 40, 8. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Fukuda, A.; Hisatake, K. Mechanisms of the Metabolic Shift during Somatic Cell Reprogramming. Int. J. Mol. Sci. 2019, 20, 2254. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial–Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The Metabolome of Induced Pluripotent Stem Cells Reveals Metabolic Changes Occurring in Somatic Cell Reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef] [PubMed]
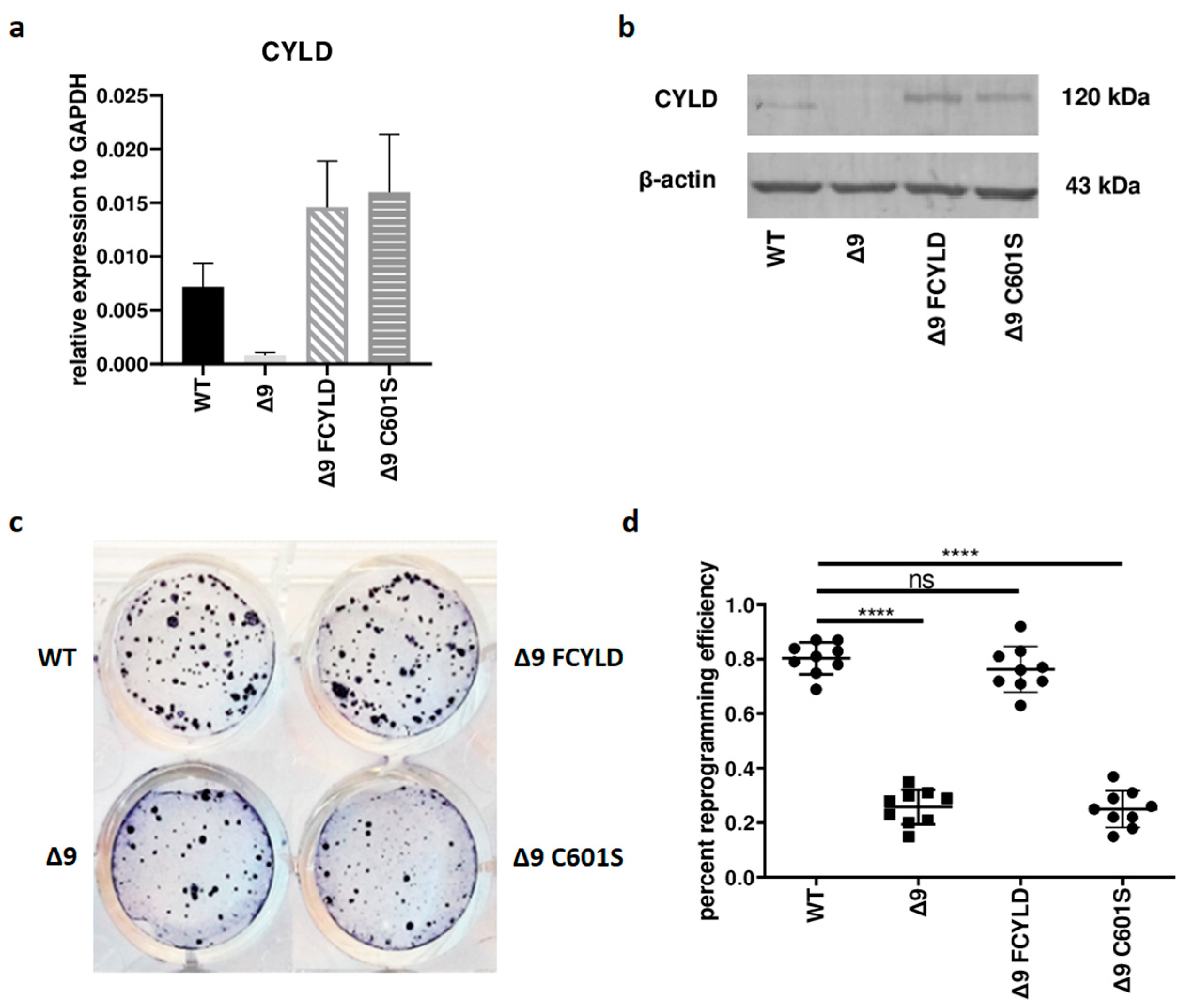
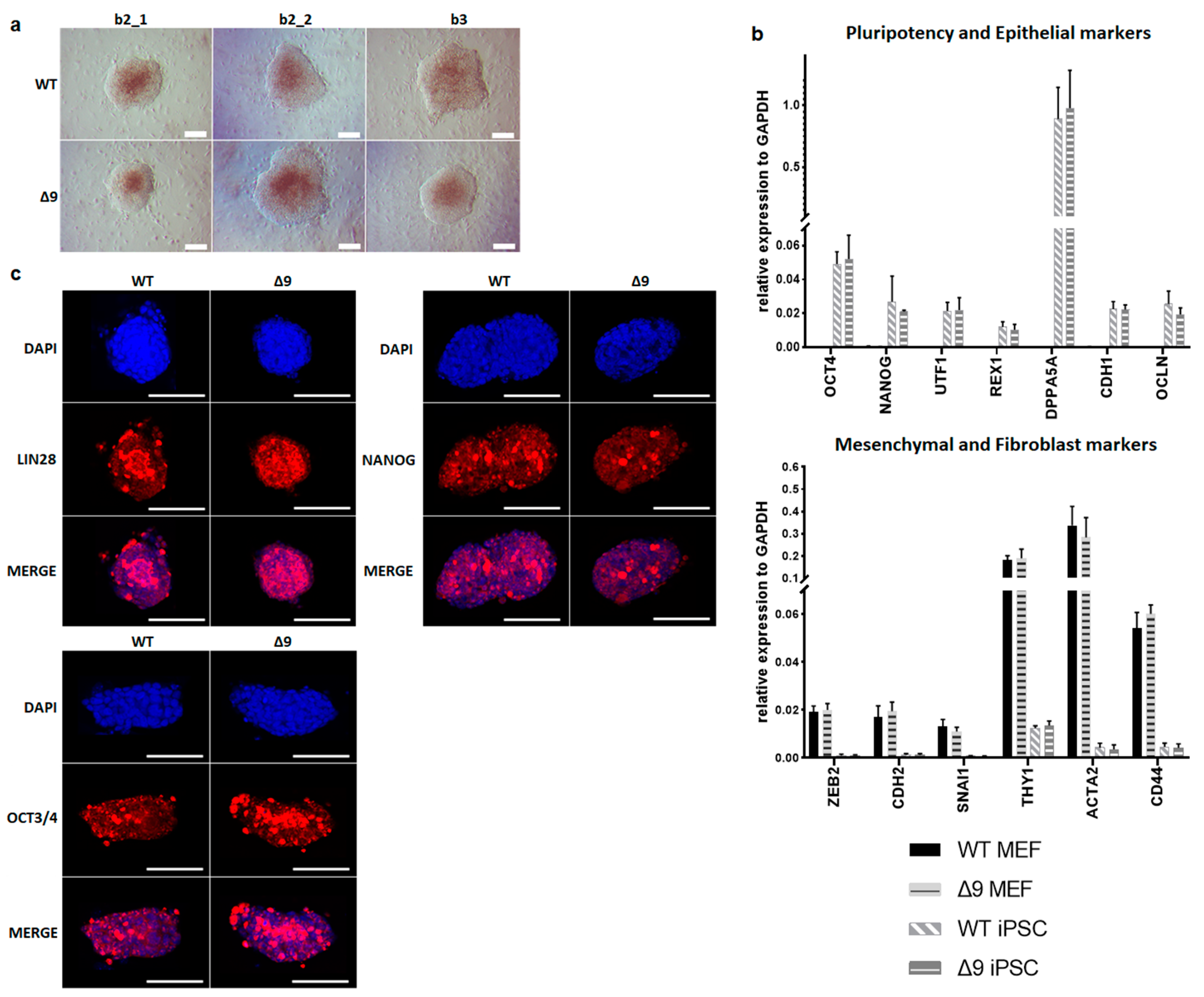
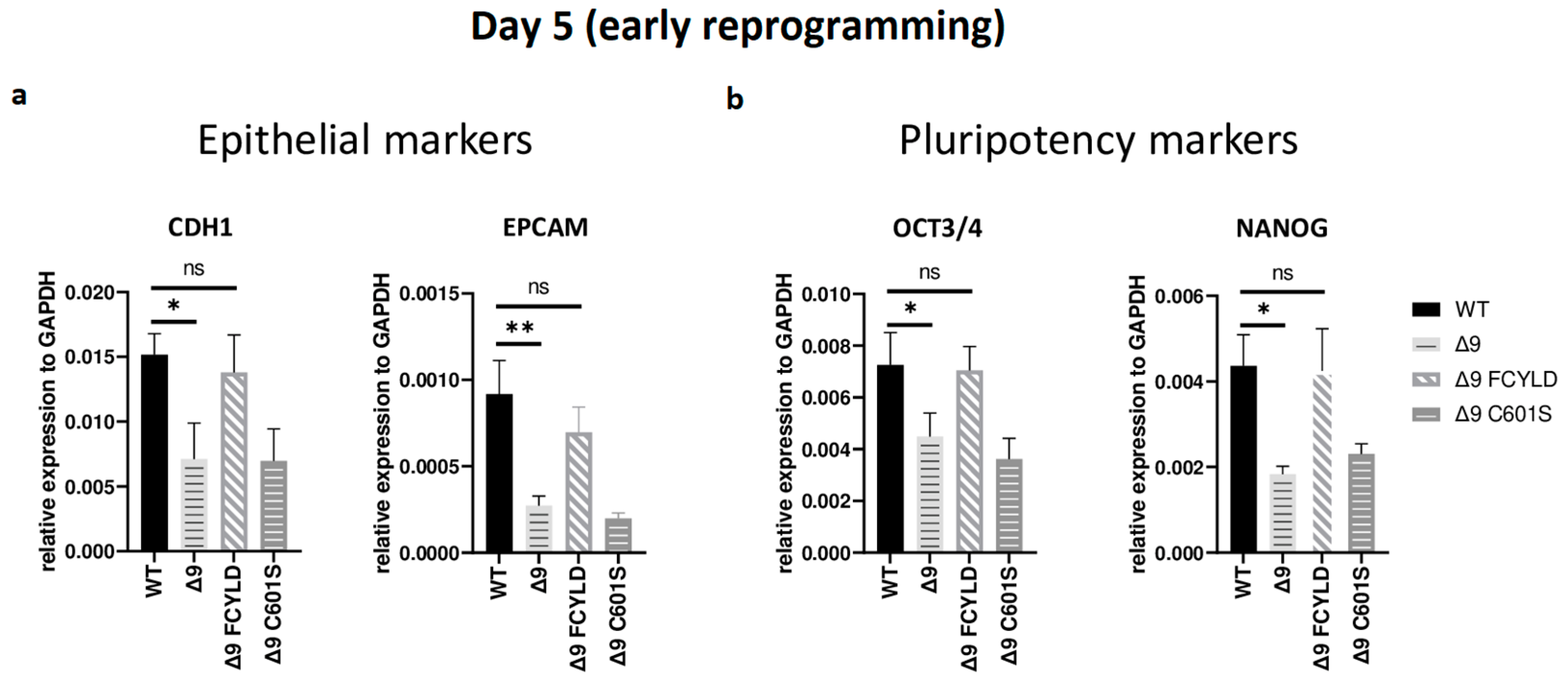
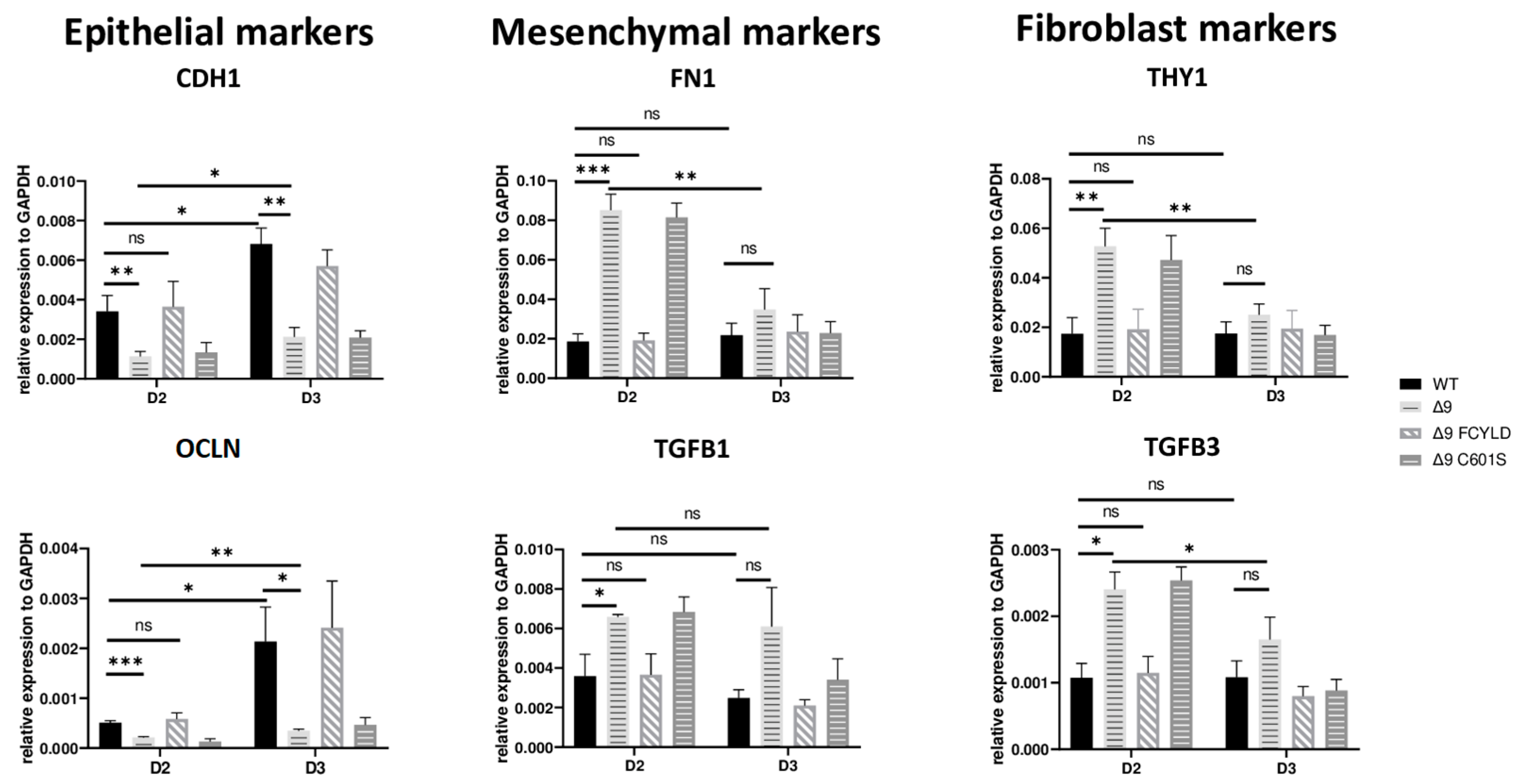
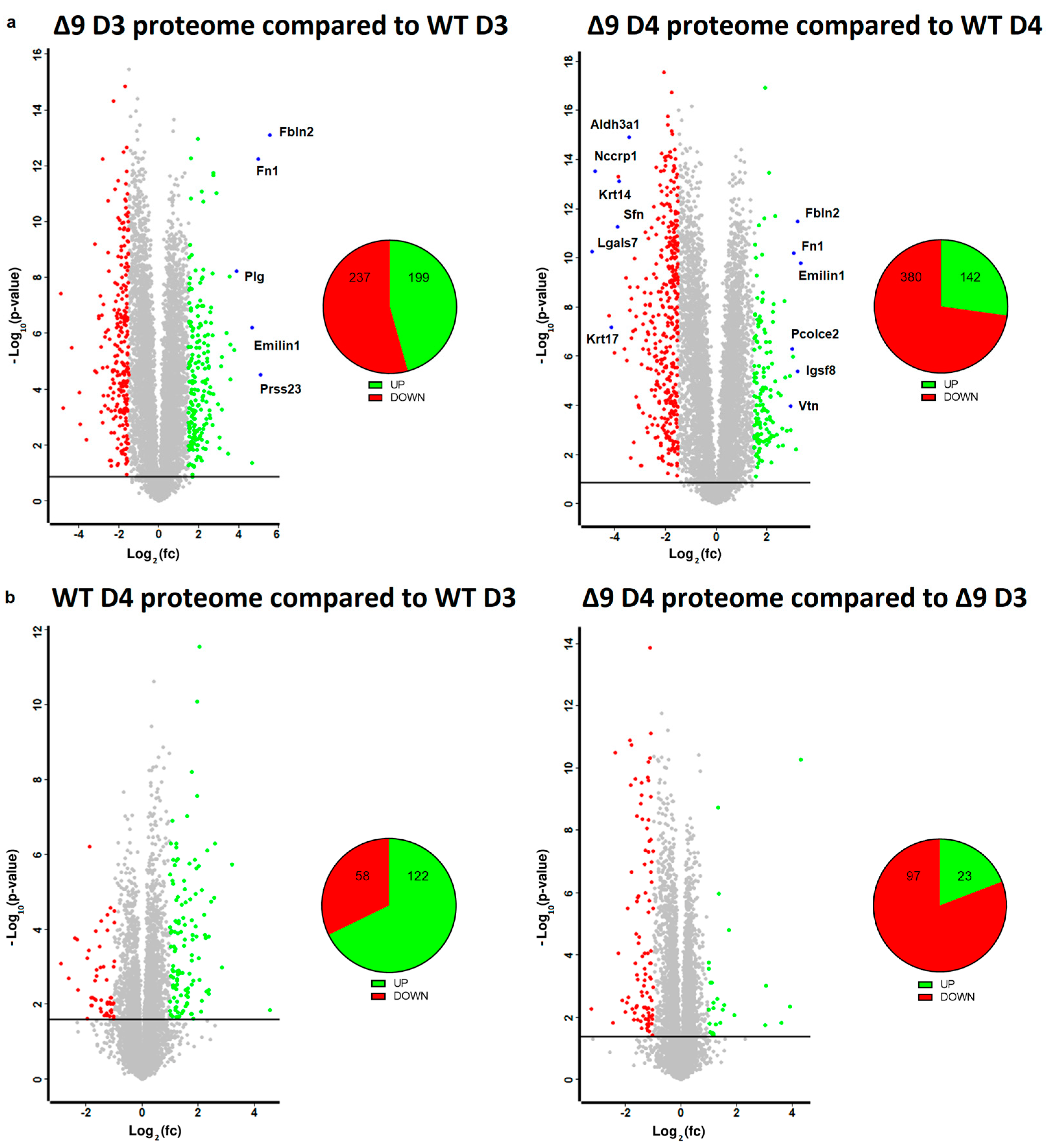
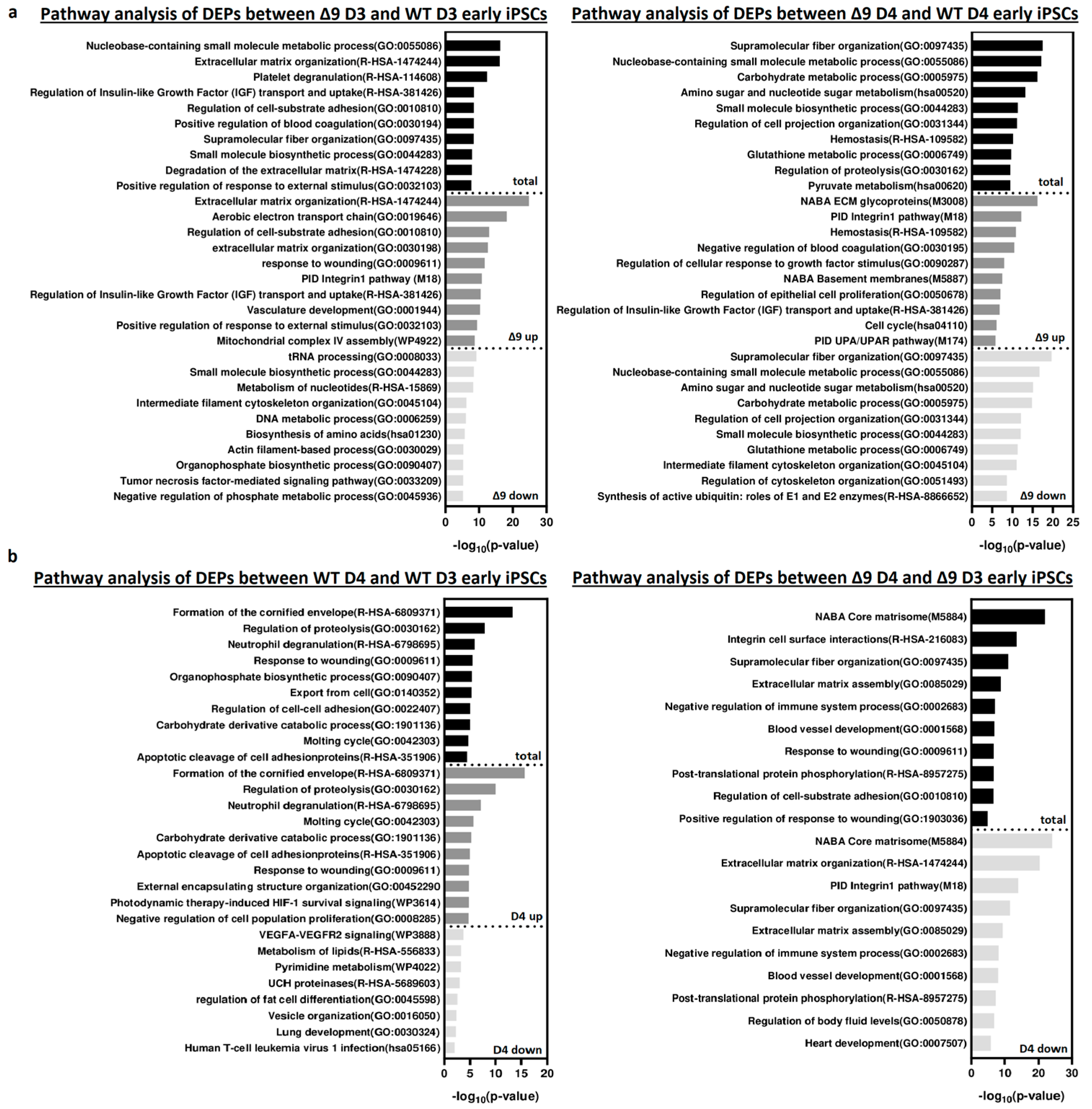
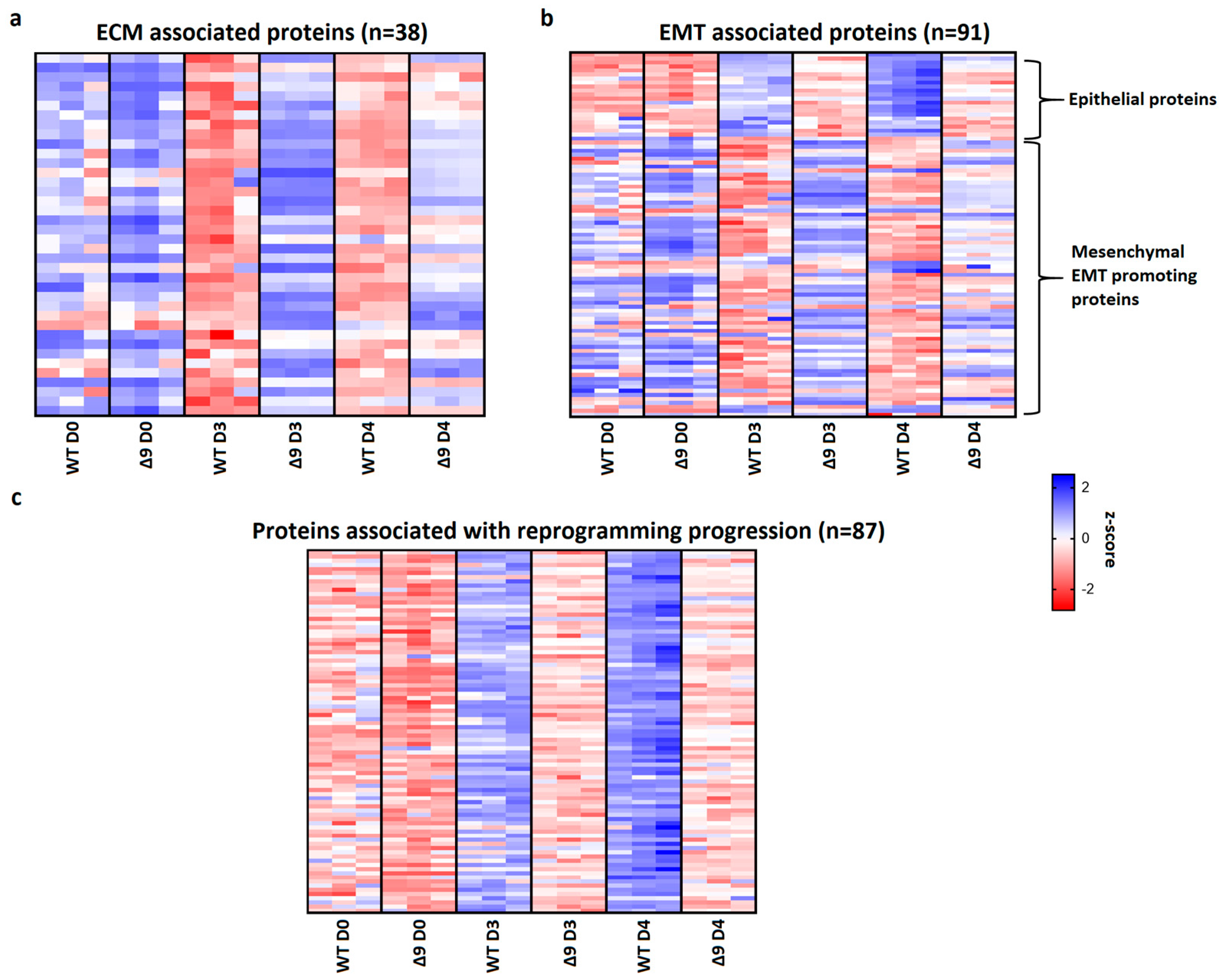
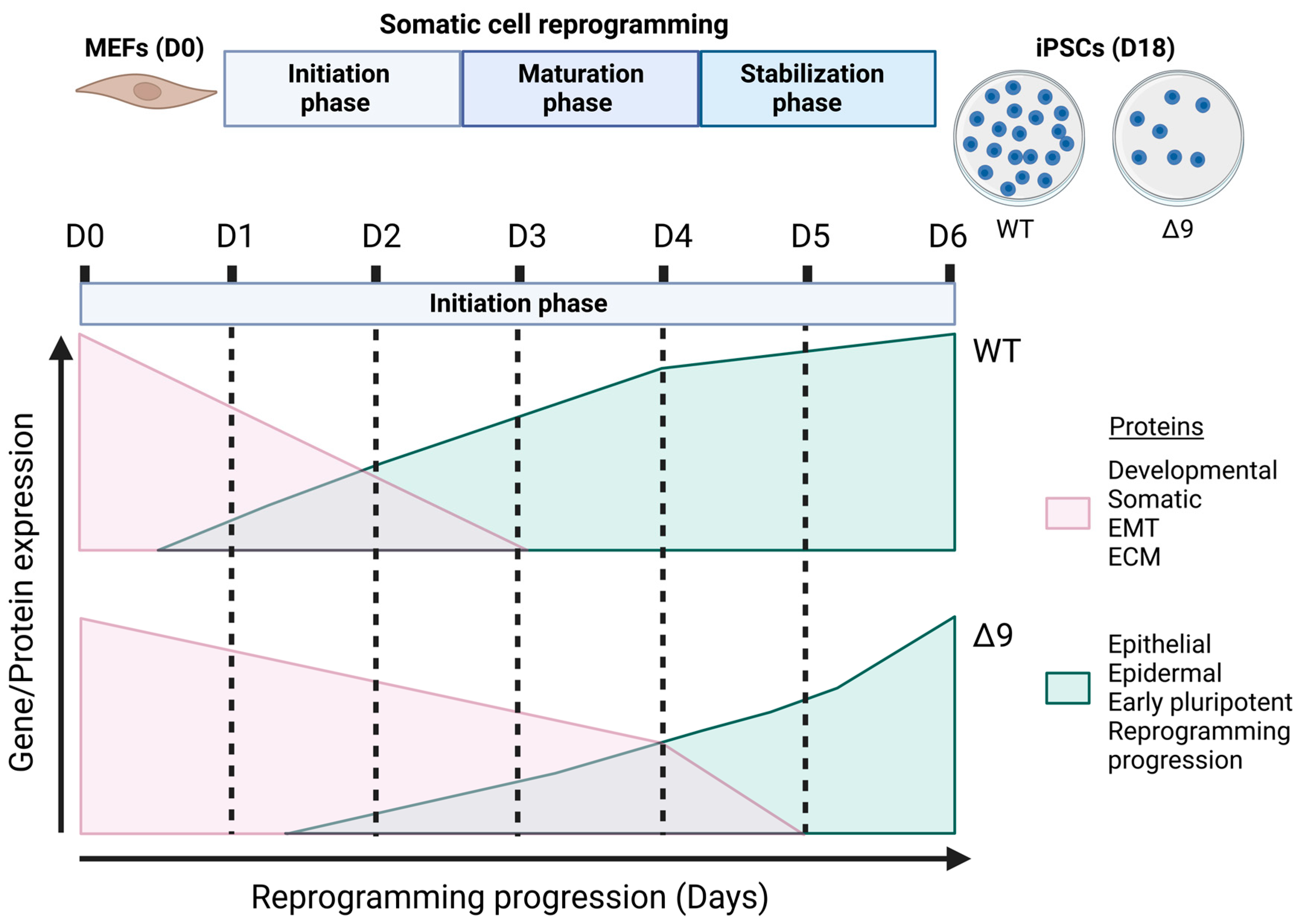
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).