

Heterogeneity in the Metastatic Microenvironment: JunB-Expressing Microglia Cells as Potential Drivers of Melanoma Brain Metastasis Progression

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Melanoma- or Microglia-Conditioned Medium
2.3. Cytokines
2.4. Animals
2.5. Intracardiac Inoculation of Tumor Cells and Immunostaining
2.6. RNA Preparation and Reverse Transcription Quantitative Real-Time PCR (RT-qPCR)
2.7. Western Blotting
2.8. ELISA Assay
2.9. Flow Cytometry
2.10. Construction of JunBhi and JunBlo Microglia Cells
2.11. RNA-Sequencing Analysis
2.12. Reverse-Phase Protein Analysis (RPPA)
2.13. Inhibitors
2.14. Viability Assay (XTT)
2.15. Apoptosis Assay
2.16. Spheroid (3D) Cultures
2.17. Wound-Healing Assay
2.18. Phagocytosis Assay
2.19. Nitric Oxide (NO) Production
2.20. Biostatistic Analysis
3. Results
3.1. Melanoma Impacts Microglial Molecular Expression
3.1.1. Melanoma-Conditioned Medium (MCM) Alters the Gene Expression Profile of Microglia Cells
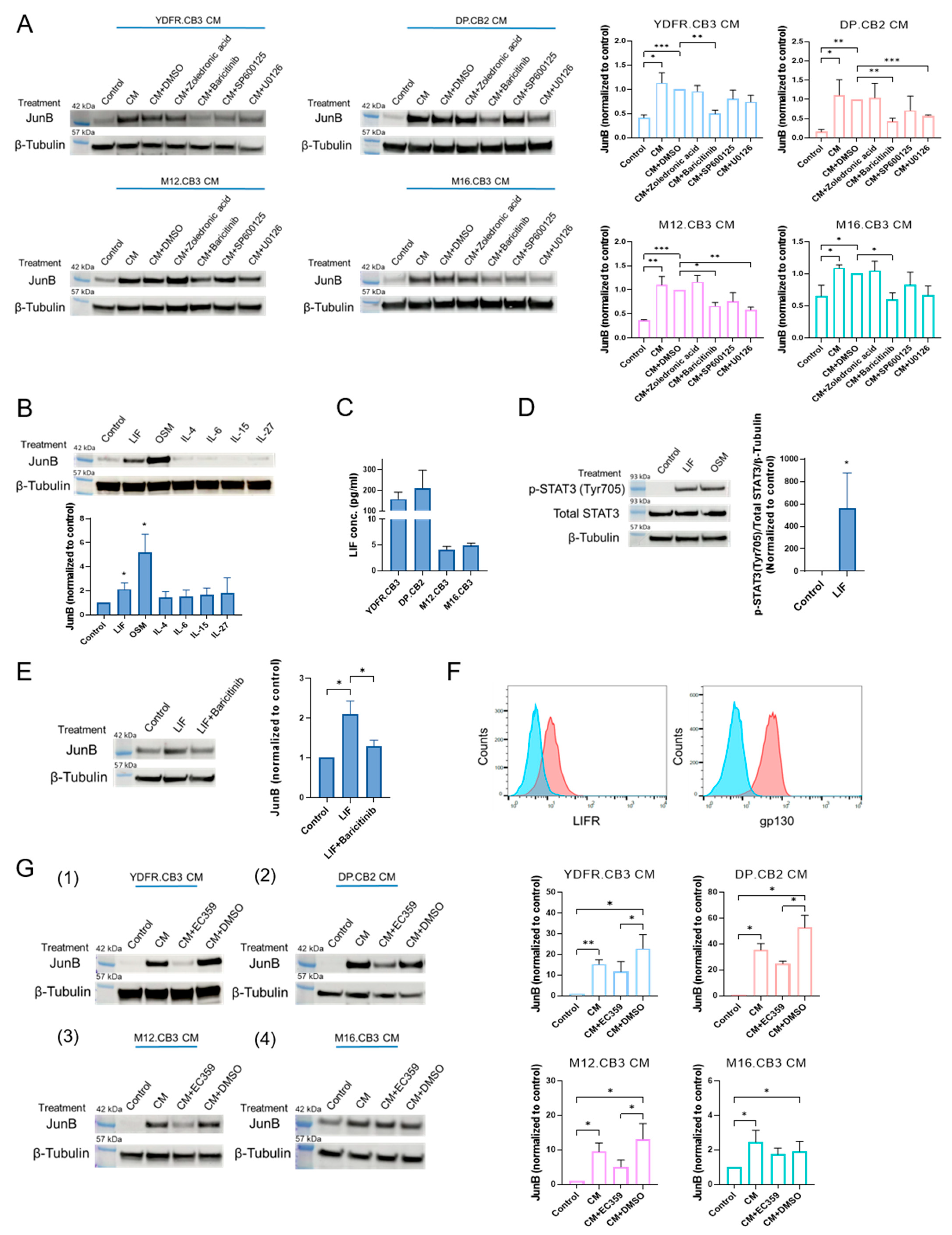
3.1.2. MCM Upregulates JunB Expression in Microglia through the JAK/STAT Signaling Pathway
3.1.3. Leukemia Inhibitory Factor Secreted by Melanoma Cells Upregulates Microglial JunB via the JAK/STAT Signaling Pathway
3.2. Melanoma-Associated Microglia Cells Expressing High JunB Levels Exhibit a Pro-Tumorigenic, Immunosuppressive Phenotype
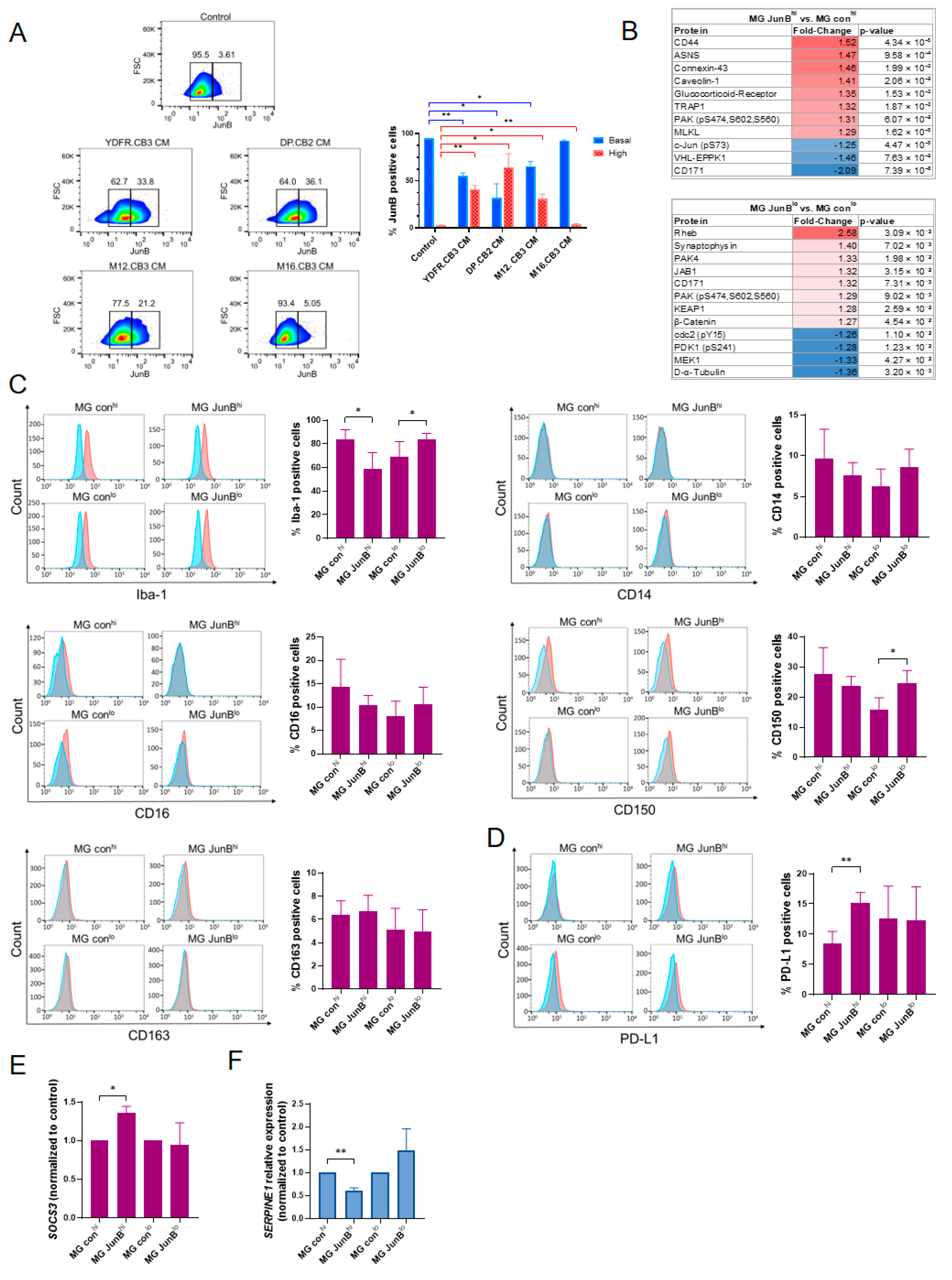
3.2.1. Factors Secreted by Melanoma Cells Induce JunB Expression Heterogeneity in Microglial Cells
3.2.2. Generation of Stable Microglia Variants Expressing High or Low Levels of JunB
3.2.3. JunBhi and JunBlo Microglia Cells Differ in Their Proteomic Expression Profile
3.2.4. JunBhi and JunBlo Microglia Differentially Express Microglial Activation Markers and Immunosuppressive Molecules
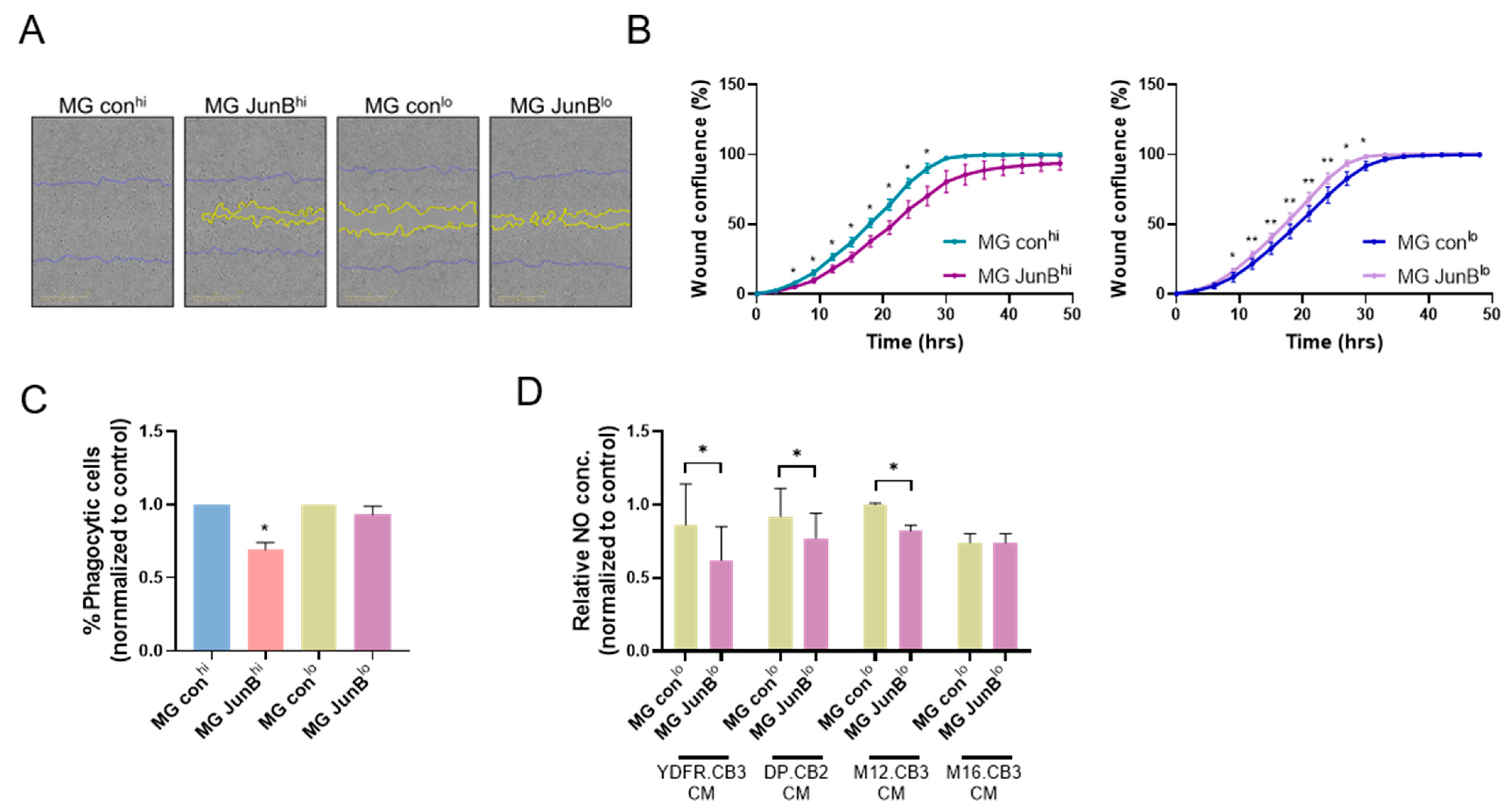
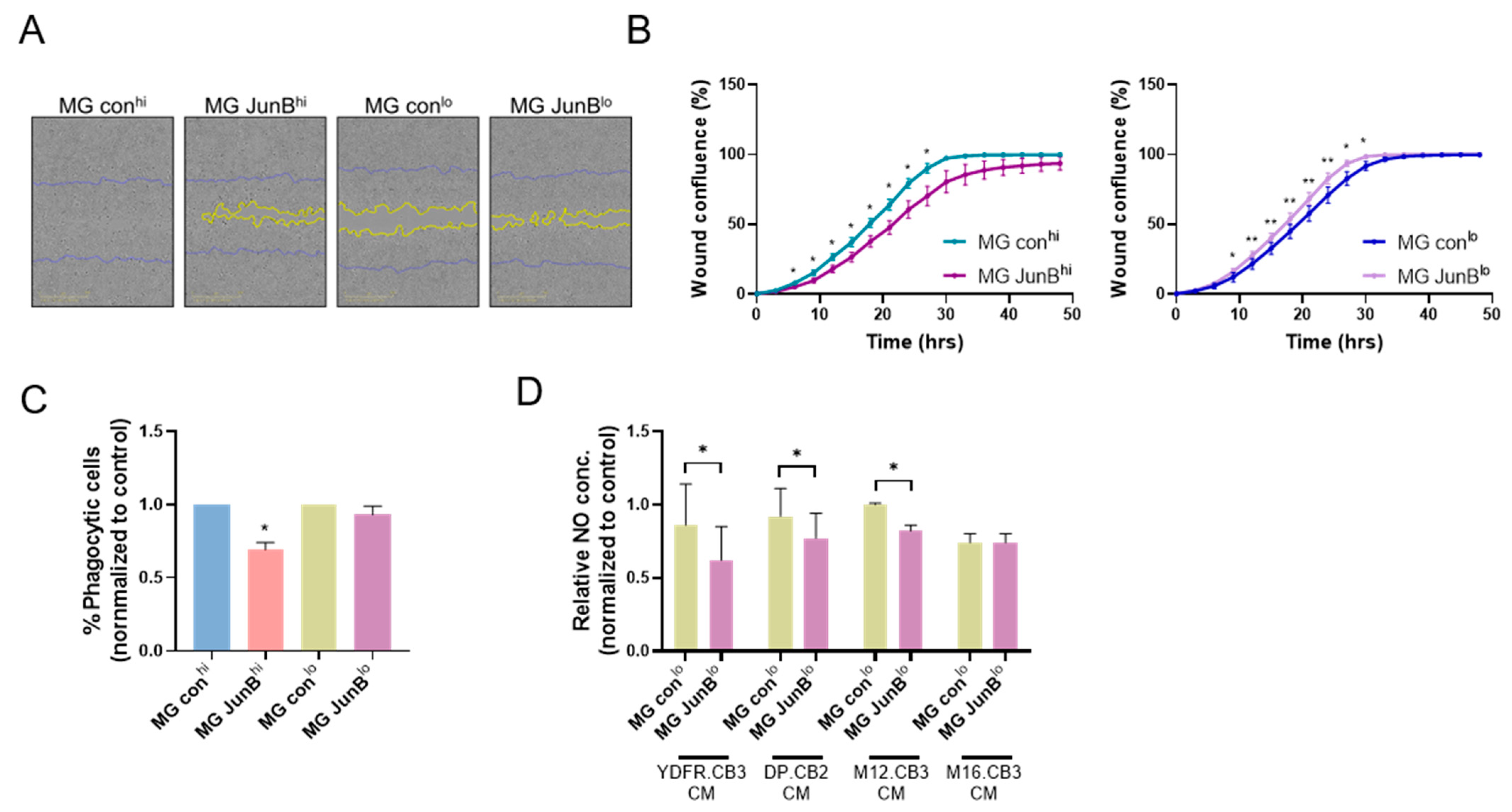
3.2.5. JunBhi and JunBlo Microglia Cells Differ in Their Migratory Capacity and Phagocytic Activity
3.2.6. JunBlo Cells Exhibit Increased Consumption of Melanoma-Derived Nitric Oxide (NO)
3.3. The Involvement of Melanoma-Associated Microglia Cells Expressing High or Low JunB Levels in the Progression of Brain-Metastasizing Melanoma
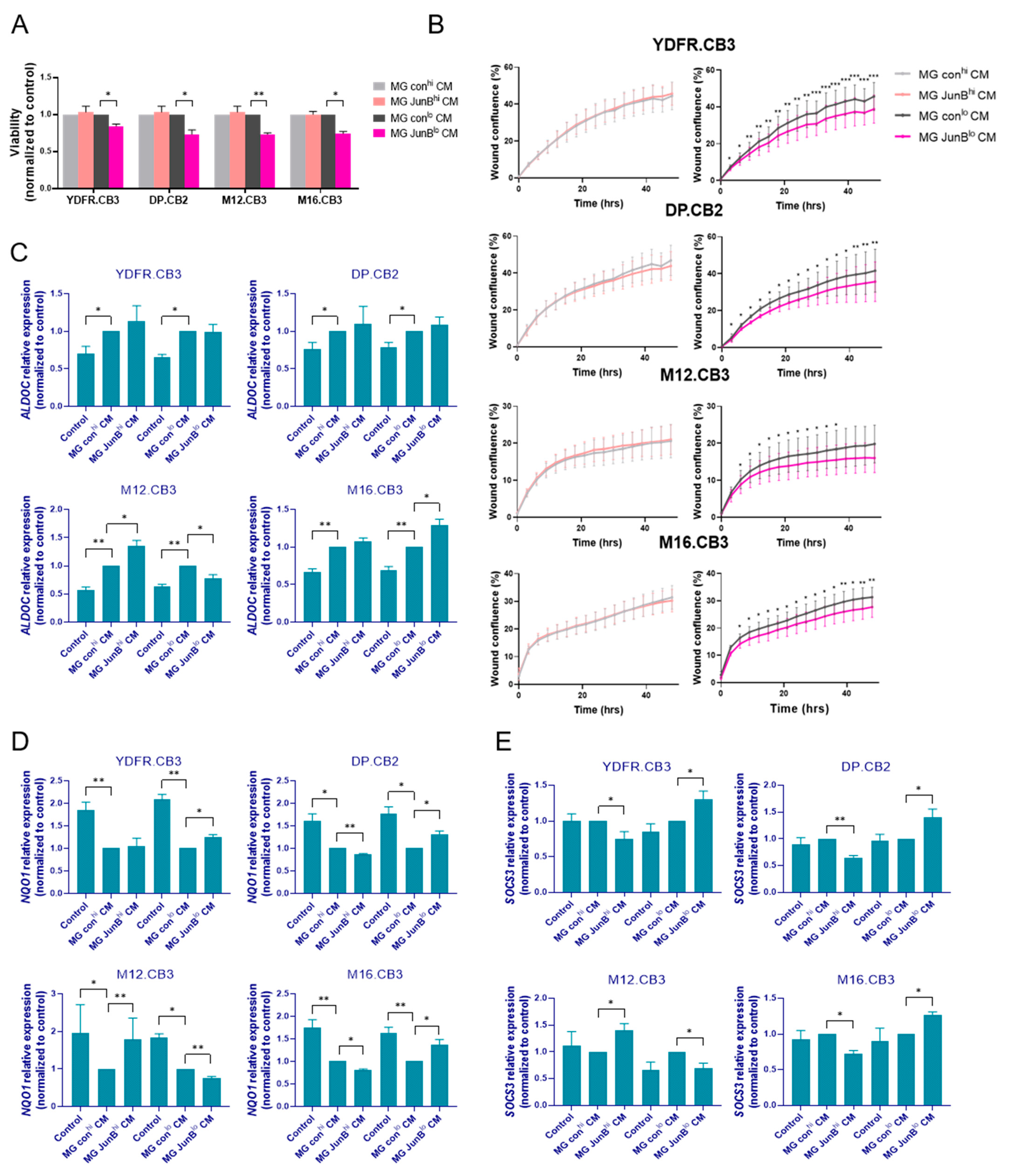
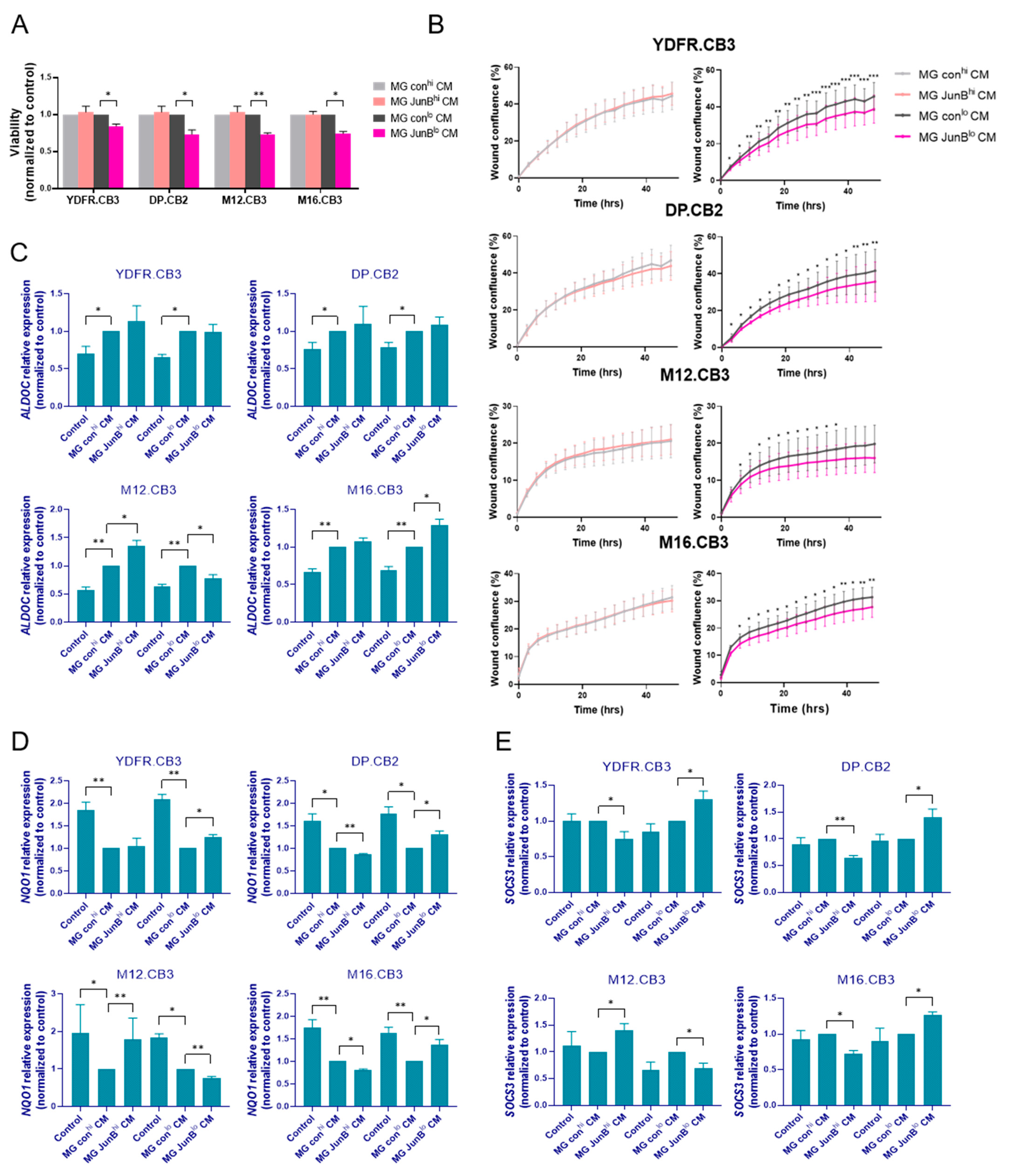
3.3.1. Factors Released from JunBlo Microglia Restrain the Proliferation and Migration of MBM Cells
3.3.2. Factors Released from JunBhi and JunBlo Microglia Regulate ALDOC, NQO1, and SOCS3 Gene Expression in MBM Cells
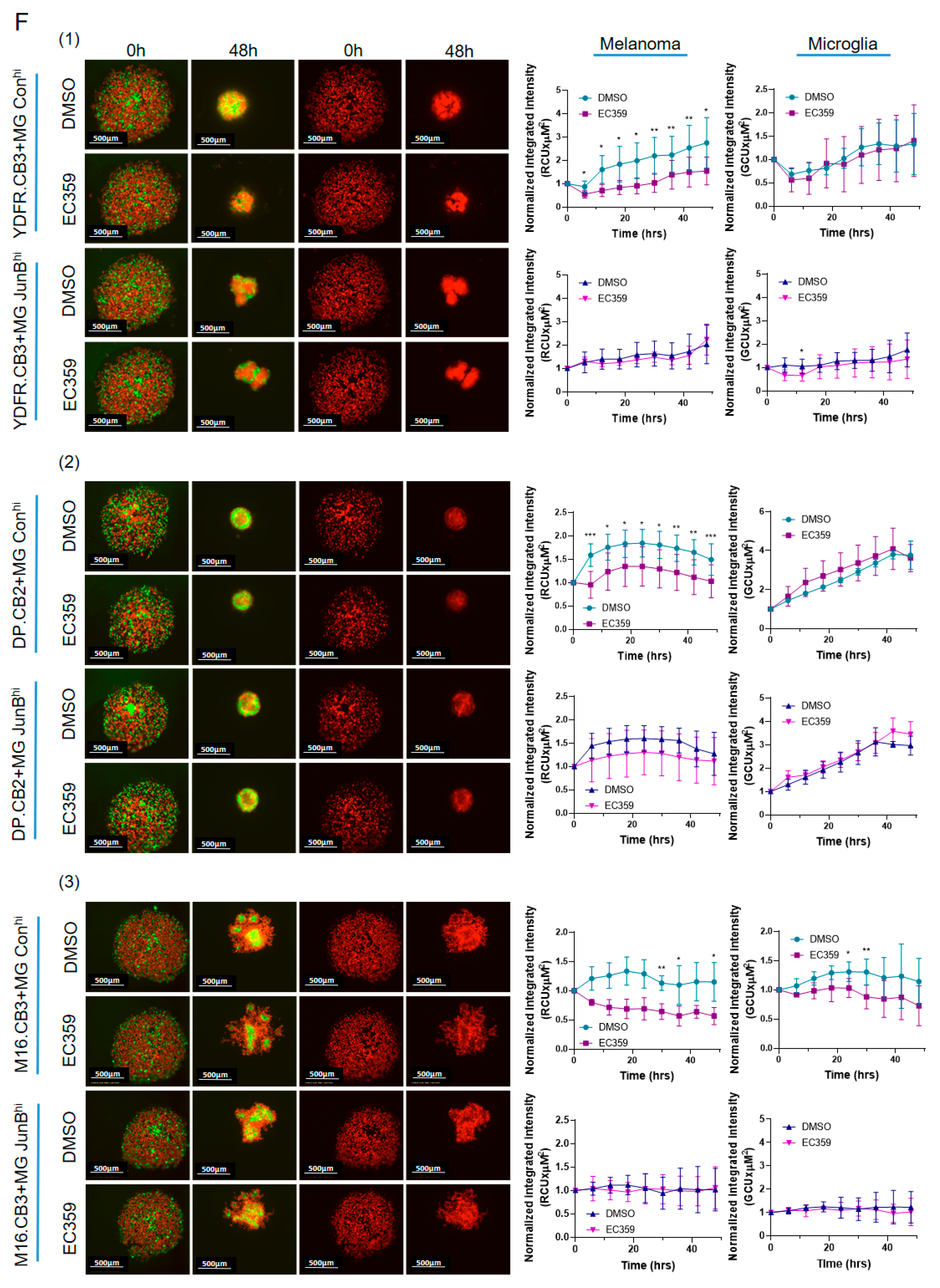
3.3.3. Microglial JunB Positively Regulates Melanoma Cell Proliferation in Melanoma–Microglia Spheroids via LIFR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, J.V.; Tawbi, H.; Margolin, K.A.; Amravadi, R.; Bosenberg, M.; Brastianos, P.K.; Chiang, V.L.; de Groot, J.; Glitza, I.C.; Herlyn, M.; et al. Melanoma central nervous system metastases: Current approaches, challenges, and opportunities. Pigment. Cell Melanoma Res. 2016, 29, 627–642. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Panagi, M.; Stylianopoulos, T.; Papageorgis, P. The Role of Tumor Microenvironment in Cancer Metastasis: Molecular Mechanisms and Therapeutic Opportunities. Cancers 2021, 13, 2053. [Google Scholar] [CrossRef] [PubMed]
- Izraely, S.; Sagi-Assif, O.; Klein, A.; Meshel, T.; Ben-Menachem, S.; Zaritsky, A.; Ehrlich, M.; Prieto, V.G.; Bar-Eli, M.; Pirker, C.; et al. The metastatic microenvironment: Claudin-1 suppresses the malignant phenotype of melanoma brain metastasis. Int. J. Cancer 2015, 136, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Schwartz, H.; Sagi-Assif, O.; Meshel, T.; Izraely, S.; Ben Menachem, S.; Bengaiev, R.; Ben-Shmuel, A.; Nahmias, C.; Couraud, P.O.; et al. Astrocytes facilitate melanoma brain metastasis via secretion of IL-23. J. Pathol. 2015, 236, 116–127. [Google Scholar] [CrossRef]
- Izraely, S.; Ben-Menachem, S.; Sagi-Assif, O.; Telerman, A.; Zubrilov, I.; Ashkenazi, O.; Meshel, T.; Maman, S.; Orozco, J.I.J.; Salomon, M.P.; et al. The metastatic microenvironment: Melanoma-microglia cross-talk promotes the malignant phenotype of melanoma cells. Int. J. Cancer 2019, 144, 802–817. [Google Scholar] [CrossRef] [PubMed]
- Izraely, S.; Ben-Menachem, S.; Malka, S.; Sagi-Assif, O.; Bustos, M.A.; Adir, O.; Meshel, T.; Chelladurai, M.; Ryu, S.; Ramos, R.I.; et al. The Vicious Cycle of Melanoma-Microglia Crosstalk: Inter-Melanoma Variations in the Brain-Metastasis-Promoting IL-6/JAK/STAT3 Signaling Pathway. Cells 2023, 12, 1513. [Google Scholar] [CrossRef] [PubMed]
- Becco, P.; Gallo, S.; Poletto, S.; Frascione, M.P.M.; Crotto, L.; Zaccagna, A.; Paruzzo, L.; Caravelli, D.; Carnevale-Schianca, F.; Aglietta, M. Melanoma Brain Metastases in the Era of Target Therapies: An Overview. Cancers 2020, 12, 1640. [Google Scholar] [CrossRef]
- Aronheim, A.; Zandi, E.; Hennemann, H.; Elledge, S.J.; Karin, M. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol. Cell Biol. 1997, 17, 3094–3102. [Google Scholar] [CrossRef]
- Yoshitomi, Y.; Ikeda, T.; Saito-Takatsuji, H.; Yonekura, H. Emerging Role of AP-1 Transcription Factor JunB in Angiogenesis and Vascular Development. Int. J. Mol. Sci. 2021, 22, 2804. [Google Scholar] [CrossRef]
- Piechaczyk, M.; Farras, R. Regulation and function of JunB in cell proliferation. Biochem. Soc. Trans. 2008, 36, 864–867. [Google Scholar] [CrossRef]
- Katagiri, T.; Kameda, H.; Nakano, H.; Yamazaki, S. Regulation of T cell differentiation by the AP-1 transcription factor JunB. Immunol. Med. 2021, 44, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Gazon, H.; Barbeau, B.; Mesnard, J.M.; Peloponese, J.M., Jr. Hijacking of the AP-1 Signaling Pathway during Development of ATL. Front. Microbiol. 2017, 8, 2686. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, G. Dissecting Immunosuppressive Cell Communication Patterns Reveals JunB Proto-Oncogene (JUNB) Shaping a Non-Inflamed Tumor Microenvironment. Front. Genet. 2022, 13, 883583. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E. AP-1—The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell Signal 2010, 22, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Hyakusoku, H.; Sano, D.; Takahashi, H.; Hatano, T.; Isono, Y.; Shimada, S.; Ito, Y.; Myers, J.N.; Oridate, N. JunB promotes cell invasion, migration and distant metastasis of head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 6. [Google Scholar] [CrossRef]
- Fujimura, T.; Kakizaki, A.; Furudate, S.; Kambayashi, Y.; Aiba, S. Tumor-associated macrophages in skin: How to treat their heterogeneity and plasticity. J. Dermatol. Sci. 2016, 83, 167–173. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Singh, N.; Benoit, M.R.; Zhou, J.; Das, B.; Davila-Velderrain, J.; Kellis, M.; Tsai, L.H.; Hu, X.; Yan, R. BACE-1 inhibition facilitates the transition from homeostatic microglia to DAM-1. Sci. Adv. 2022, 8, eabo1286. [Google Scholar] [CrossRef]
- Andersen, R.S.; Anand, A.; Harwood, D.S.L.; Kristensen, B.W. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers 2021, 13, 4255. [Google Scholar] [CrossRef]
- Izraely, S.; Ben-Menachem, S.; Sagi-Assif, O.; Meshel, T.; Marzese, D.M.; Ohe, S.; Zubrilov, I.; Pasmanik-Chor, M.; Hoon, D.S.B.; Witz, I.P. ANGPTL4 promotes the progression of cutaneous melanoma to brain metastasis. Oncotarget 2017, 8, 75778–75796. [Google Scholar] [CrossRef]
- Izraely, S.; Ben-Menachem, S.; Sagi-Assif, O.; Meshel, T.; Malka, S.; Telerman, A.; Bustos, M.A.; Ramos, R.I.; Pasmanik-Chor, M.; Hoon, D.S.B.; et al. The melanoma brain metastatic microenvironment: Aldolase C partakes in shaping the malignant phenotype of melanoma cells—A case of inter-tumor heterogeneity. Mol. Oncol. 2021, 15, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Izraely, S.; Klein, A.; Sagi-Assif, O.; Meshel, T.; Tsarfaty, G.; Hoon, D.S.; Witz, I.P. Chemokine-chemokine receptor axes in melanoma brain metastasis. Immunol. Lett. 2010, 130, 107–114. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]
- Izraely, S.; Sagi-Assif, O.; Klein, A.; Meshel, T.; Tsarfaty, G.; Pasmanik-Chor, M.; Nahmias, C.; Couraud, P.O.; Ateh, E.; Bryant, J.L.; et al. The metastatic microenvironment: Brain-residing melanoma metastasis and dormant micrometastasis. Int. J. Cancer 2012, 131, 1071–1082. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal 2017, 15, 23. [Google Scholar] [CrossRef]
- Jatiani, S.S.; Baker, S.J.; Silverman, L.R.; Reddy, E.P. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: Approaches for targeted therapies. Genes. Cancer 2010, 1, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Pang, L.; Dunterman, M.; Lesniak, M.S.; Heimberger, A.B.; Chen, P. Macrophages and microglia in glioblastoma: Heterogeneity, plasticity, and therapy. J. Clin. Investig. 2023, 133, e163446. [Google Scholar] [CrossRef] [PubMed]
- Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 689. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, S.S.; Mills, G.B. New era of integrated cancer biomarker discovery using reverse-phase protein arrays. Drug Metab. Pharmacokinet. 2016, 31, 35–45. [Google Scholar] [CrossRef]
- Johnson, P.; Ruffell, B. CD44 and its role in inflammation and inflammatory diseases. Inflamm. Allergy Drug Targets 2009, 8, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Kim, H.P.; Song, R.; Choi, A.M. Caveolin-1 confers antiinflammatory effects in murine macrophages via the MKK3/p38 MAPK pathway. Am. J. Respir. Cell Mol. Biol. 2006, 34, 434–442. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Brown, D.E.; Brewer, J.A.; Vogt, S.K.; Muglia, L.J. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 2007, 109, 4313–4319. [Google Scholar] [CrossRef]
- Park, S.H.; Hong, M.; Lee, H.S.; Ye, B.D.; Hwang, S.W.; Jung, S.; Baek, J.; Moon, J.W.; Kim, B.M.; Oh, S.H.; et al. Association of TRAP1 with infliximab-induced mucosal healing in Crohn’s disease. J. Gastroenterol. Hepatol. 2019, 34, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hu, J.; Wu, Y.; Luo, H.; Meng, W.; Xiao, B.; Xiao, X.; Zhou, Z.; Liu, F. Rheb (Ras Homolog Enriched in Brain 1) Deficiency in Mature Macrophages Prevents Atherosclerosis by Repressing Macrophage Proliferation, Inflammation, and Lipid Uptake. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1787–1801. [Google Scholar] [CrossRef]
- Zhu, X.; Yue, L.; Fan, C.; Liu, Y.; Wang, Y.; Zhao, H. Mechanism of Cdk5-synaptophysin-SNARE pathway in acute and chronic inflammatory pain. Am. J. Transl. Res. 2021, 13, 1075–1084. [Google Scholar]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.F.; Baccarella, A.; Pancholi, N.; Pufall, M.A.; Herbert, D.R.; Kim, C.C. JUNB is a key transcriptional modulator of macrophage activation. J. Immunol. 2015, 194, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yao, H.; Liu, W.; Ya, B.; Cheng, H.; Xing, Z.; Wu, Y. Microglia Polarization in Alzheimer’s Disease: Mechanisms and a Potential Therapeutic Target. Front. Aging Neurosci. 2021, 13, 772717. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Satoskar, A.; Faubion, W.; Howie, D.; Okamoto, S.; Feske, S.; Gullo, C.; Clarke, K.; Sosa, M.R.; Sharpe, A.H.; et al. The cell surface receptor SLAM controls T cell and macrophage functions. J. Exp. Med. 2004, 199, 1255–1264. [Google Scholar] [CrossRef]
- Qin, H.; Holdbrooks, A.T.; Liu, Y.; Reynolds, S.L.; Yanagisawa, L.L.; Benveniste, E.N. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J. Immunol. 2012, 189, 3439–3448. [Google Scholar] [CrossRef]
- Qin, H.; Yeh, W.I.; De Sarno, P.; Holdbrooks, A.T.; Liu, Y.; Muldowney, M.T.; Reynolds, S.L.; Yanagisawa, L.L.; Fox, T.H., 3rd; Park, K.; et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 5004–5009. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, J.H.; Kim, J.H.; Lee, W.H.; Lee, M.S.; Suk, K. Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J. Neuroinflamm. 2012, 9, 149. [Google Scholar] [CrossRef]
- Cui, K.; Ardell, C.L.; Podolnikova, N.P.; Yakubenko, V.P. Distinct Migratory Properties of M1, M2, and Resident Macrophages Are Regulated by alpha(D)beta(2) and alpha(M)beta(2) Integrin-Mediated Adhesion. Front. Immunol. 2018, 9, 2650. [Google Scholar] [CrossRef]
- Gao, W.J.; Liu, J.X.; Liu, M.N.; Yao, Y.D.; Liu, Z.Q.; Liu, L.; He, H.H.; Zhou, H. Macrophage 3D migration: A potential therapeutic target for inflammation and deleterious progression in diseases. Pharmacol. Res. 2021, 167, 105563. [Google Scholar] [CrossRef]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V. The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef]
- Salvucci, O.; Carsana, M.; Bersani, I.; Tragni, G.; Anichini, A. Antiapoptotic role of endogenous nitric oxide in human melanoma cells. Cancer Res. 2001, 61, 318–326. [Google Scholar] [PubMed]
- Nalwaya, N.; Deen, W.M. Nitric oxide, oxygen, and superoxide formation and consumption in macrophage cultures. Chem. Res. Toxicol. 2005, 18, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Krakhmal, N.V.; Zavyalova, M.V.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V.M. Cancer Invasion: Patterns and Mechanisms. Acta Naturae 2015, 7, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadeh, Z.; Cesmeli, S.; Biray Avci, C. Crucial players in glycolysis: Cancer progress. Gene 2020, 726, 144158. [Google Scholar] [CrossRef]
- Tang, Q.; Jiang, J.; Liu, J. CCR5 Blockade Suppresses Melanoma Development Through Inhibition of IL-6-Stat3 Pathway via Upregulation of SOCS3. Inflammation 2015, 38, 2049–2056. [Google Scholar] [CrossRef]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Kumari, N.; Choi, S.H. Tumor-associated macrophages in cancer: Recent advancements in cancer nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef]
- You, H.; Baluszek, S.; Kaminska, B. Supportive roles of brain macrophages in CNS metastases and assessment of new approaches targeting their functions. Theranostics 2020, 10, 2949–2964. [Google Scholar] [CrossRef]
- Liu, X.; Liu, R.; Croker, B.A.; Lawlor, K.E.; Smyth, G.K.; Wicks, I.P. Distinctive pro-inflammatory gene signatures induced in articular chondrocytes by oncostatin M and IL-6 are regulated by Suppressor of Cytokine Signaling-3. Osteoarthr. Cartil. 2015, 23, 1743–1754. [Google Scholar] [CrossRef]
- Kamohara, H.; Sakamoto, K.; Ishiko, T.; Masuda, Y.; Abe, T.; Ogawa, M. Leukemia inhibitory factor induces apoptosis and proliferation of human carcinoma cells through different oncogene pathways. Int. J. Cancer 1997, 72, 687–695. [Google Scholar] [CrossRef]
- Deng, Y.; Tang, S.; Cheng, J.; Zhang, X.; Jing, D.; Lin, Z.; Zhou, J. Integrated analysis reveals Atf3 promotes neuropathic pain via orchestrating JunB mediated release of inflammatory cytokines in DRG macrophage. Life Sci. 2023, 329, 121939. [Google Scholar] [CrossRef]
- Cai, J.; Liang, J.; Zhang, Y.; Shen, L.; Lin, H.; Hu, T.; Zhan, S.; Xie, M.; Liang, S.; Xian, M.; et al. Cyclo-(Phe-Tyr) as a novel cyclic dipeptide compound alleviates ischemic/reperfusion brain injury via JUNB/JNK/NF-kappaB and SOX5/PI3K/AKT pathways. Pharmacol. Res. 2022, 180, 106230. [Google Scholar] [CrossRef]
- Du, W.; Bos, P.D. Tracing bone marrow-derived microglia in brain metastatic tumors. Methods Enzymol. 2020, 635, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Kawana, H.; Karaki, H.; Higashi, M.; Miyazaki, M.; Hilberg, F.; Kitagawa, M.; Harigaya, K. CD44 suppresses TLR-mediated inflammation. J. Immunol. 2008, 180, 4235–4245. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Karin, M. JunB differs from c-Jun in its DNA-binding and dimerization domains, and represses c-Jun by formation of inactive heterodimers. Genes. Dev. 1993, 7, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.K.; Potteti, H.R.; Tamatam, C.R.; Elangovan, I.; Reddy, S.P. c-Jun Is Required for Nuclear Factor-kappaB-Dependent, LPS-Stimulated Fos-Related Antigen-1 Transcription in Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2016, 55, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, N.; Jordan, J.; Paul, S.; Reid, S.; Baenkler, H.W.; Sonnewald, S.; Bauerle, T.; Vera, J.; Schett, G.; Bozec, A. The AP-1 Transcription Factor c-Jun Promotes Arthritis by Regulating Cyclooxygenase-2 and Arginase-1 Expression in Macrophages. J. Immunol. 2017, 198, 3605–3614. [Google Scholar] [CrossRef]
- McFarland, B.C.; Marks, M.P.; Rowse, A.L.; Fehling, S.C.; Gerigk, M.; Qin, H.; Benveniste, E.N. Loss of SOCS3 in myeloid cells prolongs survival in a syngeneic model of glioma. Oncotarget 2016, 7, 20621–20635. [Google Scholar] [CrossRef]
- Manenti, S.; Orrico, M.; Masciocchi, S.; Mandelli, A.; Finardi, A.; Furlan, R. PD-1/PD-L Axis in Neuroinflammation: New Insights. Front. Neurol. 2022, 13, 877936. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.R.; Chang, T.W.; Lee, C.T.; Shen, C.J.; Chang, W.C.; Chen, B.K. ARNT deficiency represses pyruvate dehydrogenase kinase 1 to trigger ROS production and melanoma metastasis. Oncogenesis 2021, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yao, X.; Zhang, C.; Liu, Y.; Wei, L.; Huang, Q.; Wang, M.; Zhang, Y.; Hu, D.; Wu, W. PTK6 inhibits autophagy to promote uveal melanoma tumorigenesis by binding to SOCS3 and regulating mTOR phosphorylation. Cell Death Dis. 2023, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Hopperton, K.E.; Mohammad, D.; Trepanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Bispo da Silva, A.; Cerqueira Coelho, P.L.; Alves Oliveira Amparo, J.; Alves de Almeida Carneiro, M.M.; Pereira Borges, J.M.; Dos Santos Souza, C.; Dias Costa, M.F.; Mecha, M.; Guaza Rodriguez, C.; Amaral da Silva, V.D.; et al. The flavonoid rutin modulates microglial/macrophage activation to a CD150/CD206 M2 phenotype. Chem. Biol. Interact. 2017, 274, 89–99. [Google Scholar] [CrossRef]
- Mehrle, S.; Schmidt, J.; Buchler, M.W.; Watzl, C.; Marten, A. Enhancement of anti-tumor activity in vitro and in vivo by CD150 and SAP. Mol. Immunol. 2008, 45, 796–804. [Google Scholar] [CrossRef]
- Keane, L.; Antignano, I.; Riechers, S.P.; Zollinger, R.; Dumas, A.A.; Offermann, N.; Bernis, M.E.; Russ, J.; Graelmann, F.; McCormick, P.N.; et al. mTOR-dependent translation amplifies microglia priming in aging mice. J. Clin. Investig. 2021, 131, e132727. [Google Scholar] [CrossRef]
- Claret, F.X.; Hibi, M.; Dhut, S.; Toda, T.; Karin, M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature 1996, 383, 453–457. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Zhu, B.M.; Ishida, Y.; Robinson, G.W.; Pacher-Zavisin, M.; Yoshimura, A.; Murphy, P.M.; Hennighausen, L. SOCS3 negatively regulates the gp130-STAT3 pathway in mouse skin wound healing. J. Investig. Dermatol. 2008, 128, 1821–1829. [Google Scholar] [CrossRef]
- Liu, J.; Qu, X.; Shao, L.; Hu, Y.; Yu, X.; Lan, P.; Guo, Q.; Han, Q.; Zhang, J.; Zhang, C. Pim-3 enhances melanoma cell migration and invasion by promoting STAT3 phosphorylation. Cancer Biol. Ther. 2018, 19, 160–168. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yang, Y.; Xu, J.; Liao, Y.; Liu, L.; Deng, L.; Xiong, X. IL22 drives cutaneous melanoma cell proliferation, migration and invasion through activation of miR-181/STAT3/AKT axis. J. Cancer 2020, 11, 2679–2687. [Google Scholar] [CrossRef] [PubMed]
- Horvathova, J.; Moravcik, R.; Matuskova, M.; Sisovsky, V.; Bohac, A.; Zeman, M. Inhibition of Glycolysis Suppresses Cell Proliferation and Tumor Progression In Vivo: Perspectives for Chronotherapy. Int. J. Mol. Sci. 2021, 22, 4390. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, T.; Verdone, J.E.; Huang, J.; Kahlert, U.D.; Hernandez, J.R.; Torga, G.; Zarif, J.C.; Epstein, T.; Gatenby, R.; McCartney, A.; et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2015, 6, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Moshe, A.; Izraely, S.; Sagi-Assif, O.; Malka, S.; Ben-Menachem, S.; Meshel, T.; Pasmanik-Chor, M.; Hoon, D.S.B.; Witz, I.P. Inter-Tumor Heterogeneity-Melanomas Respond Differently to GM-CSF-Mediated Activation. Cells 2020, 9, 1683. [Google Scholar] [CrossRef]
- Neuditschko, B.; Janker, L.; Niederstaetter, L.; Brunmair, J.; Krivanek, K.; Izraely, S.; Sagi-Assif, O.; Meshel, T.; Keppler, B.K.; Del Favero, G.; et al. The Challenge of Classifying Metastatic Cell Properties by Molecular Profiling Exemplified with Cutaneous Melanoma Cells and Their Cerebral Metastasis from Patient Derived Mouse Xenografts. Mol. Cell Proteomics 2020, 19, 478–489. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adir, O.; Sagi-Assif, O.; Meshel, T.; Ben-Menachem, S.; Pasmanik-Chor, M.; Hoon, D.S.B.; Witz, I.P.; Izraely, S. Heterogeneity in the Metastatic Microenvironment: JunB-Expressing Microglia Cells as Potential Drivers of Melanoma Brain Metastasis Progression. Cancers 2023, 15, 4979. https://doi.org/10.3390/cancers15204979
Adir O, Sagi-Assif O, Meshel T, Ben-Menachem S, Pasmanik-Chor M, Hoon DSB, Witz IP, Izraely S. Heterogeneity in the Metastatic Microenvironment: JunB-Expressing Microglia Cells as Potential Drivers of Melanoma Brain Metastasis Progression. Cancers. 2023; 15(20):4979. https://doi.org/10.3390/cancers15204979
Chicago/Turabian StyleAdir, Orit, Orit Sagi-Assif, Tsipi Meshel, Shlomit Ben-Menachem, Metsada Pasmanik-Chor, Dave S. B. Hoon, Isaac P. Witz, and Sivan Izraely. 2023. "Heterogeneity in the Metastatic Microenvironment: JunB-Expressing Microglia Cells as Potential Drivers of Melanoma Brain Metastasis Progression" Cancers 15, no. 20: 4979. https://doi.org/10.3390/cancers15204979
APA StyleAdir, O., Sagi-Assif, O., Meshel, T., Ben-Menachem, S., Pasmanik-Chor, M., Hoon, D. S. B., Witz, I. P., & Izraely, S. (2023). Heterogeneity in the Metastatic Microenvironment: JunB-Expressing Microglia Cells as Potential Drivers of Melanoma Brain Metastasis Progression. Cancers, 15(20), 4979. https://doi.org/10.3390/cancers15204979