
Colorectal Cancer Is Borrowing Blueprints from Intestinal Ontogenesis



{kind=link}
Abstract
:Simple Summary
Abstract
1. Implications of Tumor Heterogeneity in the Initiation and Progression of Colorectal Cancer
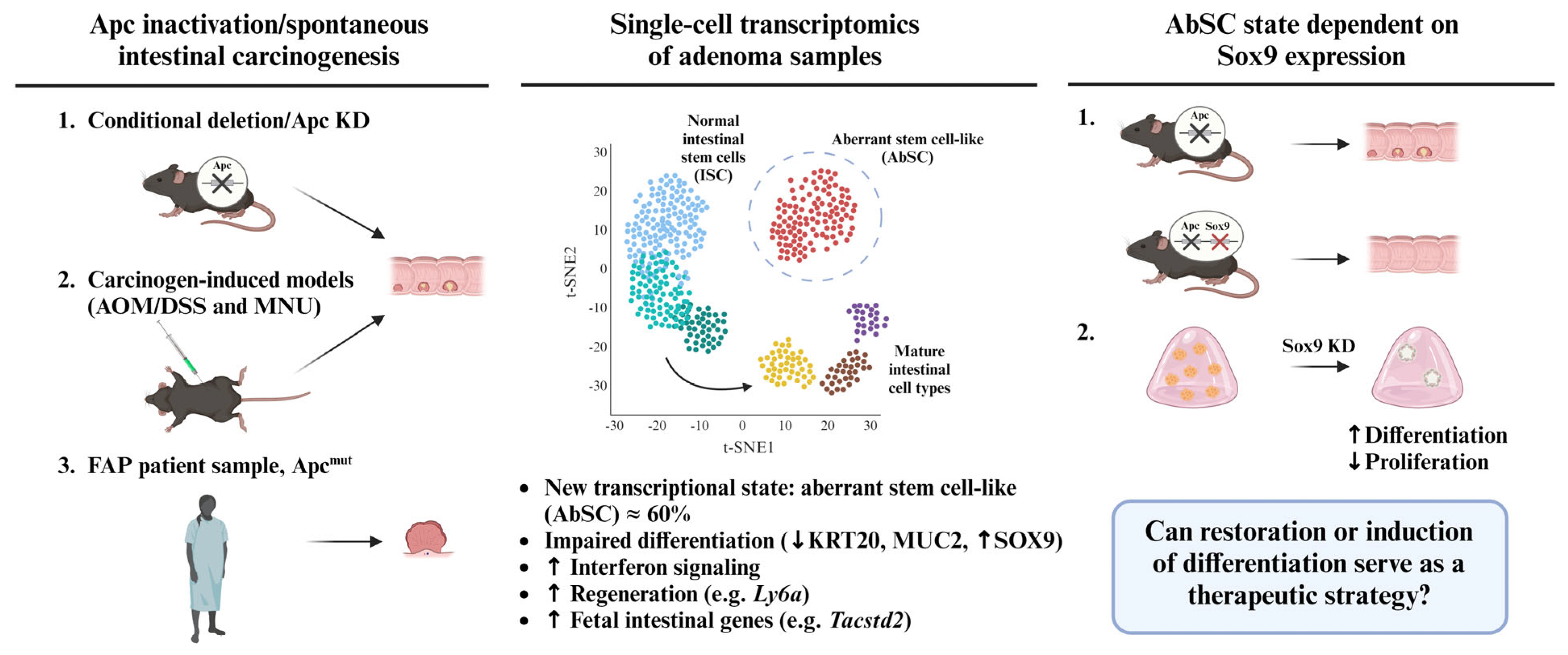
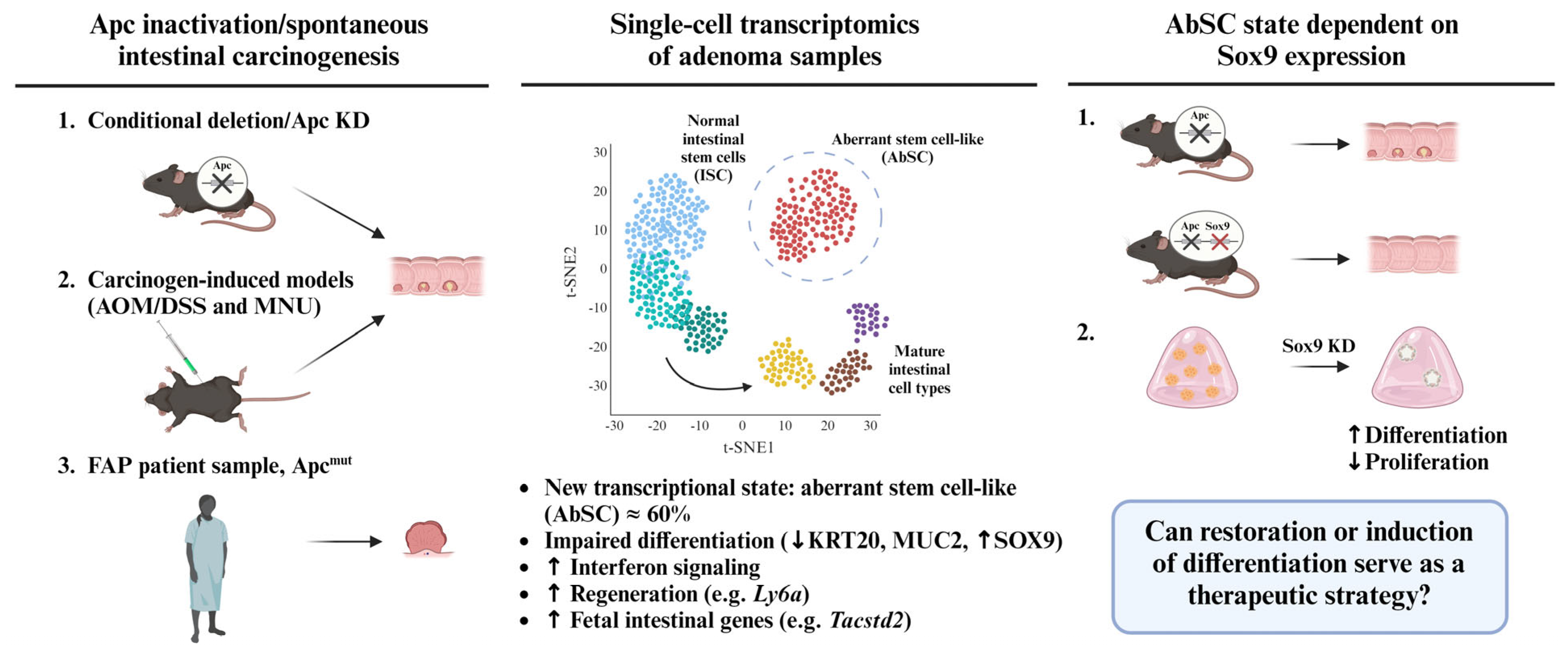
2. An Aberrant Stem Cell (AbSC) Population Characterized by a Fetal-like Gene Signature Drives the Initiation of Intestinal Neoplasia
3. Sox9 Expression Regulates Developmental Reprogramming and Maintenance of AbSCs
4. Chemical Induction of Germ Layer Commitment Programs in Aberrant Stem Cells Is a Promising Approach to Treat Colorectal Cancer and Other Malignancies
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frank, M.H.; Wilson, B.J.; Gold, J.S.; Frank, N.Y. Clinical Implications of Colorectal Cancer Stem Cells in the Age of Single-Cell Omics and Targeted Therapies. Gastroenterology 2021, 160, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e315. [Google Scholar] [CrossRef]
- Haebe, J.R.; Bergin, C.J.; Sandouka, T.; Benoit, Y.D. Emerging role of G9a in cancer stemness and promises as a therapeutic target. Oncogenesis 2021, 10, 76. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Brewin, J.; Horne, G.; Chevassut, T. Genomic landscapes and clonality of de novo AML. N. Engl. J. Med. 2013, 369, 1472–1473. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Bergin, C.J.; Zouggar, A.; Haebe, J.R.; Masibag, A.N.; Desrochers, F.M.; Reilley, S.Y.; Agrawal, G.; Benoit, Y.D. G9a controls pluripotent-like identity and tumor-initiating function in human colorectal cancer. Oncogene 2021, 40, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; van Galen, P.; Pedley, N.M.; Lima-Fernandes, E.; Frelin, C.; Davis, T.; Cao, L.; Baiazitov, R.; Du, W.; Sydorenko, N.; et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lima-Fernandes, E.; Murison, A.; da Silva Medina, T.; Wang, Y.; Ma, A.; Leung, C.; Luciani, G.M.; Haynes, J.; Pollett, A.; Zeller, C.; et al. Targeting bivalency de-represses Indian Hedgehog and inhibits self-renewal of colorectal cancer-initiating cells. Nat. Commun. 2019, 10, 1436. [Google Scholar] [CrossRef]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef]
- de Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Oost, K.C.; Kester, L.; Morgner, J.; Bornes, L.; Bruens, L.; Spaargaren, L.; Azkanaz, M.; Schelfhorst, T.; Beerling, E.; et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell 2020, 26, 569–578.e567. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Bala, P.; Rennhack, J.P.; Aitymbayev, D.; Morris, C.; Moyer, S.M.; Duronio, G.N.; Doan, P.; Li, Z.; Liang, X.; Hornick, J.L.; et al. Aberrant cell state plasticity mediated by developmental reprogramming precedes colorectal cancer initiation. Sci. Adv. 2023, 9, eadf0927. [Google Scholar] [CrossRef]
- Stoyanova, T.; Goldstein, A.S.; Cai, H.; Drake, J.M.; Huang, J.; Witte, O.N. Regulated proteolysis of Trop2 drives epithelial hyperplasia and stem cell self-renewal via β-catenin signaling. Genes. Dev. 2012, 26, 2271–2285. [Google Scholar] [CrossRef]
- Riera, K.M.; Jang, B.; Min, J.; Roland, J.T.; Yang, Q.; Fesmire, W.T.; Camilleri-Broet, S.; Ferri, L.; Kim, W.H.; Choi, E.; et al. Trop2 is upregulated in the transition to dysplasia in the metaplastic gastric mucosa. J. Pathol. 2020, 251, 336–347. [Google Scholar] [CrossRef]
- Sprangers, J.; Zaalberg, I.C.; Maurice, M.M. Organoid-based modeling of intestinal development, regeneration, and repair. Cell Death Differ. 2021, 28, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Duronio, G.N.; Yang, Y.; Bala, P.; Hebbar, P.; Spisak, S.; Sahgal, P.; Singh, H.; Zhang, Y.; Xie, Y.; et al. An Enhancer-Driven Stem Cell-Like Program Mediated by SOX9 Blocks Intestinal Differentiation in Colorectal Cancer. Gastroenterology 2022, 162, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Orlando, L.; Benoit, Y.D.; Reid, J.C.; Nakanishi, M.; Boyd, A.L.; García-Rodriguez, J.L.; Tanasijevic, B.; Doyle, M.S.; Luchman, A.; Restall, I.J.; et al. Chemical genomics reveals targetable programs of human cancers rooted in pluripotency. Cell Chem. Biol. 2023, 30, 780–794. [Google Scholar] [CrossRef]
- Masibag, A.N.; Bergin, C.J.; Haebe, J.R.; Zouggar, A.; Shah, M.S.; Sandouka, T.; Mendes da Silva, A.; Desrochers, F.M.; Fournier-Morin, A.; Benoit, Y.D. Pharmacological targeting of Sam68 functions in colorectal cancer stem cells. iScience 2021, 24, 103442. [Google Scholar] [CrossRef]
- Werbowetski-Ogilvie, T.E.; Bossé, M.; Stewart, M.; Schnerch, A.; Ramos-Mejia, V.; Rouleau, A.; Wynder, T.; Smith, M.J.; Dingwall, S.; Carter, T.; et al. Characterization of human embryonic stem cells with features of neoplastic progression. Nat. Biotechnol. 2009, 27, 91–97. [Google Scholar] [CrossRef]
- Sachlos, E.; Risueño, R.M.; Laronde, S.; Shapovalova, Z.; Lee, J.H.; Russell, J.; Malig, M.; McNicol, J.D.; Fiebig-Comyn, A.; Graham, M.; et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012, 149, 1284–1297. [Google Scholar] [CrossRef]
- Benoit, Y.D.; Mitchell, R.R.; Risueño, R.M.; Orlando, L.; Tanasijevic, B.; Boyd, A.L.; Aslostovar, L.; Salci, K.R.; Shapovalova, Z.; Russell, J.; et al. Sam68 Allows Selective Targeting of Human Cancer Stem Cells. Cell Chem. Biol. 2017, 24, 833–844.e839. [Google Scholar] [CrossRef]
- Nilsson, L.M.; Green, L.C.; Muralidharan, S.V.; Demir, D.; Welin, M.; Bhadury, J.; Logan, D.T.; Walse, B.; Nilsson, J.A. Cancer Differentiating Agent Hexamethylene Bisacetamide Inhibits BET Bromodomain Proteins. Cancer Res. 2016, 76, 2376–2383. [Google Scholar] [CrossRef]
- Gangat, N.; Tefferi, A. Venetoclax-based chemotherapy in acute and chronic myeloid neoplasms: Literature survey and practice points. Blood Cancer J. 2020, 10, 122. [Google Scholar] [CrossRef]
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021, 11, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Calvo, R.; Drabkin, H.A. Embryonic genes in cancer. Ann. Oncol. 2000, 11 (Suppl. 3), 207–218. [Google Scholar] [CrossRef] [PubMed]
- Monk, M.; Holding, C. Human embryonic genes re-expressed in cancer cells. Oncogene 2001, 20, 8085–8091. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef]
- Lin, V.T.G.; Pruitt, H.C.; Samant, R.S.; Shevde, L.A. Developing Cures: Targeting Ontogenesis in Cancer. Trends Cancer 2017, 3, 126–136. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Billingsley, J.L.; Yevdokimova, V.; Ayoub, K.; Benoit, Y.D. Colorectal Cancer Is Borrowing Blueprints from Intestinal Ontogenesis. Cancers 2023, 15, 4928. https://doi.org/10.3390/cancers15204928
Billingsley JL, Yevdokimova V, Ayoub K, Benoit YD. Colorectal Cancer Is Borrowing Blueprints from Intestinal Ontogenesis. Cancers. 2023; 15(20):4928. https://doi.org/10.3390/cancers15204928
Chicago/Turabian StyleBillingsley, Jacob L., Veronika Yevdokimova, Kristina Ayoub, and Yannick D. Benoit. 2023. "Colorectal Cancer Is Borrowing Blueprints from Intestinal Ontogenesis" Cancers 15, no. 20: 4928. https://doi.org/10.3390/cancers15204928
APA StyleBillingsley, J. L., Yevdokimova, V., Ayoub, K., & Benoit, Y. D. (2023). Colorectal Cancer Is Borrowing Blueprints from Intestinal Ontogenesis. Cancers, 15(20), 4928. https://doi.org/10.3390/cancers15204928