
Identification of Gut Microbiota Profile Associated with Colorectal Cancer in Saudi Population

 , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Assessment of Diet Quality and Fat Intake
2.3. DNA Extraction and PCR Amplification
2.4. Library Construction
2.5. 16SrRNA Bioinformatics Analysis
2.6. Microbial Data Analysis
2.7. Statistical Analysis
3. Results
3.1. Clinical Samples
3.2. Comparing Diet Quality: Exploring the Differences between CRC Patients and Healthy Controls
3.3. Microbiome Study of Fecal Samples from CRC Patients and Healthy Controls
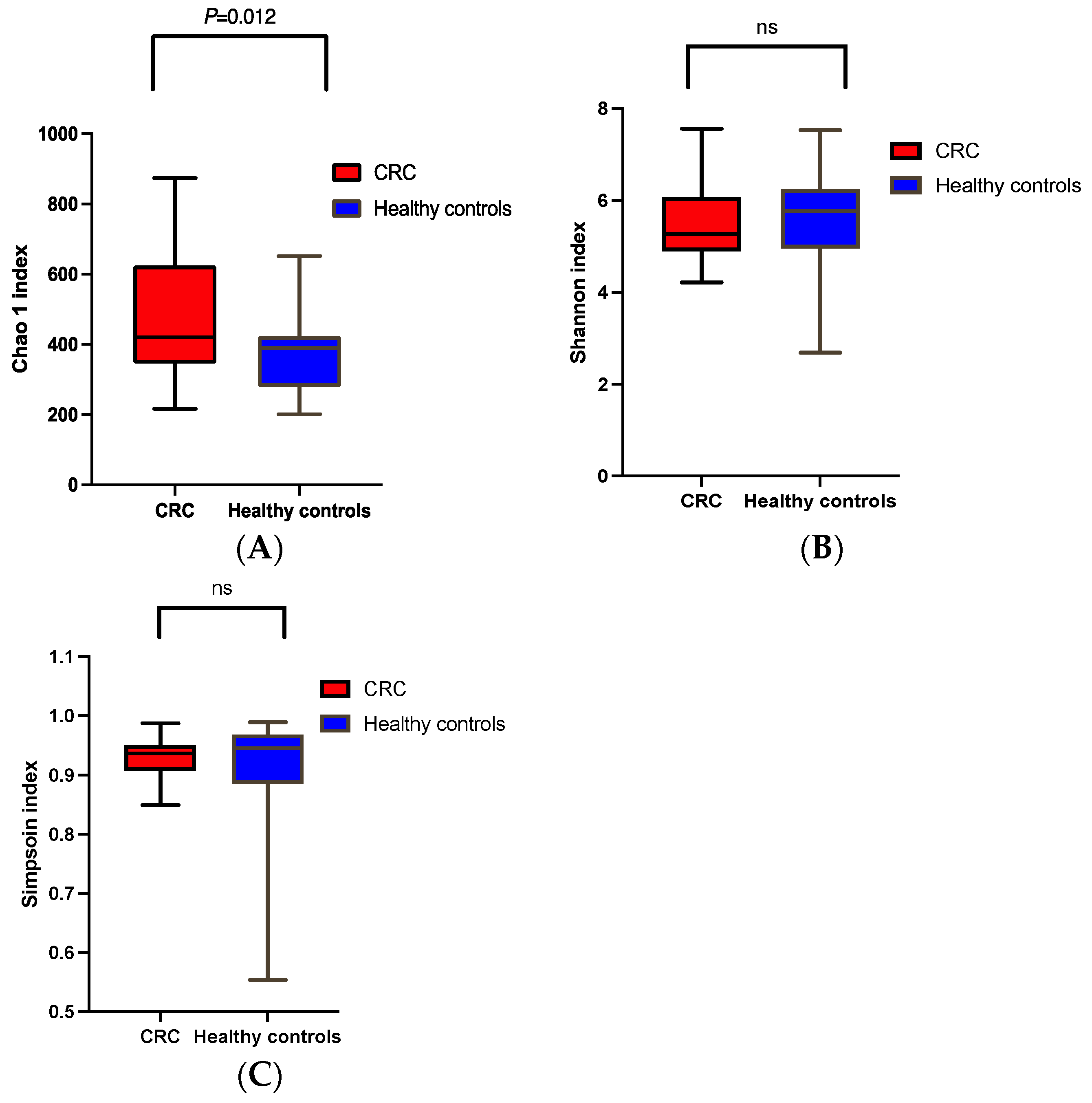
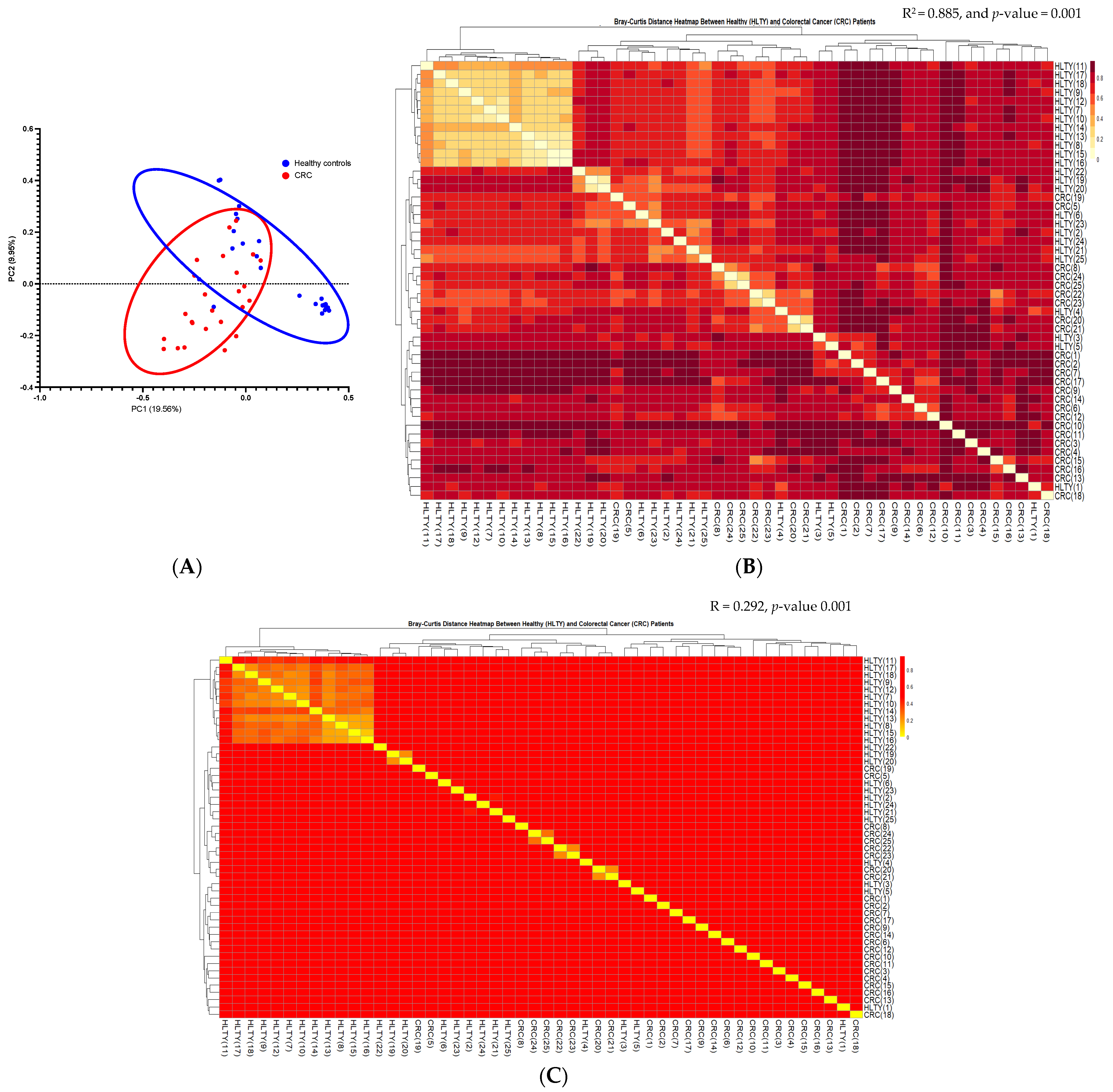
α and β Diversities of CRC vs. the Healthy Controls
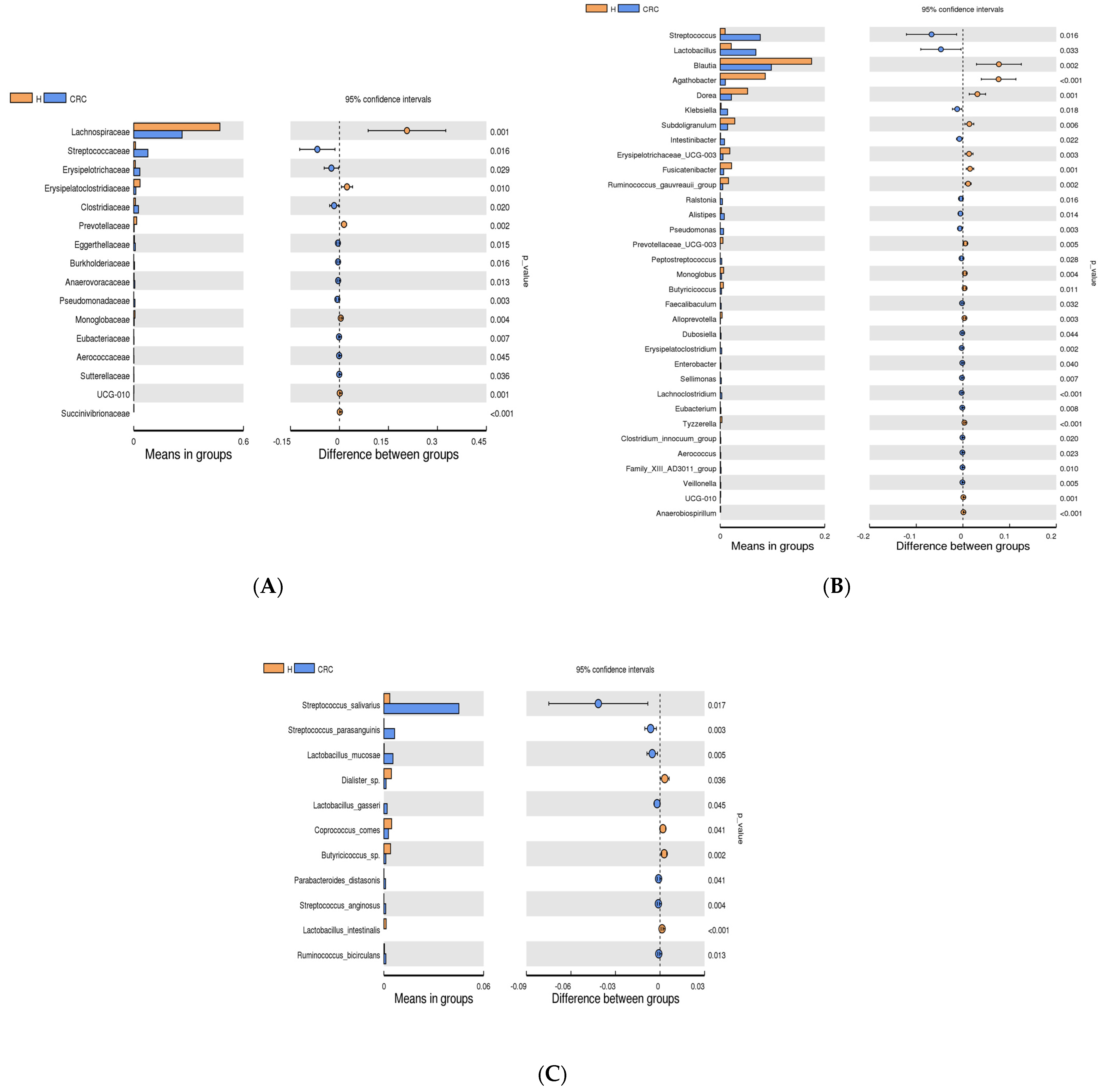
3.4. Microbial Profile of CRC and Healthy Controls
3.4.1. The Most Abundant Bacteria in CRC vs. the Healthy Controls
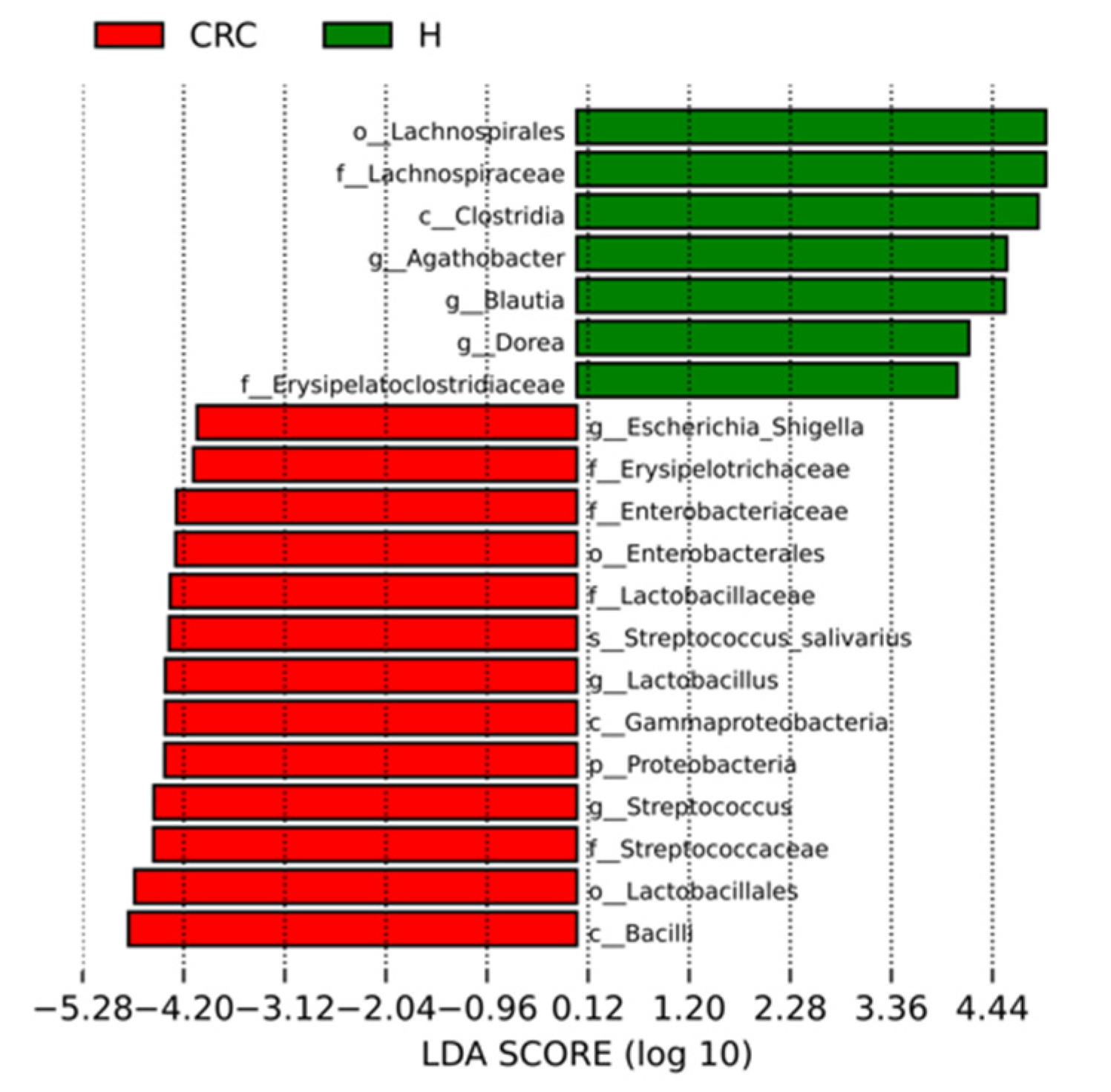
3.4.2. Characterization of the Microbiomes of CRC Patients and Healthy Participants via LEfSe Analysis and LDA Based on OTUs
3.4.3. Diet Quality and Fat Intake Association with Most Abundant Bacteria Genera among CRC
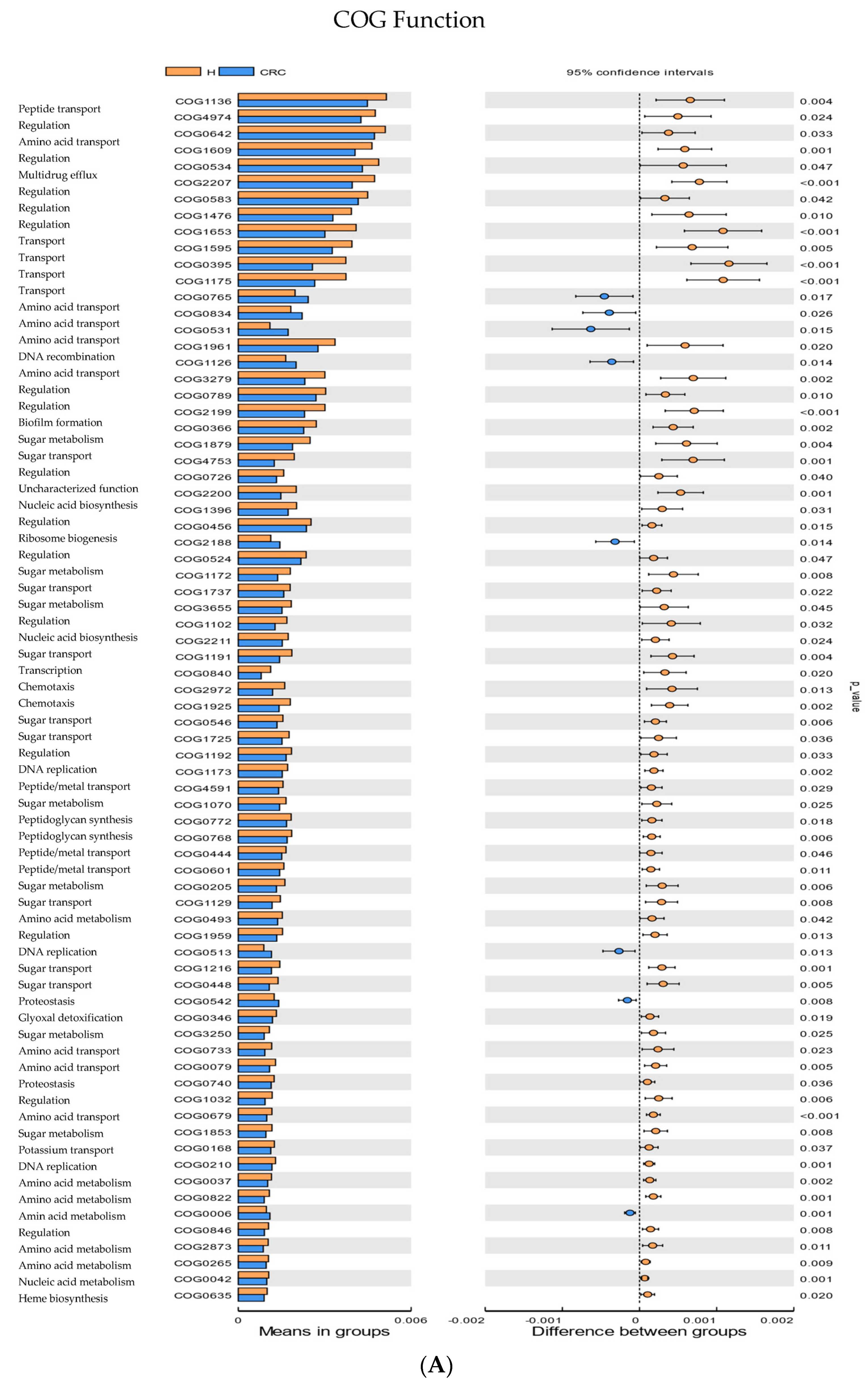
3.5. Functional Enrichment Analysis and Pathway Abundance Differences in CRC Compared to Healthy Controls
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Alyabsi, M.; Algarni, M.; Alshammari, K. Trends in Colorectal Cancer Incidence Rates in Saudi Arabia (2001–2016) Using Saudi National Registry: Early- versus Late-Onset Disease. Front. Oncol. 2021, 11, 730689. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, E.; Fathi, W.; Hussain, F.; Hashmi, S.K. The Increasing Trends in Cases of the Most Common Cancers in Saudi Arabia. J. Epidemiol. Glob. Health 2020, 10, 258–262. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Hofseth, L.J.; Hebert, J.R.; Chanda, A.; Chen, H.; Love, B.L.; Pena, M.M.; Murphy, E.A.; Sajish, M.; Sheth, A.; Buckhaults, P.J.; et al. Publisher Correction: Early-onset colorectal cancer: Initial clues and current views. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 517. [Google Scholar] [CrossRef]
- Wild, C.P.; Scalbert, A.; Herceg, Z. Measuring the exposome: A powerful basis for evaluating environmental exposures and cancer risk. Environ. Mol. Mutagen. 2013, 54, 480–499. [Google Scholar] [CrossRef]
- Crosbie, A.B.; Roche, L.M.; Johnson, L.M.; Pawlish, K.S.; Paddock, L.E.; Stroup, A.M. Trends in colorectal cancer incidence among younger adults-Disparities by age, sex, race, ethnicity, and subsite. Cancer Med. 2018, 7, 4077–4086. [Google Scholar] [CrossRef]
- Sánchez-Alcoholado, L.; Ramos-Molina, B.; Otero, A.; Laborda-Illanes, A.; Ordóñez, R.; Medina, J.A.; Gómez-Millán, J.; Queipo-Ortuño, M.I. The Role of the Gut Microbiome in Colorectal Cancer Development and Therapy Response. Cancers 2020, 12, 1406. [Google Scholar] [CrossRef]
- Kazmierczak-Siedlecka, K.; Daca, A.; Fic, M.; van de Wetering, T.; Folwarski, M.; Makarewicz, W. Therapeutic methods of gut microbiota modification in colorectal cancer management—Fecal microbiota transplantation, prebiotics, probiotics, and synbiotics. Gut Microbes 2020, 11, 1518–1530. [Google Scholar] [CrossRef]
- Fong, W.; Li, Q.; Yu, J. Gut microbiota modulation: A novel strategy for prevention and treatment of colorectal cancer. Oncogene 2020, 39, 4925–4943. [Google Scholar] [CrossRef]
- Liu, Y.; Lau, H.C.-H.; Cheng, W.Y.; Yu, J. Gut microbiome in colorectal cancer: Clinical diagnosis and treatment. Genom. Proteom. Bioinform. 2023, 21, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Fang, L.; Lee, M.H. Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol. Rep. 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial Driver–passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The intestinal microbiota in colorectal cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61, 1500902. [Google Scholar] [CrossRef]
- Kim, K.H.; Park, D.; Jia, B.; Baek, J.H.; Hahn, Y.; Jeon, C.O. Identification and characterization of major bile acid 7α-dehydroxylating bacteria in the human gut. mSystems 2022, 7, e00455-22. [Google Scholar] [CrossRef]
- Tong, Z.; Wang, X.; Shi, S.; Hou, T.; Gao, G.; Li, D.; Shan, Y.; Zhang, C. Development of lactate-related gene signature and prediction of overall survival and chemosensitivity in patients with colorectal cancer. Cancer Med. 2023, 12, 10105–10122. [Google Scholar] [CrossRef]
- Genua, F.; Mirković, B.; Mullee, A.; Levy, M.; Gallagher, W.M.; Vodicka, P.; Hughes, D.J. Association of circulating short chain fatty acid levels with colorectal adenomas and colorectal cancer. Clin. Nutr. ESPEN 2021, 46, 297–304. [Google Scholar] [CrossRef]
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014, 5, 675–680. [Google Scholar] [CrossRef]
- Jalandra, R.; Dalal, N.; Yadav, A.K.; Verma, D.; Sharma, M.; Singh, R.; Khosla, A.; Kumar, A.; Solanki, P.R. Emerging role of trimethylamine-N-oxide (TMAO) in colorectal cancer. Appl. Microbiol. Biotechnol. 2021, 105, 7651–7660. [Google Scholar] [CrossRef]
- Cleghorn, C.L.; Harrison, R.A.; Ransley, J.K.; Wilkinson, S.; Thomas, J.; Cade, J.E. Can a dietary quality score derived from a short-form FFQ assess dietary quality in UK adult population surveys? Public Health Nutr. 2016, 19, 2915–2923. [Google Scholar] [CrossRef] [PubMed]
- Mumena, W.A.; Ateek, A.A.; Alamri, R.K.; Alobaid, S.A.; Alshallali, S.H.; Afifi, S.Y.; Aljohani, G.A.; Kutbi, H.A. Fast-food consumption, dietary quality, and dietary intake of adolescents in Saudi Arabia. Int. J. Environ. Res. Public Health 2022, 19, 15083. [Google Scholar] [CrossRef]
- Mumena, W.A.; Alnezari, A.I.; Safar, H.I.; Alharbi, N.S.; Alahmadi, R.B.; Qadhi, R.I.; Faqeeh, S.F.; Kutbi, H.A. Media use, dietary intake, and diet quality of adolescents in Saudi Arabia. Pediatr. Res. 2023, 94, 789–795. [Google Scholar] [CrossRef]
- Yang, T.C.; Sahota, P.; Pickett, K.E.; Bryant, M. Association of food security status with overweight and dietary intake: Exploration of White British and Pakistani-origin families in the Born in Bradford cohort. Nutr. J. 2018, 17, 48. [Google Scholar] [CrossRef]
- Bredin, C.; Naimimohasses, S.; Norris, S.; Wright, C.; Hancock, N.; Hart, K.; Moore, J.B. Development and relative validation of a short food frequency questionnaire for assessing dietary intakes of non-alcoholic fatty liver disease patients. Eur. J. Nutr. 2020, 59, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Merino, J.; Joshi, A.D.; Nguyen, L.H.; Leeming, E.R.; Mazidi, M.; Drew, D.A.; Gibson, R.; Graham, M.S.; Lo, C.-H.; Capdevila, J.; et al. Diet quality and risk and severity of COVID-19: A prospective cohort study. Gut 2021, 70, 2096–2104. [Google Scholar] [CrossRef]
- Rohrmann, S.; Klein, G. Development and validation of a short food list to assess the intake of total fat, saturated, mono-unsaturated, polyunsaturated fatty acids and cholesterol. Eur. J. Public Health 2003, 13, 262–268. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, H.; Gao, X.; Wang, J. The intratumor microbiota signatures associate with subtype, tumor stage, and survival status of esophageal carcinoma. Front. Oncol. 2021, 11, 754788. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Poutahidis, T.; Erdman, S.E. Commensal bacteria modulate the tumor microenvironment. Cancer Lett. 2016, 380, 356–358. [Google Scholar] [CrossRef]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2015, 7, 17–44. [Google Scholar] [CrossRef]
- Moradi-Lakeh, M.; El Bcheraoui, C.; Afshin, A.; Daoud, F.; AlMazroa, M.A.; Al Saeedi, M.; Basulaiman, M.; Memish, Z.A.; Al Rabeeah, A.A.; Mokdad, A.H. Diet in Saudi Arabia: Findings from a nationally representative survey. Public Health Nutr. 2017, 20, 1075–1081. [Google Scholar] [CrossRef]
- Angelakis, E.; Yasir, M.; Bachar, D.; Azhar, E.I.; Lagier, J.-C.; Bibi, F.; Jiman-Fatani, A.A.; Alawi, M.; Bakarman, M.A.; Robert, C.; et al. Gut microbiome and dietary patterns in different Saudi populations and monkeys. Sci. Rep. 2016, 6, 32191. [Google Scholar] [CrossRef]
- Liu, X.; Mao, B.; Gu, J.; Wu, J.; Cui, S.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Blautia—A new functional genus with potential probiotic properties? Gut Microbes 2021, 13, 1875796. [Google Scholar] [CrossRef] [PubMed]
- Andriulli, A.; Bevilacqua, A.; Palmieri, O.; Latiano, A.; Fontana, R.; Gioffreda, D.; Castellana, S.; Mazza, T.; Panza, A.; Menzaghi, C.; et al. Healthy and pro-inflammatory gut ecology plays a crucial role in the digestion and tolerance of a novel Gluten Friendly™ bread in celiac subjects: A randomized, double blind, placebo control in vivo study. Food Funct. 2022, 13, 1299–1315. [Google Scholar] [CrossRef] [PubMed]
- Van Hul, M.; Le Roy, T.; Prifti, E.; Dao, M.C.; Paquot, A.; Zucker, J.-D.; Delzenne, N.M.; Muccioli, G.G.; Clément, K.; Cani, P.D. From correlation to causality: The case of Subdoligranulum. Gut Microbes 2020, 12, 1849998. [Google Scholar] [CrossRef]
- Prasoodanan PK, V.; Sharma, A.K.; Mahajan, S.; Dhakan, D.B.; Maji, A.; Scaria, J.; Sharma, V.K. Western and non-western gut microbiomes reveal new roles of Prevotella in carbohydrate metabolism and mouth–gut axis. NPJ Biofilms Microbiomes 2021, 7, 77. [Google Scholar] [CrossRef]
- Kim, C.C.; Lunken, G.R.; Kelly, W.J.; Patchett, M.L.; Jordens, Z.; Tannock, G.W.; Sims, I.M.; Bell, T.J.; Hedderley, D.; Henrissat, B.; et al. Correction: Genomic insights from Monoglobus pectinilyticus: A pectin-degrading specialist bacterium in the human colon. ISME J. 2023, 17, 1520. [Google Scholar] [CrossRef]
- Rajilić–Stojanović, M.; Biagi, E.; Heilig, H.G.; Kajander, K.; Kekkonen, R.A.; Tims, S.; de Vos, W.M. Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology 2011, 141, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.K.; Vasquez, R.; Kim, S.H.; Hwang, I.-C.; Song, J.H.; Park, J.H.; Kim, I.H.; Kang, D.-K. Multispecies probiotics alter fecal short-chain fatty acids and lactate levels in weaned pigs by modulating gut microbiota. J. Anim. Sci. Technol. 2021, 63, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Medawar, E.; Haange, S.-B.; Rolle-Kampczyk, U.; Engelmann, B.; Dietrich, A.; Thieleking, R.; Wiegank, C.; Fries, C.; Horstmann, A.; Villringer, A.; et al. Gut microbiota link dietary fiber intake and short-chain fatty acid metabolism with eating behavior. Transl. Psychiatry 2021, 11, 500. [Google Scholar] [CrossRef]
- Kelly, T.N.; Bazzano, L.A.; Ajami, N.J.; He, H.; Zhao, J.; Petrosino, J.F.; Correa, A.; He, J. Gut microbiome associates with lifetime cardiovascular disease risk profile among Bogalusa heart study participants. Circ. Res. 2016, 119, 956–964. [Google Scholar] [CrossRef]
- Burakova, I.; Smirnova, Y.; Gryaznova, M.; Syromyatnikov, M.; Chizhkov, P.; Popov, E.; Popov, V. The effect of short-term consumption of lactic acid bacteria on the gut microbiota in obese people. Nutrients 2022, 14, 3384. [Google Scholar] [CrossRef]
- Ecklu-Mensah, G.; Choo-Kang, C.; Maseng, M.G.; Donato, S.; Bovet, P.; Viswanathan, B.; Bedu-Addo, K.; Plange-Rhule, J.; Boateng, P.O.; Forrester, T.E.; et al. Gut microbiota and fecal short chain fatty acids differ with adiposity and country of origin: The METS-microbiome study. Nat. Commun. 2023, 14, 5160. [Google Scholar] [CrossRef]
- Kelesidis, T. Bloodstream infection with Anaerobiospirillum succiniciproducens: A potentially lethal infection. South. Med. J. 2011, 104, 205–214. [Google Scholar] [CrossRef]
- Malnick, H. Anaerobiospirillum thomasii sp. nov., an anaerobic spiral bacterium isolated from the feces of cats and dogs and from diarrheal feces of humans, and emendation of the genus Anaerobiospirillum. Int. J. Syst. Evol. Microbiol. 1997, 47, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Torralba, M.G.; Moncera, K.J.; DiLello, L.; Petrini, J.; Nelson, K.E.; Pieper, R. Gastro-intestinal and oral microbiome signatures associated with healthy aging. GeroScience 2019, 41, 907–921. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.-J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, S.; Crawley, A.B.; Theriot, C.M.; Barrangou, R. The Lactobacillus bile salt hydrolase repertoire reveals niche-specific adaptation. mSphere 2018, 3, e00140-18. [Google Scholar] [CrossRef]
- Kotelevets, L.; Chastre, E. Extracellular Vesicles in Colorectal Cancer: From Tumor Growth and Metastasis to Biomarkers and Nanomedications. Cancers 2023, 15, 1107. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.; Masuda, N. Characterization of NADP-dependent 7 beta-hydroxysteroid dehydrogenases from Peptostreptococcus productus and Eubacterium aerofaciens. Appl. Environ. Microbiol. 1982, 43, 1057–1063. [Google Scholar] [CrossRef]
- Wang, Y.; Wan, X.; Wu, X.; Zhang, C.; Liu, J.; Hou, S. Eubacterium rectale contributes to colorectal cancer initiation via promoting colitis. Gut Pathog. 2021, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Fan, J.; Li, T.; Yan, X.; Jiang, Y. Nuciferine protects against high-fat diet-induced hepatic steatosis via modulation of gut microbiota and bile acid metabolism in rats. J. Agric. Food Chem. 2022, 70, 12014–12028. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Louis, P.; Flint, H.J. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl. Environ. Microbiol. 2004, 70, 5810–5817. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.; Cato, E.P.; Holdeman, L.V. Fermentation patterns of some Clostridium species. Int. J. Syst. Evol. Microbiol. 1966, 16, 383–416. [Google Scholar] [CrossRef]
- Abedi, E.; Hashemi, S.M.B. Lactic acid production—Producing microorganisms and substrates sources-state of art. Heliyon 2020, 6, e04974. [Google Scholar] [CrossRef]
- Bruno, M.E.C.; Rogier, E.W.; Frantz, A.L.; Stefka, A.T.; Thompson, S.N.; Kaetzel, C.S. Regulation of the polymeric immunoglobulin receptor in intestinal epithelial cells by Enterobacteriaceae: Implications for mucosal homeostasis. Immunol. Investig. 2010, 39, 356–382. [Google Scholar] [CrossRef] [PubMed]
- Llibre, A.; Grudzinska, F.S.; O’Shea, M.K.; Duffy, D.; Thickett, D.R.; Mauro, C.; Scott, A. Lactate cross-talk in host–pathogen interactions. Biochem. J. 2021, 478, 3157–3178. [Google Scholar] [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Stecher, B. The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. Metab. Bact. Pathog. 2015, 3, 297–320. [Google Scholar] [CrossRef]
- Mira-Pascual, L.; Cabrera-Rubio, R.; Ocon, S.; Costales, P.; Parra, A.; Suarez, A.; Moris, F.; Rodrigo, L.; Mira, A.; Collado, M.C. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J. Gastroenterol. 2015, 50, 167–179. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Shang, Y.; Wu, B.; Yang, E.; Luo, Y.; Li, X. Intestinal bacteria detected in cancer and adjacent tissue from patients with colorectal cancer. Oncol. Lett. 2019, 17, 1115–1127. [Google Scholar] [CrossRef]
- Huang, W.K.; Chang, J.C.; See, L.C.; Tu, H.T.; Chen, J.S.; Liaw, C.C.; Lin, Y.C.; Yang, T.S. Higher rate of colorectal cancer among patients with pyogenic liver abscess with Klebsiella pneumoniae than those without: An 11-year follow-up study. Color. Dis. 2012, 14, e794–e801. [Google Scholar] [CrossRef] [PubMed]
- Strakova, N.; Korena, K.; Karpiskova, R. Klebsiella pneumoniae producing bacterial toxin colibactin as a risk of colorectal cancer development—A systematic review. Toxicon 2021, 197, 126–135. [Google Scholar] [CrossRef]
- Biarc, J.; Nguyen, I.S.; Pini, A.; Gossé, F.; Richert, S.; Thiersé, D.; Van Dorsselaer, A.; Leize-Wagner, E.; Raul, F.; Klein, J.-P.; et al. Carcinogenic properties of proteins with pro-inflammatory activity from Streptococcus infantarius (formerly S. bovis). Carcinogenesis 2004, 25, 1477–1484. [Google Scholar] [CrossRef]
- Pasquereau-Kotula, E.; Martins, M.; Aymeric, L.; Dramsi, S. Significance of Streptococcus gallolyticus subsp. gallolyticus association with colorectal cancer. Front. Microbiol. 2018, 9, 614. [Google Scholar]
- Boleij, A.; Muytjens, C.M.J.; Bukhari, S.I.; Cayet, N.; Glaser, P.; Hermans, P.W.M.; Swinkels, D.W.; Bolhuis, A.; Tjalsma, H. Novel clues on the specific association of Streptococcus gallolyticus subsp. gallolyticus with colorectal cancer. J. Infect. Dis. 2011, 203, 1101–1109. [Google Scholar]
- Abdulamir, A.S.; Hafidh, R.R.; Bakar, F. Molecular detection, quantification, and isolation of Streptococcus gallolyticus bacteria colonizing colorectal tumors: Inflammation-driven potential of carcinogenesis via IL-1, COX-2, and IL-8. Mol. Cancer 2010, 9, 249. [Google Scholar] [CrossRef]
- Nicolescu, C.M.; Bumbac, M.; Buruleanu, C.L.; Popescu, E.C.; Stanescu, S.G.; Georgescu, A.A.; Toma, S.M. Biopolymers Produced by Lactic Acid Bacteria: Characterization and Food Application. Polymers 2023, 15, 1539. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Montassier, E.; Abrahante, J.; Priya, S.; Niccum, D.E.; Khoruts, A.; Starr, T.K.; Knights, D.; Blekhman, R. Colorectal cancer mutational profiles correlate with defined microbial communities in the tumor microenvironment. PLoS Genet. 2018, 14, e1007376. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Gharaibeh, R.Z.; Mühlbauer, M.; Perez-Chanona, E.; Uronis, J.M.; McCafferty, J.; Fodor, A.A.; Jobin, C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. 2014, 5, 4724. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Bleich, R.M.; Arthur, J.C. Revealing a microbial carcinogen. Science 2019, 363, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Déchelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Putze, J.; Hennequin, C.; Nougayrède, J.-P.; Zhang, W.; Homburg, S.; Karch, H.; Bringer, M.-A.; Fayolle, C.; Carniel, E.; Rabsch, W.; et al. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect. Immun. 2009, 77, 4696–4703. [Google Scholar] [CrossRef]
- de Graaf, F.K.; Tieze, G.A.; Bonga, S.W.; Stouthamer, A.H. Purification and genetic determination of bacteriocin production in Enterobacter cloacae. J. Bacteriol. 1968, 95, 631–640. [Google Scholar] [CrossRef]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The genus Alistipes: Gut bacteria with emerging implications to inflammation, cancer, and mental health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Chaplin, A.V.; Khokhlova, E.V.; Shcherbakova, V.A.; Motuzova, O.V.; Bozhenko, V.K.; Kafarskaia, L.I.; Efimov, B.V. Alistipes inops sp. nov. and Coprobacter secundus sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2015, 65 Pt 12, 4580–4588. [Google Scholar] [CrossRef]
- Walker, A.; Pfitzner, B.; Harir, M.; Schaubeck, M.; Calasan, J.; Heinzmann, S.S.; Turaev, D.; Rattei, T.; Endesfelder, D.; zu Castell, W.; et al. Sulfonolipids as novel metabolite markers of Alistipes and Odoribacter affected by high-fat diets. Sci. Rep. 2017, 7, 11047. [Google Scholar] [CrossRef]
- Kobayashi, T.; Honke, K.; Kamio, K.; Sakakibara, N.; Gasa, S.; Miyao, N.; Tsukamoto, T.; Ishizuka, I.; Miyazaki, T.; Makita, A. Sulfolipids and glycolipid sulfotransferase activities in human renal cell carcinoma cells. Br. J. Cancer 1993, 67, 76–80. [Google Scholar] [CrossRef]
- Seal, J.B.; Alverdy, J.C.; Zaborina, O.; An, G. Agent-based dynamic knowledge representation of Pseudomonas aeruginosa virulence activation in the stressed gut: Towards characterizing host-pathogen interactions in gut-derived sepsis. Theor. Biol. Med. Model. 2011, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.P.; Adley, C.C. Ralstonia spp.: Emerging global opportunistic pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Selgrad, M.; Malfertheiner, P.; Fini, L.; Goel, A.; Boland, C.R.; Ricciardiello, L. The role of viral and bacterial pathogens in gastrointestinal cancer. J. Cell. Physiol. 2008, 216, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Vuotto, F.; Berthon, C.; Lemaitre, N.; Duhamel, A.; Balkaran, S.; Le Ray, E.; Micol, J.B.; Faure, K.; Alfandari, S. Risk factors, clinical features, and outcome of Pseudomonas aeruginosa bacteremia in patients with hematologic malignancies: A case-control study. Am. J. Infect. Control 2013, 41, 527–530. [Google Scholar] [CrossRef]
- Andremont, A.; Marang, B.; Tancrède, C.; Baume, D.; Hill, C. Antibiotic treatment and intestinal colonization by Pseudomonas aeruginosa in cancer patients. Antimicrob. Agents Chemother. 1989, 33, 1400–1402. [Google Scholar] [CrossRef]
- Liang, J.Q.; Li, T.; Nakatsu, G.; Chen, Y.-X.; Yau, T.O.; Chu, E.; Wong, S.; Szeto, C.H.; Ng, S.C.; Chan, F.K.L.; et al. A novel faecal Lachnoclostridium marker for the non-invasive diagnosis of colorectal adenoma and cancer. Gut 2020, 69, 1248–1257. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Zagato, E.; Pozzi, C.; Bertocchi, A.; Schioppa, T.; Saccheri, F.; Guglietta, S.; Fosso, B.; Melocchi, L.; Nizzoli, G.; Troisi, J.; et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat. Microbiol. 2020, 5, 511–524. [Google Scholar] [CrossRef]
- Sui, H.; Zhang, L.; Gu, K.; Chai, N.; Ji, Q.; Zhou, L.; Wang, Y.; Ren, J.; Yang, L.; Zhang, B.; et al. YYFZBJS ameliorates colorectal cancer progression in ApcMin/+ mice by remodeling gut microbiota and inhibiting regulatory T-cell generation. Cell Commun. Signal. 2020, 18, 113. [Google Scholar] [CrossRef]
- Koh, G.Y.; Kane, A.V.; Wu, X.; Crott, J.W. Parabacteroides distasonis attenuates tumorigenesis, modulates inflammatory markers and promotes intestinal barrier integrity in azoxymethane-treated A/J mice. Carcinogenesis 2020, 41, 909–917. [Google Scholar] [CrossRef]
- Fuhren, J.; Schwalbe, M.; Boekhorst, J.; Rösch, C.; Schols, H.A.; Kleerebezem, M. Dietary calcium phosphate strongly impacts gut microbiome changes elicited by inulin and galacto-oligosaccharides consumption. Microbiome 2021, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Hong, X.; Wang, J.; Sun, T.; Yu, T.; Yu, Y.; Fang, J.; Xiong, H. Metformin elicits antitumour effect by modulation of the gut microbiota and rescues Fusobacterium nucleatum-induced colorectal tumourigenesis. EBioMedicine 2020, 61, 103037. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, L.; Yuan, X.; Li, Y.; Shi, J.; Zhang, H.; Zhao, Y.; Han, L.; Wang, H.; Yan, Y.; et al. Pre-administration of berberine exerts chemopreventive effects in AOM/DSS-induced colitis-associated carcinogenesis mice via modulating inflammation and intestinal microbiota. Nutrients 2022, 14, 726. [Google Scholar] [CrossRef]
- Crowe, W.; Pan, X.; Mackle, J.; Harris, A.; Hardiman, G.; Elliott, C.T.; Green, B.D. Dietary inclusion of nitrite-containing frankfurter exacerbates colorectal cancer pathology and alters metabolism in APCmin mice. NPJ Sci. Food 2022, 6, 60. [Google Scholar] [CrossRef]
- Crowe, W.; Elliott, C.T.; Green, B.D. A review of the in vivo evidence investigating the role of nitrite exposure from processed meat consumption in the development of colorectal cancer. Nutrients 2019, 11, 2673. [Google Scholar] [CrossRef] [PubMed]
- Iadsee, N.; Chuaypen, N.; Techawiwattanaboon, T.; Jinato, T.; Patcharatrakul, T.; Malakorn, S.; Petchlorlian, A.; Praditpornsilpa, K.; Patarakul, K. Identification of a novel gut microbiota signature associated with colorectal cancer in Thai population. Sci. Rep. 2023, 13, 6702. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Liu, G.; Lin, Y.; Guo, C.L.; Han, J.; Chu, E.S.H.; Shi, C.; Li, Y.; Zhang, H.; Hu, C.; et al. Altered gut microbiome composition by appendectomy contributes to colorectal cancer. Oncogene 2023, 42, 530–540. [Google Scholar] [CrossRef]
- Gryaznova, M.; Dvoretskaya, Y.; Burakova, I.; Syromyatnikov, M.; Popov, E.; Kokina, A.; Mikhaylov, E.; Popov, V. Dynamics of changes in the gut microbiota of healthy mice fed with lactic acid bacteria and bifidobacteria. Microorganisms 2022, 10, 1020. [Google Scholar] [CrossRef]
- Zhang, S.-M.; Huang, S.-L. The Commensal Anaerobe Veillonella dispar Reprograms Its Lactate Metabolism and Short-Chain Fatty Acid Production during the Stationary Phase. Microbiol. Spectr. 2023, 11, e03558-22. [Google Scholar] [CrossRef]
- Kelly, J.R.; Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G.; Hyland, N.P. Breaking down the barriers: The gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell. Neurosci. 2015, 9, 392. [Google Scholar] [CrossRef]
- Ringel, Y. The gut microbiome in irritable bowel syndrome and other functional bowel disorders. Gastroenterol. Clin. N. Am. 2017, 46, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Caminero, A.; McCarville, J.L.; Galipeau, H.J.; Deraison, C.; Bernier, S.P.; Constante, M.; Rolland, C.; Meisel, M.; Murray, J.A.; Yu, X.B.; et al. Duodenal bacterial proteolytic activity determines sensitivity to dietary antigen through protease-activated receptor-2. Nat. Commun. 2019, 10, 1198. [Google Scholar] [CrossRef] [PubMed]
- Steck, N.; Hoffmann, M.; Sava, I.G.; Kim, S.C.; Hahne, H.; Tonkonogy, S.L.; Mair, K.; Krueger, D.; Pruteanu, M.; Shanahan, F.; et al. Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 2011, 141, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Sheflin, A.M.; Whitney, A.K.; Weir, T.L. Cancer-promoting effects of microbial dysbiosis. Curr. Oncol. Rep. 2014, 16, 406. [Google Scholar] [CrossRef]
- Rivas-Domínguez, A.; Pastor, N.; Martínez-López, L.; Colón-Pérez, J.; Bermúdez, B.; Orta, M.L. The role of DNA damage response in dysbiosis-induced colorectal cancer. Cells 2021, 10, 1934. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Healthy Controls N = 25 (50%) | CRC Patients N = 25 (50%) |
|---|---|---|
| Sex | ||
| Male | 21 (84.0) | 15 (60.0) |
| Female | 4 (16.0) | 10 (40.0) |
| Age (yrs) | ||
| Mean ± SD | 47.4 ± 11.7 | 54.3 ± 14.2 |
| Weight status | ||
| Underweight | 0 (0.00) | 2 (8.00) |
| Healthy weight | 16 (64.0) | 12 (48.0) |
| Overweight | 2 (8.00) | 9 (36.0) |
| Obesity | 7 (28.0) | 2 (9.00) |
| Cancer stage | ||
| I | 0 (0.00) | |
| II | 0 (0.00) | |
| III | 13 (52.0) | |
| IV | 12 (48.0) | |
| Metastasis | 4 (16.0) | |
| Cancer location | ||
| Sigmoid | 3 (12.0) | |
| Colon | 13 (52.0) | |
| Rectum | 9 (36.0) |
|
Total
(n = 25) |
Healthy Controls
(n = 15) | CRC Patients (n = 10) | p-Value | |
|---|---|---|---|---|
| Diet quality score |
9.48 ± 1.11
10.0 (9.00–11.0) |
9.67 ± 1.11
10.0 (9.00–11.00) |
10.1 ± 1.10
10.0 (9.00–11.00) | 0.495 |
| Fruits, servings/day |
1.60 ± 0.50
2.00 (1.00–2.00) |
1.53 ± 0.52
2.00 (1.00–2.00) |
1.70 ± 0.48
2.00 (1.00–2.00) | 0.495 |
| Vegetables, servings/day |
1.44 ± 0.58
1.00 (1.00–2.00) |
1.33 ± 0.49
1.00 (1.00–2.00) |
1.60 ± 0.70
1.50 (1.00–2.00) | 0.428 |
| Total fat, g/day |
83.00 ± 41.40
76.00 (52.50–104) |
83.30 ± 36.10
77.0 (54.00–100) |
82.60 ± 50.60
69.0 (39.00–140) | 0.531 |
| Saturated fat, g/day |
29.50 ± 15.40
25.00 (16.50–39.5) |
29.10 ± 14.00
25.0 0(18.00–30.0) |
30.00 ± 18.00
23.50 (13.50–51.00) | 0.683 |
| Monounsaturated fat, g/day |
30.80 ± 17.30
26.00 (16.50–42.50) |
30.40 ± 13.30
28.00 (22.00–38.00) |
31.50 ± 22.80
25.50 (11.00–51.80) | 0.683 |
| Polyunsaturated fat, g/day |
14.30 ± 8.500
12.00 (7.50–20.00) |
14.10 ± 6.55
13.00 (9.00–20.00) |
14.60 ± 11.20
11.00 (5.00–24.50) | 0.567 |
| Fat from plant food sources, g/day |
39.40 ± 27.0
30.00 (15.00–57.00) |
37.9 ± 20.3
36.0 (22.00–57.00) |
41.6 ± 35.9
23.0 (13.00–66.50) | 0.723 |
| Fat from animal food sources, g/day |
43.60 ± 22.30
43.00 (27.50–50.00) |
45.2 ± 20.20
43.0 (30.00–49.00) |
41.20 ± 26.10
35.5 (21.00–68.00) | 0.397 |
|
Odds Ratio
(OR) | 95% Confidence Interval | p- Value | |
|---|---|---|---|
| Diet quality score | 0.40 | 0.06 to 2.70 | 0.348 |
| Fruits, servings/day | 0.11 | 0.003 to 4.34 | 0.236 |
| Vegetables, servings/day | 0.46 | 0.02 to 9.18 | 0.608 |
| Total fat, g/day | 0.99 | 0.94 to 1.03 | 0.558 |
| Saturated fat, g/day | 1.01 | 0.92 to 1.12 | 0.798 |
| Monounsaturated fat, g/day | 0.94 | 0.82 to 1.07 | 0.340 |
| Polyunsaturated fat, g/day | 0.90 | 0.70 to 1.14 | 0.370 |
| Fat from plant food sources, g/day | 0.97 | 0.89 to 1.05 | 0.422 |
| Fat from animal food sources, g/day | 1.00 | 0.92 to 1.07 | 0.912 |
| Bacteria | Healthy Controls | CRC Patients | ||||
|---|---|---|---|---|---|---|
| B (SE) | p-Value | R-Square | B (SE) | p-Value | R-Square | |
| Streptococcus | 0.00 (0.00) | 0.859 | 0.00 | −0.05 (0.04) | 0.203 | 0.19 |
| Lactobacillus | 0.00 (0.01) | 0.757 | 0.01 | 0.02 (0.02) | 0.468 | 0.07 |
| Klebsiella | * | * | * | * | * | * |
| Intestinibacter | * | * | * | * | * | * |
| Ralstonia | * | * | * | * | * | * |
| Alistipes | 0.00 (0.00) | 0.159 | 0.15 | 0.01 (0.00) | 0.029 * | 0.47 |
| Pseudomonas | * | * | * | 1.567 × 10−5 (0.00) | 0.692 | 0.02 |
| Peptostreptococcus | * | * | * | 0.00 (0.00) | 0.853 | 0.01 |
| Faecalibaculum | * | * | * | * | * | * |
| Dubosiella | * | * | * | * | * | * |
| Erysipelatoclostridium | * | * | * | * | * | * |
| Enterobacter | * | * | * | * | * | * |
| Sellimonas | * | * | * | * | * | * |
| Lachnoclostridium | * | * | * | 2.618 × 10−5 (0.00) | 0.063 | 0.37 |
| Eubacterium | 5.560 × 10−5 (0.00) | 0.225 | 0.11 | −1.137 × 10−5 (0.00) | 0.320 | 0.12 |
| Clostridium | 0.00 (0.00) | 0.061 | 0.25 | 8.491 × 10−5 (0.00) | 0.839 | 0.01 |
| Aerococcus | * | * | * | * | * | * |
| Family XIII AD3011 group | * | * | * | * | * | * |
| Veillonella | * | * | * | 0.00 (0.00) | 0.820 | 0.01 |
| Bacteria | Healthy Controls | CRC Patients | ||||
|---|---|---|---|---|---|---|
| B (SE) | p-Value | R-Square | B (SE) | p-Value | R-Square | |
| Streptococcus | 4.981 × 10−5 (0.00) | 0.327 | 0.07 | 0.00 (0.00) | 0.303 | 0.13 |
| Lactobacillus | 0.00 (0.00) | 0.457 | 0.04 | 0.00 (0.00) | 0.467 | 0.07 |
| Klebsiella | * | * | * | * | * | * |
| Intestinibacter | * | * | * | * | * | * |
| Ralstonia | * | * | * | * | * | * |
| Alistipes | −5.251 × 10−6 | 0.818 | 0.00 | −5.948 × 10−5 (0.00) | 0.456 | 0.07 |
| Pseudomonas | * | * | * | −4.883 × 10−7 (0.00) | 0.569 | 0.04 |
| Peptostreptococcus | * | * | * | −4.403 × 10−5 (0.00) | 0.218 | 0.18 |
| Faecalibaculum | * | * | * | * | * | * |
| Dubosiella | * | * | * | * | * | * |
| Erysipelatoclostridium | * | * | * | * | * | * |
| Enterobacter | * | * | * | * | * | * |
| Sellimonas | * | * | * | * | * | * |
| Lachnoclostridium | * | * | * | −6.919 × 10−8 (0.00) | 0.840 | 0.01 |
| Eubacterium | −3.448 × 10−7 (0.00) | 0.813 | 0.00 | −2.281 × 10−7 (0.00) | 0.361 | 0.11 |
| Clostridium | 2.200 × 10−8 (0.00) | 0.992 | 0.00 | 1.296 × 10−5 (0.00) | 0.124 | 0.27 |
| Aerococcus | * | * | * | * | * | * |
| Family XIII AD3011 group | * | * | * | * | * | * |
| Veillonella | * | * | * | 1.724 × 10−5 (0.00) | 0.090 | 0.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhhazmi, A.A.; Almutawif, Y.A.; Mumena, W.A.; Alhazmi, S.M.; Abujamel, T.S.; Alhusayni, R.M.; Aloufi, R.; Al-Hejaili, R.R.; Alhujaily, R.; Alrehaili, L.M.; et al. Identification of Gut Microbiota Profile Associated with Colorectal Cancer in Saudi Population. Cancers 2023, 15, 5019. https://doi.org/10.3390/cancers15205019
Alhhazmi AA, Almutawif YA, Mumena WA, Alhazmi SM, Abujamel TS, Alhusayni RM, Aloufi R, Al-Hejaili RR, Alhujaily R, Alrehaili LM, et al. Identification of Gut Microbiota Profile Associated with Colorectal Cancer in Saudi Population. Cancers. 2023; 15(20):5019. https://doi.org/10.3390/cancers15205019
Chicago/Turabian StyleAlhhazmi, Areej A., Yahya A. Almutawif, Walaa A. Mumena, Shaima M. Alhazmi, Turki S. Abujamel, Ruba M. Alhusayni, Raghad Aloufi, Razan R. Al-Hejaili, Rahaf Alhujaily, Lama M. Alrehaili, and et al. 2023. "Identification of Gut Microbiota Profile Associated with Colorectal Cancer in Saudi Population" Cancers 15, no. 20: 5019. https://doi.org/10.3390/cancers15205019
APA StyleAlhhazmi, A. A., Almutawif, Y. A., Mumena, W. A., Alhazmi, S. M., Abujamel, T. S., Alhusayni, R. M., Aloufi, R., Al-Hejaili, R. R., Alhujaily, R., Alrehaili, L. M., Alsaedy, R. A., Khoja, R. H., Ahmed, W., Abdelmohsen, M. F., & Mohammed-Saeid, W. (2023). Identification of Gut Microbiota Profile Associated with Colorectal Cancer in Saudi Population. Cancers, 15(20), 5019. https://doi.org/10.3390/cancers15205019