
Emphasis on Adipocyte Transformation: Anti-Inflammatory Agents to Prevent the Development of Cancer-Associated Adipocytes
Abstract
Simple Summary
Abstract
1. Introduction
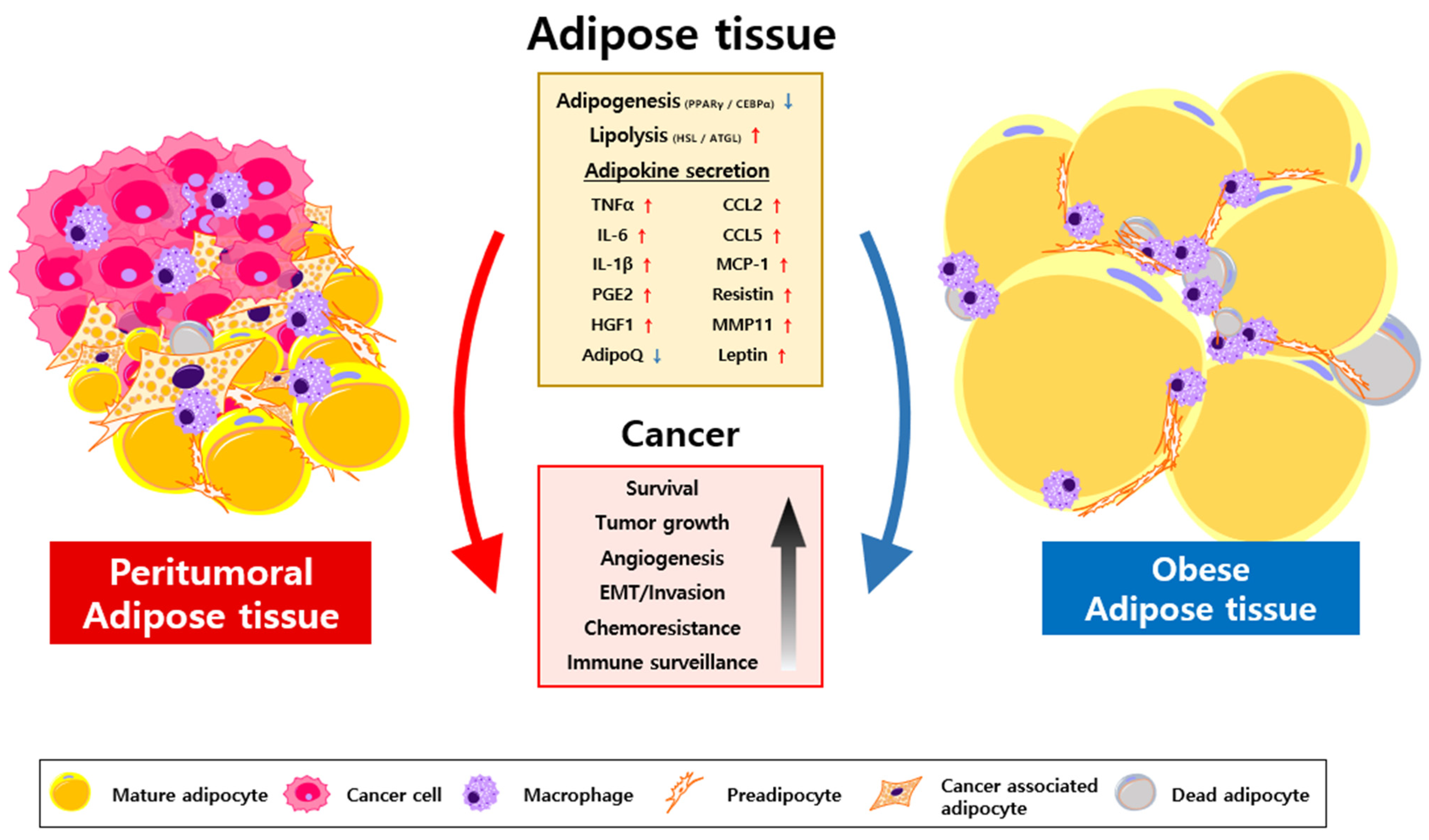
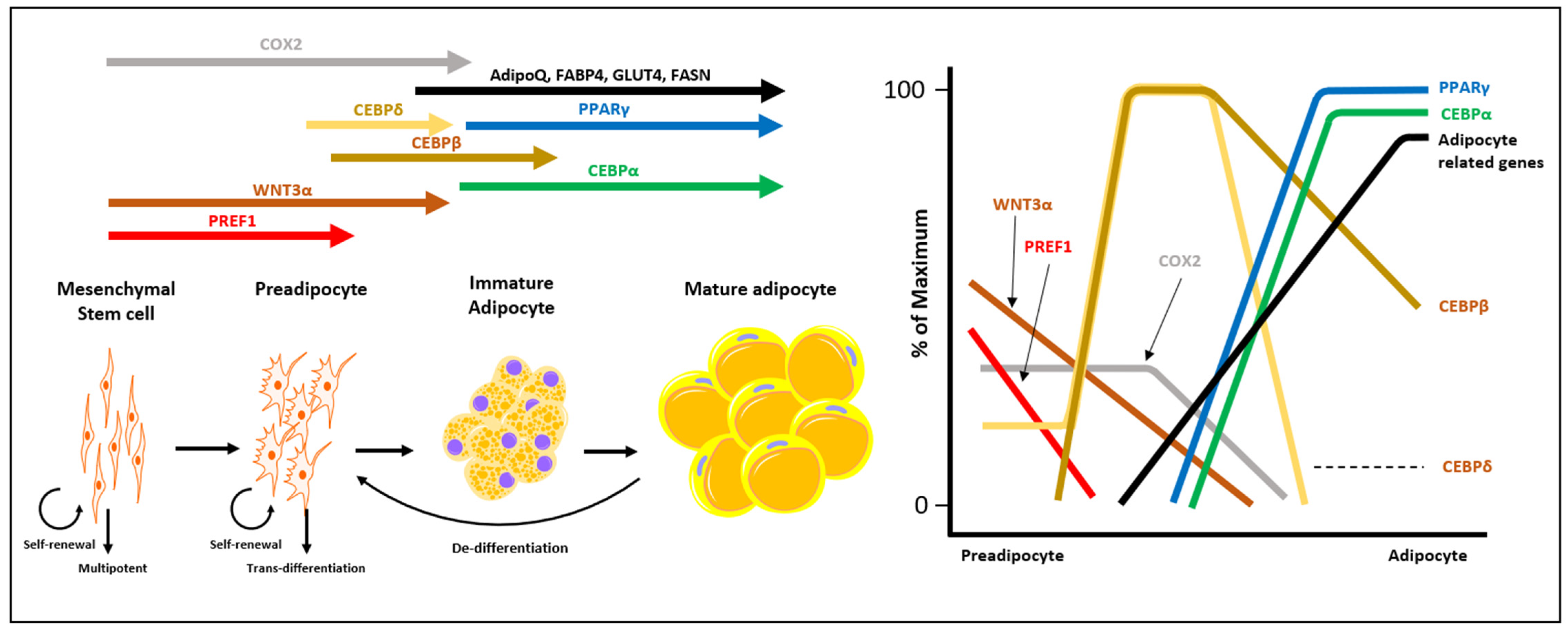
2. Characterization of CAA
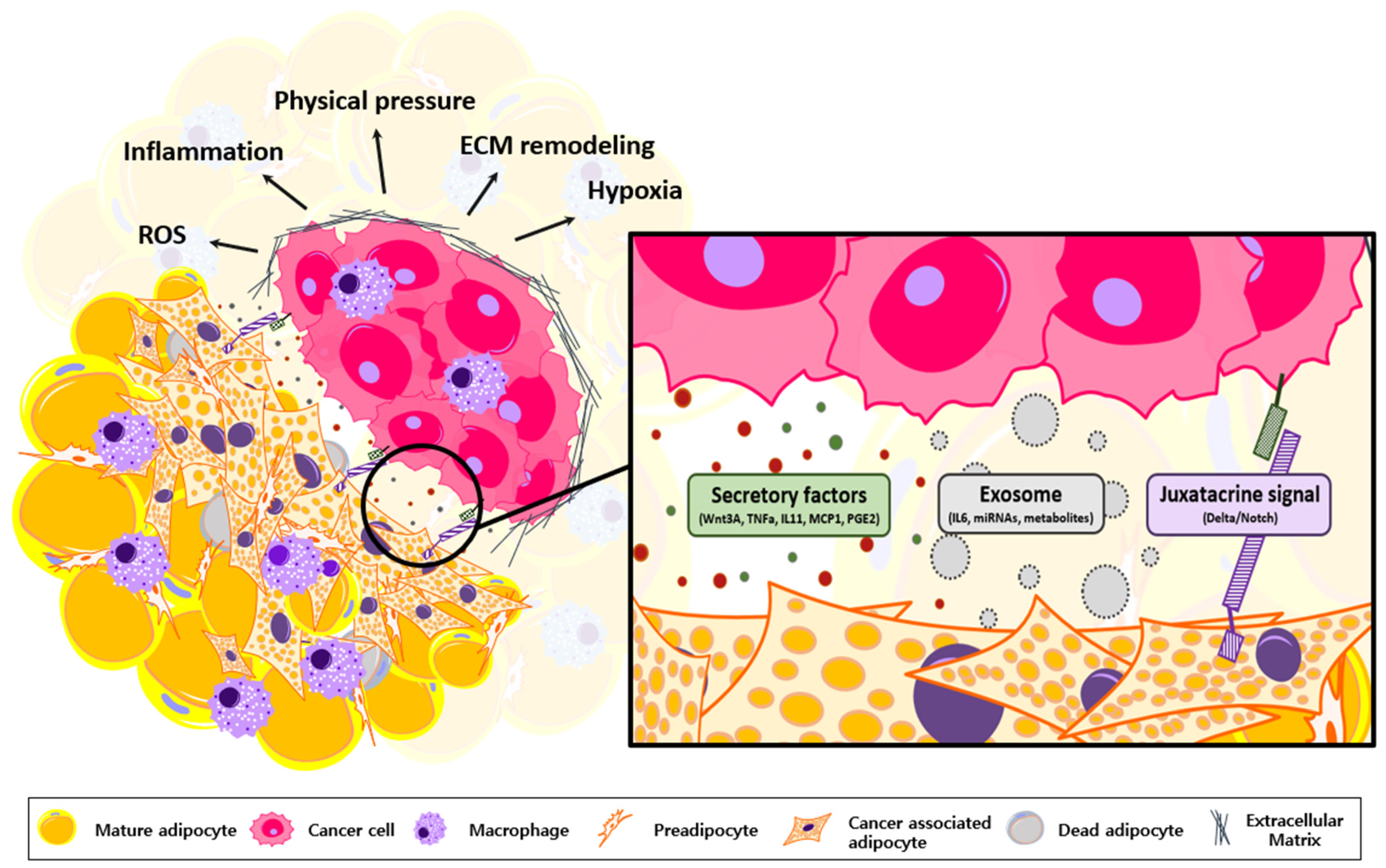
3. Oncogenic Roles of Transformed Adipocytes
4. Wnt and Notch-Signaling to Induce CAA Transformation
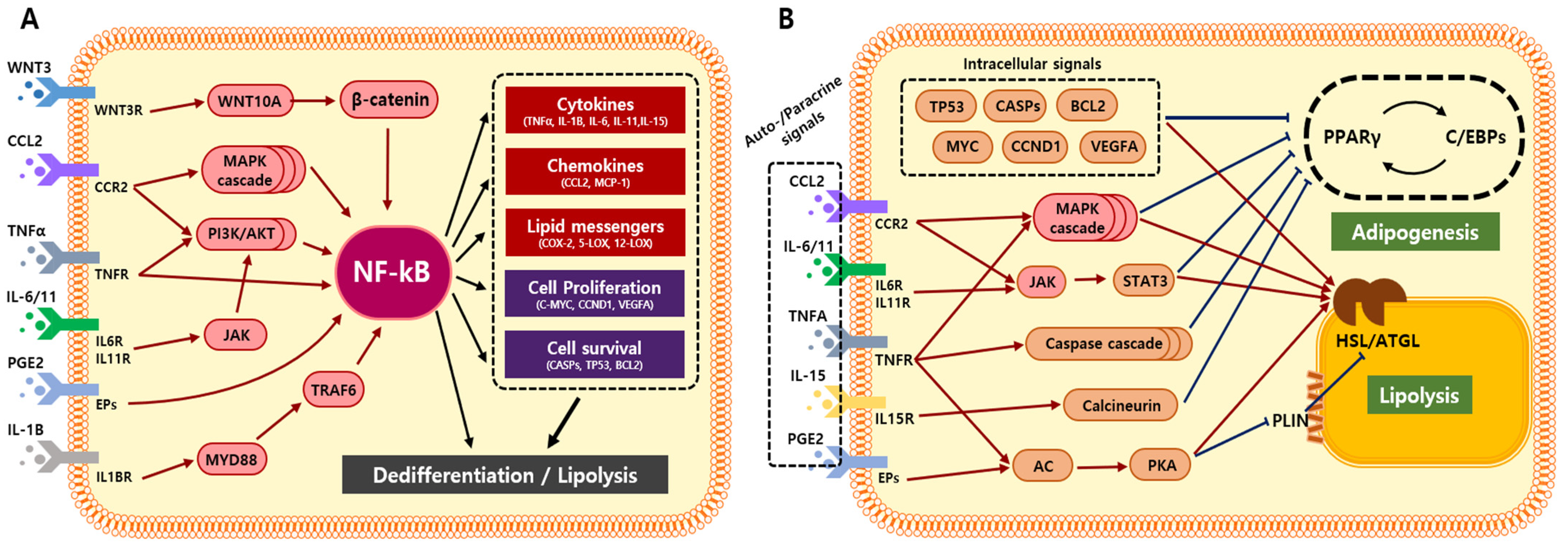
5. Adipocyte Transformation via Cancer-Derived Inflammatory Factors
6. Adipocyte Transformation via NF-ĸB-Mediated Inflammation
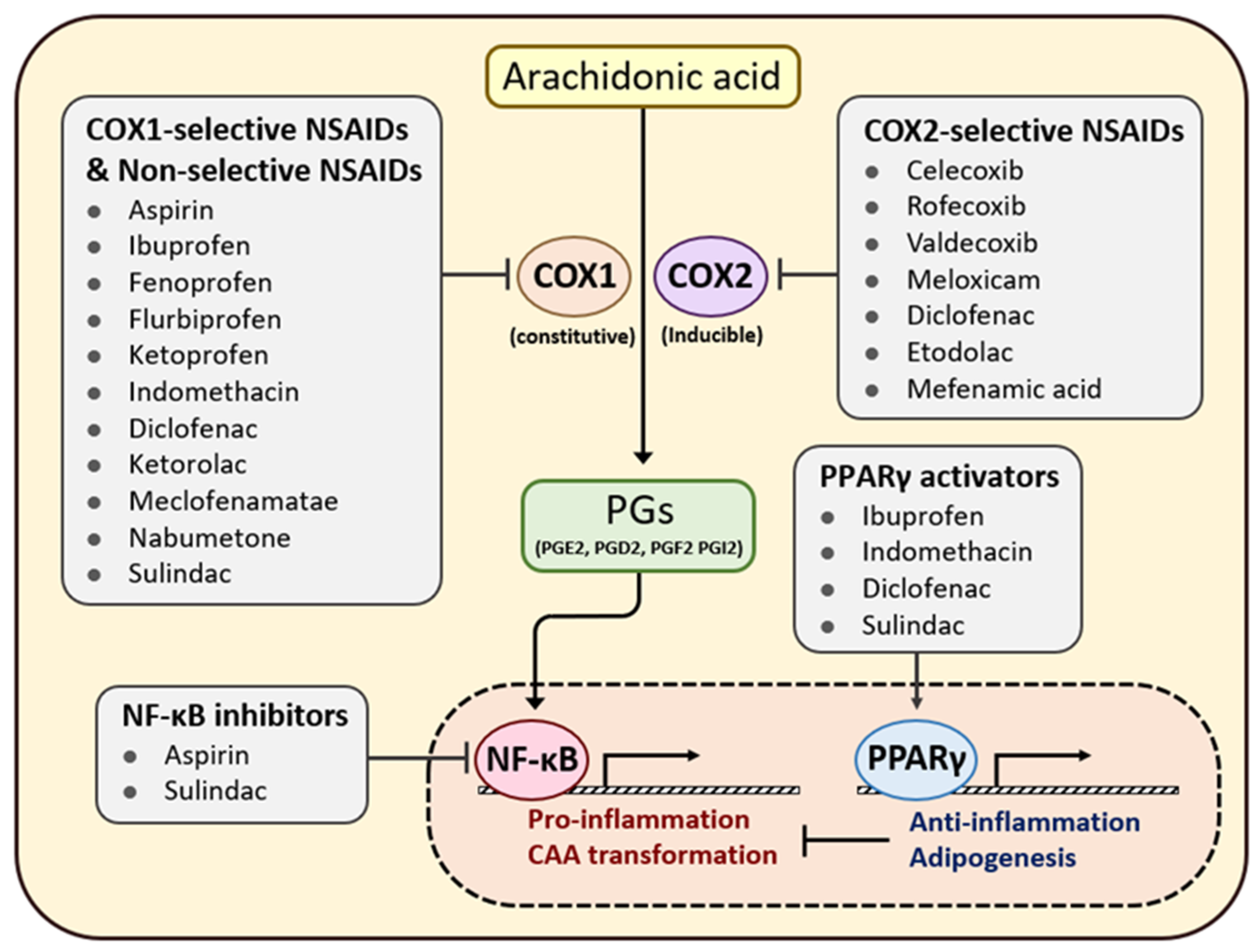
7. Efficacy of Anti-Inflammatory Agents in Patients with Cancer
8. Anti-Inflammatory Agents in Obese Patients with Cancer
9. Future Perspective
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAA | cancer-associated adipocyte |
| TME | tumor microenvironment |
| COX-2 | cyclooxygenase-2 |
| NSAID | nonsteroidal anti-inflammatory drug |
| FA | fatty acid |
| BMI | body mass index |
| PG | prostaglandin |
| PPAR | peroxisome proliferator-activated receptor |
| C/EBP | CCAAT enhancer binding protein |
| IL | interleukin |
| TNFα | tumor necrosis factor α |
| FFA | free fatty acid |
| FAO | fatty acid oxidation |
| TG | triacylglycerol |
| MMP | matrix metalloproteinase |
| CCL2 | chemokine ligand 2 |
| VEGF | vascular endothelial growth factor |
| ECM | extracellular matrix |
| EMT | epithelial-mesenchymal transition |
| IGF | insulin-like growth factor |
| IGFBP | IGF binding protein |
| AP1 | activator protein-1 |
| MSC | mesenchymal stem cell |
| CiDA | compression-induced dedifferentiated adipocytes |
| LPS | liposarcoma |
| AP2 | adipocyte protein 2 |
| TGFβ | transforming growth factor β |
| MCP1 | monocyte chemoattractant protein-1 |
| PPRE | PPAR response element |
| PLIN1 | perilipin1 |
| HSL | hormone sensitive lipase |
| CCR2 | C-C chemokine receptor type 2 |
| cAMP | cyclic AMP |
| PKA | protein kinase A |
| ATGL | adipose triglyceride lipase |
| JAK | Janus kinase |
| STAT | signal transducer and activator of transcription |
| EV | extracellular vesicle |
| FAP | familial adenomatous polyposis |
| OR | odds ratio |
| CI | confidence interval |
| HR | hazard ratio |
References
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocr. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagai, R. Adipose tissue inflammation in obesity and metabolic syndrome. Discov. Med. 2009, 8, 55–60. [Google Scholar] [PubMed]
- Lafontan, M. Adipose tissue and adipocyte dysregulation. Diabetes Metab. 2014, 40, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The Role of Tumor Stroma in Cancer Progression and Prognosis Emphasis on Carcinoma-Associated Fibroblasts and Non-small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.H.; He, S.B. Multi-faceted role of cancer-associated adipocytes in the tumor microenvironment. Mol. Med. Rep. 2021, 24, 866. [Google Scholar] [CrossRef]
- Martin-Perez, M.; Urdiroz-Urricelqui, U.; Bigas, C.; Benitah, S.A. The role of lipids in cancer progression and metastasis. Cell Metab. 2022, 34, 1675–1699. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhu, Y.; Hepler, C.; Zhang, Q.; Park, J.; Gliniak, C.; Henry, G.H.; Crewe, C.; Bu, D.; Zhang, Z.; et al. Adipocyte mesenchymal transition contributes to mammary tumor progression. Cell Rep. 2022, 40, 111362. [Google Scholar] [CrossRef]
- Attane, C.; Muller, C. Drilling for Oil: Tumor-Surrounding Adipocytes Fueling Cancer. Trends. Cancer 2020, 6, 593–604. [Google Scholar] [CrossRef]
- Zoico, E.; Darra, E.; Rizzatti, V.; Budui, S.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Menegazzi, M.; Cinti, S.; et al. Adipocytes WNT5a mediated dedifferentiation: A possible target in pancreatic cancer microenvironment. Oncotarget 2016, 7, 20223–20235. [Google Scholar] [CrossRef]
- Gustafson, B.; Smith, U. Activation of Canonical Wingless-type MMTV Integration Site Family (Wnt) Signaling in Mature Adipocytes Increases beta-Catenin Levels and Leads to Cell Dedifferentiation and Insulin Resistance. J. Biol. Chem. 2010, 285, 14031–14041. [Google Scholar] [CrossRef]
- De Pergola, G.; Silvestris, F. Obesity as a major risk factor for cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Laurencikiene, J.; van Harmelen, V.; Nordstrom, E.A.; Dicker, A.; Blomqvist, L.; Naslund, E.; Langin, D.; Arner, P.; Ryden, M. NF-kappa B is important for TNF-alpha-induced lipolysis in human adipocytes. J. Lipid Res. 2007, 48, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, A.M.; Foster, C.; Landry, A.; Sorisky, A. The role of interleukin 1 beta in the anti-adipogenic action of macrophages on human preadipocytes. J. Endocrinol. 2013, 217, 197–206. [Google Scholar] [CrossRef]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-Inflammatory Drugs as Anticancer Agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.Y. Role of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in Cancer Prevention and Cancer Promotion. Adv. Pharmacol. Sci. 2019, 2019, 3418975. [Google Scholar] [CrossRef]
- Rybinska, I.; Mangano, N.; Tagliabue, E.; Triulzi, T. Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. Int. J. Mol. Sci. 2021, 22, 3775. [Google Scholar] [CrossRef] [PubMed]
- Rybinska, I.; Agresti, R.; Trapani, A.; Tagliabue, E.; Triulzi, T. Adipocytes in Breast Cancer, the Thick and the Thin. Cells 2020, 9, 560. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Amor, S.; Iglesias-de la Cruz, M.C.; Ferrero, E.; Garcia-Villar, O.; Barrios, V.; Fernandez, N.; Monge, L.; Garcia-Villalon, A.L.; Granado, M. Peritumoral adipose tissue as a source of inflammatory and angiogenic factors in colorectal cancer. Int. J. Colorectal. Dis. 2016, 31, 365–375. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuede, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-Derived Fibroblasts Promote Tumor Progression and Contribute to the Desmoplastic Reaction in Breast Cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef]
- Dumas, J.F.; Brisson, L. Interaction between adipose tissue and cancer cells: Role for cancer progression. Cancer Metastasis Rev. 2021, 40, 31–46. [Google Scholar] [CrossRef]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk, N.; Lecoeur, B.; Bettaieb, A.; Paul, C.; Vegran, F. Impact of Lipid Metabolism on Antitumor Immune Response. Cancers 2022, 14, 1850. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Zuckier, L.S.; Ghesani, N.V. Dominant Uptake of Fatty Acid over Glucose by Prostate Cells: A Potential New Diagnostic and Therapeutic Approach. Anticancer Res. 2010, 30, 369–374. [Google Scholar] [PubMed]
- Siltari, A.; Syvala, H.; Lou, Y.R.; Gao, Y.; Murtola, T.J. Role of Lipids and Lipid Metabolism in Prostate Cancer Progression and the Tumor’s Immune Environment. Cancers 2022, 14, 4293. [Google Scholar] [CrossRef] [PubMed]
- Marino, N.; German, R.; Rao, X.; Simpson, E.; Liu, S.; Wan, J.; Liu, Y.; Sandusky, G.; Jacobsen, M.; Stoval, M.; et al. Upregulation of lipid metabolism genes in the breast prior to cancer diagnosis. NPJ Breast Cancer 2020, 6, 50. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Ren, J.; Xiao, Y.J.; Singh, L.S.; Zhao, X.X.; Zhao, Z.W.; Feng, L.; Rose, T.M.; Prestwich, G.D.; Xu, Y. Lysophosphatidic acid is constitutively produced by human peritoneal mesothelial cells and enhances adhesion, migration, and invasion of ovarian cancer cells. Cancer Res. 2006, 66, 3006–3014. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife 2020, 9, e55185. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, B.; Li, J.J.; Sun, S.; Yuan, J.P.; Sun, S.R. Cancer-associated adipocytes as immunomodulators in cancer. Biomark. Res. 2021, 9, 2. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Gnerlich, J.L.; Yao, K.; Wyrwicz, A.M.; Fitchev, P.; Crawford, S.E. Peritumoral Expression of Adipokines and Fatty Acids in Breast Cancer. Ann. Surg. Oncol. 2013, 20, 731–738. [Google Scholar] [CrossRef]
- Riondino, S.; Roselli, M.; Palmirotta, R.; Della-Morte, D.; Ferroni, P.; Guadagni, F. Obesity and colorectal cancer: Role of adipokines in tumor initiation and progression. World J. Gastroenterol. 2014, 20, 5177–5190. [Google Scholar] [CrossRef]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Brindley, D.N.; Tang, X.Y.; Meng, G.M.; Benesch, M.G.K. Role of Adipose Tissue-Derived Autotaxin, Lysophosphatidate Signaling, and Inflammation in the Progression and Treatment of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 5938. [Google Scholar] [CrossRef]
- D’Esposito, V.; Passaretti, F.; Hammarstedt, A.; Liguoro, D.; Terracciano, D.; Molea, G.; Canta, L.; Miele, C.; Smith, U.; Beguinot, F.; et al. Adipocyte-released insulin-like growth factor-1 is regulated by glucose and fatty acids and controls breast cancer cell growth in vitro. Diabetologia 2012, 55, 2811–2822. [Google Scholar] [CrossRef]
- Kelesidis, I.; Kelesidis, T.; Mantzoros, C. Adiponectin and cancer: A systematic review. Br. J. Cancer 2006, 94, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Theriau, C.F.; Sauve, O.S.; Beaudoin, M.S.; Wright, D.C.; Connor, M.K. Proliferative endocrine effects of adipose tissue from obese animals on MCF7 cells are ameliorated by resveratrol supplementation. PLoS ONE 2017, 12, e0183897. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.Y.; Li, J.J.; Sun, S.; Sun, S.R. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.S.M.; Vieira, A.R.; Aune, D.; Bandera, E.V.; Greenwood, D.C.; McTiernan, A.; Rosenblatt, D.N.; Thune, I.; Vieira, R.; Norat, T. Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Ann. Oncol. 2014, 25, 1901–1914. [Google Scholar] [CrossRef]
- Kim, H.S.; Jung, M.; Choi, S.K.; Woo, J.; Piao, Y.J.; Hwang, E.H.; Kim, H.; Kim, S.J.; Moon, W.K. IL-6-mediated cross-talk between human preadipocytes and ductal carcinoma in situ in breast cancer progression. J. Exp. Clin. Cancer Res. 2018, 37, 200. [Google Scholar] [CrossRef]
- Madsen, M.S.; Siersbaek, R.; Boergesen, M.; Nielsen, R.; Mandrup, S. Peroxisome Proliferator-Activated Receptor gamma and C/EBP alpha Synergistically Activate Key Metabolic Adipocyte Genes by Assisted Loading. Mol. Cell Biol. 2014, 34, 939–954. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.W.; Mao, A.S.; Seo, B.R.; Zhao, X.; Gupta, S.K.; Chen, M.R.; Han, Y.L.; Shih, T.Y.; Mooney, D.J.; Guo, M. Compression-induced dedifferentiation of adipocytes promotes tumor progression. Sci. Adv. 2020, 6, eaax5611. [Google Scholar] [CrossRef]
- Bi, P.P.; Yue, F.; Karki, A.; Castro, B.; Wirbisky, S.E.; Wang, C.; Durkes, A.; Elzey, B.D.; Andrisani, O.M.; Bidwell, C.A.; et al. Notch activation drives adipocyte dedifferentiation and tumorigenic transformation in mice. J. Exp. Med. 2016, 213, 2019–2037. [Google Scholar] [CrossRef]
- Jridi, I.; Cante-Barrett, K.; Pike-Overzet, K.; Staal, F.J.T. Inflammation and Wnt Signaling: Target for Immunomodulatory Therapy? Front. Cell Dev. Biol. 2021, 8, 615131. [Google Scholar] [CrossRef]
- Fazio, C.; Ricciardiello, L. Inflammation and Notch signaling: A crosstalk with opposite effects on tumorigenesis. Cell Death Dis. 2016, 7, e2515. [Google Scholar] [CrossRef]
- Koch, R.; Demant, M.; Aung, T.; Diering, N.; Cicholas, A.; Chapuy, B.; Wenzel, D.; Lahmann, M.; Guntsch, A.; Kiecke, C.; et al. Populational equilibrium through exosome-mediated Wnt signaling in tumor progression of diffuse large B-cell lymphoma. Blood 2014, 123, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Hottiger, M.O. Crosstalk between wnt/beta-Catenin and NF-kappa B Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Smith, U. Cytokines promote Wnt signaling and inflammation and impair the normal differentiation and lipid accumulation in 3T3-L1 preadipocytes. J. Biol. Chem. 2006, 281, 9507–9516. [Google Scholar] [CrossRef]
- Wang, H.; Tian, Y.; Wang, J.R.; Phillips, K.L.E.; Binch, A.L.A.; Dunn, S.; Cross, A.; Chiverton, N.; Zheng, Z.M.; Shapiro, I.M.; et al. Inflammatory Cytokines Induce NOTCH Signaling in Nucleus Pulposus Cells IMPLICATIONS IN INTERVERTEBRAL DISC DEGENERATION. J. Biol. Chem. 2013, 288, 16761–16774. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nistico, P.; Signore, A. Relevance of immune cell and tumor microenvironment imaging in the new era of immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 89. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.K.; Wu, L.; Yan, G.F.; Chen, Y.; Zhou, M.Y.; Wu, Y.Z.; Li, Y.S. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Mukherjee, A.; Bilecz, A.J.; Lengyel, E. The adipocyte microenvironment and cancer. Cancer Metastasis Rev. 2022, 41, 575–587. [Google Scholar] [CrossRef]
- Guerrero, J.; Tobar, N.; Caceres, M.; Espinoza, L.; Escobar, P.; Dotor, J.; Smith, P.C.; Martinez, J. Soluble factors derived from tumor mammary cell lines induce a stromal mammary adipose reversion in human and mice adipose cells. Possible role of TGF-beta 1 and TNF-alpha. Breast Cancer Res. Treat. 2010, 119, 497–508. [Google Scholar] [CrossRef]
- Meng, L.; Zhou, J.F.; Sasano, H.; Suzuki, T.; Zeitoun, K.M.; Bulun, S.E. Tumor necrosis factor alpha and interleukin 11 secreted by malignant breast epithelial cells inhibit adipocyte differentiation by selectively down-regulating CCAAT/enhancer binding protein alpha and peroxisome proliferator-activated receptor gamma: Mechanism of desmoplastic reaction. Cancer Res. 2001, 61, 2250–2255. [Google Scholar] [PubMed]
- Wang, C.Q.; Zhang, X.; Luo, L.P.; Luo, Y.; Wu, D.D.; Spilca, D.; Le, Q.; Yang, X.; Alvarez, K.; Hines, W.C.; et al. COX-2 Deficiency Promotes White Adipogenesis via PGE2-Mediated Paracrine Mechanism and Exacerbates Diet-Induced Obesity. Cells 2022, 11, 1819. [Google Scholar] [CrossRef] [PubMed]
- Daas, S.I.; Rizeq, B.R.; Nasrallah, G.K. Adipose tissue dysfunction in cancer cachexia. J. Cell Physiol. 2019, 234, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Rupert, J.E.; Narasimhan, A.; Jengelley, D.H.A.; Jiang, Y.L.; Liu, J.G.; Au, E.N.; Silverman, L.M.; Sandusky, G.; Bonetto, A.; Cao, S.; et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J. Exp. Med. 2021, 218, e20190450. [Google Scholar] [CrossRef]
- Han, J.; Meng, Q.Y.; Shen, L.; Wu, G.H. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 2018, 17, 14. [Google Scholar] [CrossRef]
- Cheung, W.W.; Zheng, R.H.; Hao, S.; Wang, Z.; Gonzalez, A.; Zhou, P.; Hoffman, H.M.; Mak, R.H. The role of IL-1 in adipose browning and muscle wasting in CKD-associated cachexia. Sci. Rep. 2021, 11, 15141. [Google Scholar] [CrossRef]
- Cancel, M.; Pouillot, W.; Maheo, K.; Fontaine, A.; Crottes, D.; Fromont, G. Interplay between Prostate Cancer and Adipose Microenvironment: A Complex and Flexible Scenario. Int. J. Mol. Sci. 2022, 23, 10762. [Google Scholar] [CrossRef]
- Morris, E.V.; Edwards, C.M. The role of bone marrow adipocytes in bone metastasis. J. Bone Oncol. 2016, 5, 121–123. [Google Scholar] [CrossRef]
- Herroon, M.K.; Diedrich, J.D.; Rajagurubandara, E.; Martin, C.; Maddipati, K.R.; Kim, S.; Heath, E.I.; Granneman, J.; Podgorski, I. Prostate Tumor Cell-Derived IL1 beta Induces an Inflammatory Phenotype in Bone Marrow Adipocytes and Reduces Sensitivity to Docetaxel via Lipolysis-Dependent Mechanisms. Mol. Cancer Res. 2019, 17, 2508–2521. [Google Scholar] [CrossRef]
- Vyas, D.; Laput, G.; Vyas, A.K. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Oncotargets Ther. 2014, 7, 1015–1023. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.Y.; Chen, L.P.; Chen, Z.S.; Fu, L.W.; Huang, R.P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2016, 1865, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Calvo, R.; Serrano, L.; Coll, T.; Moullan, N.; Sanchez, R.M.; Merlos, M.; Palomer, X.; Laguna, J.C.; Michalik, L.; Wahli, W.; et al. Activation of peroxisome proliferator-activated receptor beta/delta inhibits lipopolysaccharide-induced cytokine production in adipocytes by lowering nuclear factor-kappaB activity via extracellular signal-related kinase 1/2. Diabetes 2008, 57, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Chen, A.; Lu, W.J.; Fan, W.; Li, P.P.; Oh, D.Y.; Patsouris, D. Regulation of chemokine and chemokine receptor expression by PPARgamma in adipocytes and macrophages. PLoS ONE 2012, 7, e34976. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Suzawa, M.; Kato, S. Nuclear receptors as targets for drug development: Crosstalk between peroxisome proliferator-activated receptor gamma and cytokines in bone marrow-derived mesenchymal stem cells. J. Pharmacol. Sci. 2005, 97, 184–189. [Google Scholar] [CrossRef]
- Chae, G.N.; Kwak, S.J. NF-kappaB is involved in the TNF-alpha induced inhibition of the differentiation of 3T3-L1 cells by reducing PPARgamma expression. Exp. Mol. Med. 2003, 35, 431–437. [Google Scholar] [CrossRef]
- Wang, D.; Paz-Priel, I.; Friedman, A.D. NF-kappa B p50 regulates C/EBP alpha expression and inflammatory cytokine-induced neutrophil production. J. Immunol. 2009, 182, 5757–5762. [Google Scholar] [CrossRef]
- Guilherme, A.; Tesz, G.J.; Guntur, K.V.P.; Czech, M.P. Tumor necrosis factor-alpha induces caspase-mediated cleavage of peroxisome proliferator-activated receptor gamma in adipocytes. J. Biol. Chem. 2009, 284, 17082–17091. [Google Scholar] [CrossRef]
- Ye, J. Regulation of PPARgamma function by TNF-alpha. Biochem. Biophys. Res. Commun. 2008, 374, 405–408. [Google Scholar] [CrossRef]
- Hu, W.; Ru, Z.; Zhou, Y.; Xiao, W.; Sun, R.; Zhang, S.; Gao, Y.; Li, X.; Zhang, X.; Yang, H. Lung cancer-derived extracellular vesicles induced myotube atrophy and adipocyte lipolysis via the extracellular IL-6-mediated STAT3 pathway. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Blunder, S.; Krimbacher, T.; Moosbrugger-Martinz, V.; Gruber, R.; Schmuth, M.; Dubrac, S. Keratinocyte-derived IL-1beta induces PPARG downregulation and PPARD upregulation in human reconstructed epidermis following barrier impairment. Exp. Dermatol. 2021, 30, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Banhos Danneskiold-Samsoe, N.; Sonne, S.B.; Larsen, J.M.; Hansen, A.N.; Fjaere, E.; Isidor, M.S.; Petersen, S.; Henningsen, J.; Severi, I.; Sartini, L.; et al. Overexpression of cyclooxygenase-2 in adipocytes reduces fat accumulation in inguinal white adipose tissue and hepatic steatosis in high-fat fed mice. Sci. Rep. 2019, 9, 8979. [Google Scholar] [CrossRef] [PubMed]
- Yogarajah, T.; Bee, Y.T.; Noordin, R.; Yin, K.B. Increased peroxisome proliferator-activated receptor gamma expression levels in visceral adipose tissue, and serum CCL2 and interleukin-6 levels during visceral adipose tissue accumulation. Mol. Med. Rep. 2015, 11, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Chung, Y.; Lee, J.H.; Chun, J.M.; Park, J.H. The Intricate Role of p53 in Adipocyte Differentiation and Function. Cells 2020, 9, 2621. [Google Scholar] [CrossRef] [PubMed]
- Behera, A.K.; Bhattacharya, A.; Vasudevan, M.; Kundu, T.K. p53 mediated regulation of coactivator associated arginine methyltransferase 1 (CARM1) expression is critical for suppression of adipogenesis. FEBS J. 2018, 285, 1730–1744. [Google Scholar] [CrossRef]
- He, F.; Doucet, J.A.; Stephens, J.M. Caspase-mediated degradation of PPARgamma proteins in adipocytes. Obesity 2008, 16, 1735–1741. [Google Scholar] [CrossRef]
- Wang, X.; Wu, H.; Miller, A.H. Interleukin 1alpha (IL-1alpha) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol. Psychiatry 2004, 9, 65–75. [Google Scholar] [CrossRef]
- Ko, J.; Yun, C.Y.; Lee, J.S.; Kim, J.H.; Kim, I.S. p38 MAPK and ERK activation by 9-cis-retinoic acid induces chemokine receptors CCR1 and CCR2 expression in human monocytic THP-1 cells. Exp. Mol. Med. 2007, 39, 129–138. [Google Scholar] [CrossRef]
- Zhou, F.H.; Foster, B.K.; Zhou, X.F.; Cowin, A.J.; Xian, C.J. TNF-alpha mediates p38 MAP kinase activation and negatively regulates bone formation at the injured growth plate in rats. J. Bone Miner Res. 2006, 21, 1075–1088. [Google Scholar] [CrossRef]
- Aouadi, M.; Laurent, K.; Prot, M.; Le Marchand-Brustel, Y.; Binetruy, B.; Bost, F. Inhibition of p38MAPK increases adipogenesis from embryonic to adult stages. Diabetes 2006, 55, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Fuster, G.; Ametller, E.; Costelli, P.; Pilla, F.; Busquets, S.; Figueras, M.; Argiles, J.M.; Lopez-Soriano, F.J. Interleukin-15 increases calcineurin expression in 3T3-L1 cells: Possible involvement on in vivo adipocyte differentiation. Int. J. Mol. Med. 2009, 24, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Fu, J.L.; Miao, Y.F.; Wang, C.J.; Han, Q.F.; Li, S.; Huang, S.Z.; Du, S.N.; Qiu, Y.X.; Yang, J.C.; et al. Prostaglandin E2 receptor EP3 regulates both adipogenesis and lipolysis in mouse white adipose tissue. J. Mol. Cell Biol. 2016, 8, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Inazumi, T.; Yamada, K.; Shirata, N.; Sato, H.; Taketomi, Y.; Morita, K.; Hohjoh, H.; Tsuchiya, S.; Oniki, K.; Watanabe, T.; et al. Prostaglandin E2-EP4 Axis Promotes Lipolysis and Fibrosis in Adipose Tissue Leading to Ectopic Fat Deposition and Insulin Resistance. Cell Rep. 2020, 33, 108265. [Google Scholar] [CrossRef]
- Kimmel, A.R.; Sztalryd, C. The Perilipins: Major Cytosolic Lipid Droplet-Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu. Rev. Nutr. 2016, 36, 471–509. [Google Scholar] [CrossRef]
- Ji, S.; Sun, J.; Bian, C.; Huang, X.; Ji, H. PKA/ATGL signaling pathway is involved in ER stress-mediated lipolysis in adipocytes of grass carp (Ctenopharyngodon idella). Fish Physiol. Biochem. 2022, 48, 683–691. [Google Scholar] [CrossRef]
- Zhang, H.H.; Halbleib, M.; Ahmad, F.; Manganiello, V.C.; Greenberg, A.S. Tumor necrosis factor-alpha stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes 2002, 51, 2929–2935. [Google Scholar] [CrossRef]
- Liu, C.; Ke, P.; Zhang, J.; Zhang, X.; Chen, X. Protein Kinase Inhibitor Peptide as a Tool to Specifically Inhibit Protein Kinase A. Front. Physiol. 2020, 11, 574030. [Google Scholar] [CrossRef]
- Wongrakpanich, S.; Wongrakpanich, A.; Melhado, K.; Rangaswami, J. A Comprehensive Review of Non-Steroidal Anti-Inflammatory Drug Use in The Elderly. Aging Dis. 2018, 9, 143–150. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.Y.; Yan, S.G. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef]
- Zhang, Y.; Tighe, S.; Zhu, Y.T. COX-2 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Goradel, N.H.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.R.; Smartt, H.J.M.; Greenhough, A.; Moore, A.E.; Williams, A.C.; Paraskeva, C. Colon tumour cells increase PGE(2) by regulating COX-2 and 15-PGDH to promote survival during the microenvironmental stress of glucose deprivation. Carcinogenesis 2011, 32, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.Q.; Yang, Y.X.; Che, Q.; Jiang, F.Z.; Wang, H.H.; Chen, Z.; Zhu, M.J.; Tong, H.; Zhang, H.L.; Yan, X.F.; et al. Prostaglandin E2 (PGE2) promotes proliferation and invasion by enhancing SUMO-1 activity via EP4 receptor in endometrial cancer. Tumor Biol. 2016, 37, 12203–12211. [Google Scholar] [CrossRef]
- Xia, Y.F.; Shen, S.; Verma, I.M. NF-kappa B, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Rebe, C.; Ghiringhelli, F. STAT3, a Master Regulator of Anti-Tumor Immune Response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef]
- Song, Y.C.; Lee, S.E.; Jin, Y.; Park, H.W.; Chun, K.H.; Lee, H.W. Classifying the Linkage between Adipose Tissue Inflammation and Tumor Growth through Cancer-Associated Adipocytes. Mol. Cells 2020, 43, 763–773. [Google Scholar] [CrossRef]
- Baron, J.A. Epidemiology of non-steroidal anti-inflammatory drugs and cancer. Prog. Exp. Tumor Res. 2003, 37, 1–24. [Google Scholar]
- Zhao, X.P.; Xu, Z.; Li, H.S. NSAIDs Use and Reduced Metastasis in Cancer Patients: Results from a meta-analysis. Sci. Rep. 2017, 7, 1875. [Google Scholar] [CrossRef]
- Harris, R.E.; Chlebowski, R.T.; Jackson, R.D.; Frid, D.J.; Ascenseo, J.L.; Anderson, G.; Loar, A.; Rodabough, R.J.; White, E.; McTiernan, A. Breast cancer and nonsteroidal anti-inflammatory drugs: Prospective results from the women’s health initiative. Cancer Res. 2003, 63, 6096–6101. [Google Scholar] [PubMed]
- Calaluce, R.; Earnest, D.L.; Heddens, D.; Einspahr, J.G.; Roe, D.; Bogert, C.L.; Marshall, J.R.; Alberts, D.S. Effects of piroxicam on prostaglandin E-2 levels in rectal mucosa of adenomatous polyp patients: A randomized phase IIb trial. Cancer Epidemiol. Biomark. Prev. 2000, 9, 1287–1292. [Google Scholar]
- Cruz-Correa, M.; Hylind, L.M.; Romans, K.E.; Booker, S.V.; Giardiello, F.M. Long-term treatment with sulindac in familial adenomatous polyposis: A prospective cohort study. Gastroenterology 2002, 122, 641–645. [Google Scholar] [CrossRef]
- Gill, S.; Sinicrope, F.A. Colorectal cancer prevention: Is an ounce of prevention worth a pound of cure? Semin. Oncol. 2005, 32, 24–34. [Google Scholar] [CrossRef]
- Alshafie, G.A.; Abou-Issa, H.M.; Seibert, K.; Harris, R.E. Chemotherapeutic evaluation of Celecoxib, a cyclooxygeuase-2 inhibitor, in a rat mammary tumor model. Oncol. Rep. 2000, 7, 1377–1381. [Google Scholar]
- Yao, M.; Zhou, W.; Sangha, S.; Albert, A.; Chang, A.J.; Liu, T.C.; Wolfe, M.M. Effects of nonselective cyclooxygenase inhibition with low-dose ibuprofen on tumor growth, angiogenesis, metastasis, and survival in a mouse model of colorectal cancer. Clin. Cancer Res. 2005, 11, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Kim, D.H.; Jang, J.Y.; Kang, Y.J.; Yoon, J.H.; Moon, J.O.; Chung, H.Y.; Kim, G.Y.; Choi, Y.H.; Copple, B.L.; et al. Aspirin enhances doxorubicin-induced apoptosis and reduces tumor growth in human hepatocellular carcinoma cells in vitro and in vivo. Int. J. Oncol. 2012, 40, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Friis, S.; Riis, A.H.; Erichsen, R.; Baron, J.A.; Sorensen, H.T. Low-Dose Aspirin or Nonsteroidal Anti-inflammatory Drug Use and Colorectal Cancer Risk A Population-Based, Case-Control Study. Ann. Intern. Med. 2015, 163, 347–355. [Google Scholar] [CrossRef]
- Kim, S.; Shore, D.L.; Wilson, L.E.; Sanniez, E.I.; Kim, J.H.; Taylor, J.A.; Sandler, D.P. Lifetime use of nonsteroidal anti-inflammatory drugs and breast cancer risk: Results from a prospective study of women with a sister with breast cancer. BMC Cancer 2015, 15, 960. [Google Scholar] [CrossRef]
- Brasky, T.M.; Bonner, M.R.; Moysich, K.B.; Ambrosone, C.B.; Nie, J.; Tao, M.H.; Edge, S.B.; Kallakury, B.V.S.; Marian, C.; Goerlitz, D.S.; et al. Non-steroidal anti-inflammatory drugs (NSAIDs) and breast cancer risk: Differences by molecular subtype. Cancer Cause Control 2011, 22, 965–975. [Google Scholar] [CrossRef]
- Dierssen-Sotos, T.; Gomez-Acebo, I.; de Pedro, M.; Perez-Gomez, B.; Servitja, S.; Moreno, V.; Amiano, P.; Fernandez-Villa, T.; Barricarte, A.; Tardon, A.; et al. Use of non-steroidal anti-inflammatory drugs and risk of breast cancer: The Spanish Multi-Case-control (MCC) study. BMC Cancer 2016, 16, 660. [Google Scholar] [CrossRef] [PubMed]
- Doat, S.; Cenee, S.; Tretarre, B.; Rebillard, X.; Lamy, P.J.; Bringer, J.P.; Iborra, F.; Murez, T.; Sanchez, M.; Menegaux, F. Nonsteroidal anti-inflammatory drugs (NSAIDs) and prostate cancer risk: Results from the EPICAP study. Cancer Med. 2017, 6, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Trabert, B.; Ness, R.B.; Lo-Ciganic, W.H.; Murphy, M.A.; Goode, E.L.; Poole, E.M.; Brinton, L.A.; Webb, P.M.; Nagle, C.M.; Jordan, S.J.; et al. Aspirin, Nonaspirin Nonsteroidal Anti-inflammatory Drug, and Acetaminophen Use and Risk of Invasive Epithelial Ovarian Cancer: A Pooled Analysis in the Ovarian Cancer Association Consortium. JNCI J. Natl. Cancer Inst. 2014, 106, djt431. [Google Scholar] [CrossRef] [PubMed]
- Prizment, A.E.; Folsom, A.R.; Anderson, K.E. Nonsteroidal Anti-Inflammatory Drugs and Risk for Ovarian and Endometrial Cancers in the Iowa Women’s Health Study. Cancer Epidemiol. Biomark. Prev. 2010, 19, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Je, Y.J.; Cho, E. Analgesic use and the risk of kidney cancer: A meta-analysis of epidemiologic studies. Int. J. Cancer 2014, 134, 384–396. [Google Scholar] [CrossRef]
- Harirforoosh, S.; Asghar, W.; Jamali, F. Adverse Effects of Nonsteroidal Antiinflammatory Drugs: An Update of Gastrointestinal, Cardiovascular and Renal Complications. J. Pharm. Pharm. Sci. 2013, 16, 821–847. [Google Scholar] [CrossRef]
- Cui, Y.; Deming-Halverson, S.L.; Shrubsole, M.J.; Beeghly-Fadiel, A.; Cai, H.; Fair, A.M.; Shu, X.O.; Zheng, W. Use of nonsteroidal anti-inflammatory drugs and reduced breast cancer risk among overweight women. Breast Cancer Res. Treat. 2014, 146, 439–446. [Google Scholar] [CrossRef]
- Bowers, L.W.; Maximo, I.X.F.; Brenner, A.J.; Beeram, M.; Hursting, S.D.; Price, R.S.; Tekmal, R.R.; Jolly, C.A.; deGraffenried, L.A. NSAID Use Reduces Breast Cancer Recurrence in Overweight and Obese Women: Role of Prostaglandin-Aromatase Interactions. Cancer Res. 2014, 74, 4446–4457. [Google Scholar] [CrossRef]
- Kim, S.; Baron, J.A.; Mott, L.A.; Burke, C.A.; Church, T.R.; McKeown-Eyssen, G.E.; Cole, B.F.; Haile, R.W.; Sandler, R.S. Aspirin may be more effective in preventing colorectal adenomas in patients with higher BMI (United States). Cancer Cause Control 2006, 17, 1299–1304. [Google Scholar] [CrossRef]
- Movahedi, M.; Bishop, D.T.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Obesity, Aspirin, and Risk of Colorectal Cancer in Carriers of Hereditary Colorectal Cancer: A Prospective Investigation in the CAPP2 Study. J. Clin. Oncol. 2015, 33, 3591–3597. [Google Scholar] [CrossRef]
- Webb, P.M.; Na, R.; Weiderpass, E.; Adami, H.O.; Anderson, K.E.; Bertrand, K.A.; Botteri, E.; Brasky, T.M.; Brinton, L.A.; Chen, C.; et al. Use of aspirin, other nonsteroidal anti-inflammatory drugs and acetaminophen and risk of endometrial cancer: The Epidemiology of Endometrial Cancer Consortium. Ann. Oncol. 2019, 30, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Verdoodt, F.; Friis, S.; Dehlendorff, C.; Albieri, V.; Kjaer, S.K. Non-steroidal anti-inflammatory drug use and risk of endometrial cancer: A systematic review and meta-analysis of observational studies. Gynecol. Oncol. 2016, 140, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Gender | NSAID | Frequency of Usage | BMI (kg/m2) | Number of Patients | Effect Compared to Normal | References |
|---|---|---|---|---|---|---|---|
| Breast | Female | Aspirin, ibuprofen, naproxen, indomethacin | Regular use | >25 | 5,078 | Lower OR (22%) | [127] |
| Breast | Female | Aspirin, ibuprofen, celecoxib, naproxen, meloxicam | Daily | >30 | 440 | Lower OR (52%) | [128] |
| Colorectal | Female, Male | Aspirin | Daily (325 mg) | >30 | 1,084 | Lower RR (56%) | [129] |
| Colorectal | Female, Male | Aspirin | Daily (600 mg) | >30 | 54 | Lower HR (10%) | [130] |
| Endometrial | Female | Aspirin, NA-NSAIDs | Over weekly use | >25, 30 | 87,189 | Lower OR (15%) | [131] |
| Endometrial | Female | Aspirin | Regular use | >30 | 410 | Lower OR (44%) | [132] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, H.; Song, Y.; Lee, H.-W. Emphasis on Adipocyte Transformation: Anti-Inflammatory Agents to Prevent the Development of Cancer-Associated Adipocytes. Cancers 2023, 15, 502. https://doi.org/10.3390/cancers15020502
Na H, Song Y, Lee H-W. Emphasis on Adipocyte Transformation: Anti-Inflammatory Agents to Prevent the Development of Cancer-Associated Adipocytes. Cancers. 2023; 15(2):502. https://doi.org/10.3390/cancers15020502
Chicago/Turabian StyleNa, Heeju, Yaechan Song, and Han-Woong Lee. 2023. "Emphasis on Adipocyte Transformation: Anti-Inflammatory Agents to Prevent the Development of Cancer-Associated Adipocytes" Cancers 15, no. 2: 502. https://doi.org/10.3390/cancers15020502
APA StyleNa, H., Song, Y., & Lee, H.-W. (2023). Emphasis on Adipocyte Transformation: Anti-Inflammatory Agents to Prevent the Development of Cancer-Associated Adipocytes. Cancers, 15(2), 502. https://doi.org/10.3390/cancers15020502