
Biological and Clinical Impacts of Glucose Metabolism in Pancreatic Ductal Adenocarcinoma

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
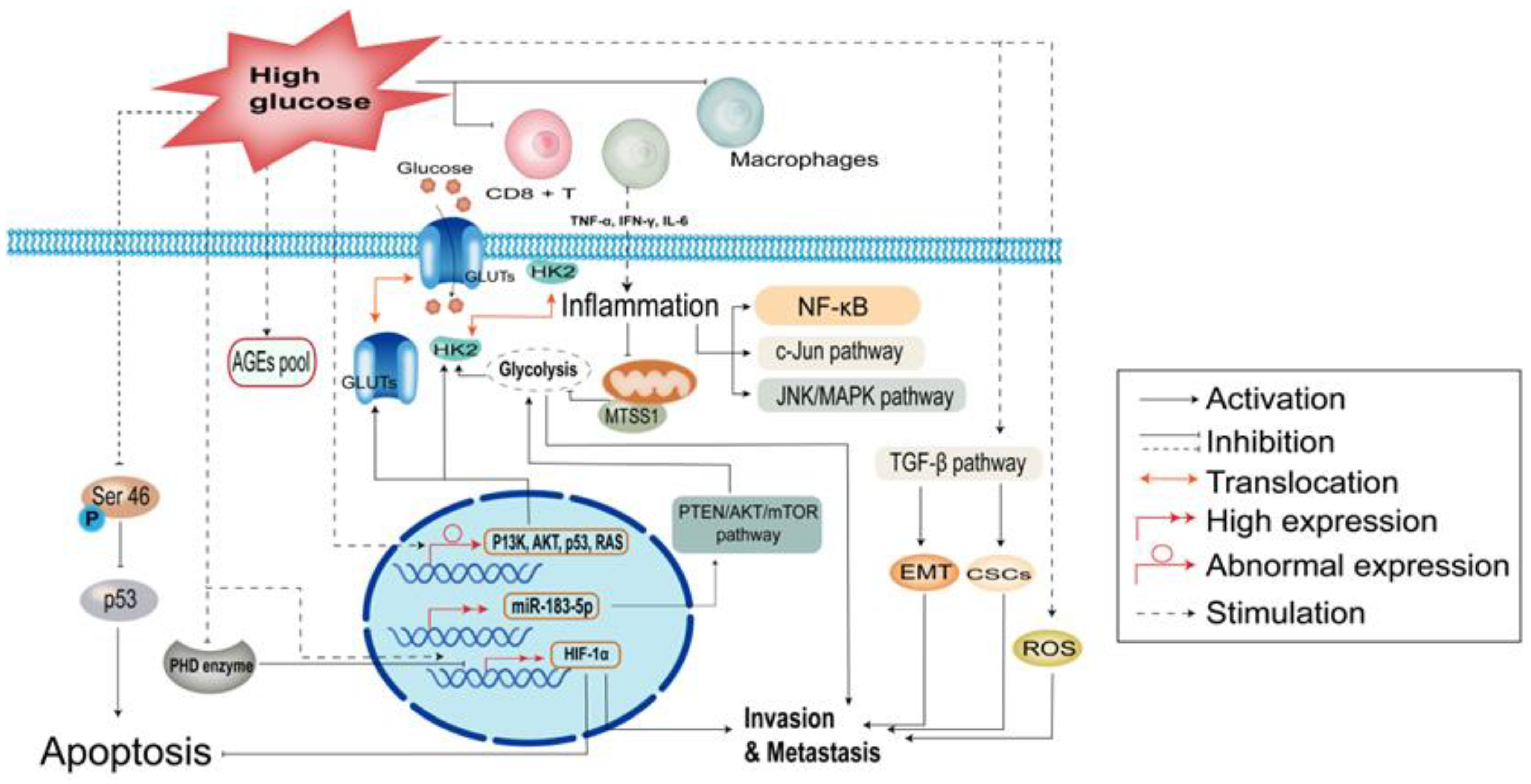
2. Biological Role of Hyperglycemia in Cancer Cells
2.1. Hyperglycemia Induces Metabolic Abnormalities
2.2. Hyperglycemia Enhances Inflammation and Immune Dysregulation
2.3. Hyperglycemia Protects Tumors against Apoptosis
2.4. Hyperglycemia Accelerates the Malignant Behavior of Cancer Cells
3. Clinical Association between PDAC and Diabetes
4. Promotion of PDAC through a High Glycolytic Phenotype
5. The TME in Pancreatic Cancer
6. Therapeutic Strategies Targeting Glycolysis
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Firpo, M.A.; Adler, D.G.; Mulvihill, S.J. Screening for pancreatic cancer: Why, how, and who? Ann. Surg. 2013, 257, 17–26. [Google Scholar] [CrossRef]
- Chari, S.T.; Leibson, C.L.; Rabe, K.G.; Timmons, L.J.; Ransom, J.; de Andrade, M.; Petersen, G.M. Pancreatic cancer-associated diabetes mellitus: Prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008, 134, 95–101. [Google Scholar] [CrossRef]
- Andersen, D.K.; Korc, M.; Petersen, G.M.; Eibl, G.; Li, D.; Rickels, M.R.; Chari, S.T.; Abbruzzese, J.L. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes 2017, 66, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, T.; Kikuta, K.; Kume, K.; Hamada, S.; Miura, S.; Yoshida, N.; Hongo, S.; Tanaka, Y.; Matsumoto, R.; Sano, T.; et al. New-Onset or Exacerbation of Diabetes Mellitus Is a Clue to the Early Diagnosis of Pancreatic Cancer. Tohoku J. Exp. Med. 2020, 252, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Duvillie, B.; Kourdoughli, R.; Druillennec, S.; Eychene, A.; Pouponnot, C. Interplay Between Diabetes and Pancreatic Ductal Adenocarcinoma and Insulinoma: The Role of Aging, Genetic Factors, and Obesity. Front. Endocrinol. 2020, 11, 563267. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hayashi, H.; Matsumura, K.; Ogata, Y.; Sato, H.; Shiraishi, Y.; Uemura, N.; Miyata, T.; Higashi, T.; Nakagawa, S.; et al. Hyperglycaemia induces metabolic reprogramming into a glycolytic phenotype and promotes epithelial-mesenchymal transitions via YAP/TAZ-Hedgehog signalling axis in pancreatic cancer. Br. J. Cancer 2022. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, J.; Jiang, N.; Wang, Z.; Liu, C. Hyperglycemia Promotes Pancreatic Cancer Initiation and Progression by Activating the Wnt/beta-Catenin Signaling Pathway. Anticancer Agents Med. Chem. 2021, 21, 2592–2602. [Google Scholar] [CrossRef]
- Menini, S.; Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G. Diabetes and Pancreatic Cancer-A Dangerous Liaison Relying on Carbonyl Stress. Cancers 2021, 13, 313. [Google Scholar] [CrossRef]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose Metabolism in Pancreatic Cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef]
- Tokajuk, A.; Krzyzanowska-Grycel, E.; Tokajuk, A.; Grycel, S.; Sadowska, A.; Car, H. Antidiabetic drugs and risk of cancer. Pharm. Rep. 2015, 67, 1240–1250. [Google Scholar] [CrossRef]
- Shlomai, G.; Neel, B.; LeRoith, D.; Gallagher, E.J. Type 2 Diabetes Mellitus and Cancer: The Role of Pharmacotherapy. J. Clin. Oncol. 2016, 34, 4261–4269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Nguyen, P.A.; Humayun, A.; Chien, S.C.; Yang, H.C.; Asdary, R.N.; Syed-Abdul, S.; Hsu, M.H.; Moldovan, M.; Yen, Y.; et al. Does long-term use of antidiabetic drugs changes cancer risk? Medicine 2019, 98, e17461. [Google Scholar] [CrossRef]
- George, S.; Jean-Baptiste, W.; Yusuf Ali, A.; Inyang, B.; Koshy, F.S.; George, K.; Poudel, P.; Chalasani, R.; Goonathilake, M.R.; Waqar, S.; et al. The Role of Type 2 Diabetes in Pancreatic Cancer. Cureus 2022, 14, e26288. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Kim, H.J.; Kim, T.; Hur, K.Y.; Kim, S.W.; Lee, M.K.; Min, Y.K.; Kim, K.W.; Chung, J.H.; Kim, J.H. Effectiveness of 3-day continuous glucose monitoring for improving glucose control in type 2 diabetic patients in clinical practice. Diabetes Metab. J. 2014, 38, 449–455. [Google Scholar] [CrossRef]
- He, X.D.; Wu, Q.; Liu, W.; Hong, T.; Li, J.J.; Miao, R.Y.; Zhao, H.T. Association of metabolic syndromes and risk factors with ampullary tumors development: A case-control study in China. World J. Gastroenterol. 2014, 20, 9541–9548. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Duan, Q.; Zhang, Z.; Li, H.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J. Cell Mol. Med. 2017, 21, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Murmu, N.; Mondal, T.; Saha, I.; Chatterjee, S.; Manna, R.; Haldar, S.; Dash, S.K.; Sarkar, T.R.; Giri, B. Multifaceted entrancing role of glucose and its analogue, 2-deoxy-D-glucose in cancer cell proliferation, inflammation, and virus infection. Biomed. Pharm. 2022, 156, 113801. [Google Scholar] [CrossRef]
- Robertson-Tessi, M.; Gillies, R.J.; Gatenby, R.A.; Anderson, A.R. Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer Res. 2015, 75, 1567–1579. [Google Scholar] [CrossRef]
- Luo, Z.; Tian, M.; Yang, G.; Tan, Q.; Chen, Y.; Li, G.; Zhang, Q.; Li, Y.; Wan, P.; Wu, J. Hypoxia signaling in human health and diseases: Implications and prospects for therapeutics. Signal Transduct. Target. 2022, 7, 218. [Google Scholar] [CrossRef]
- Ye, H.; Zhou, Q.; Zheng, S.; Li, G.; Lin, Q.; Wei, L.; Fu, Z.; Zhang, B.; Liu, Y.; Li, Z.; et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018, 9, 453. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Mol. Cancer 2016, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Byrne, F.L.; Martin, A.R.; Kosasih, M.; Caruana, B.T.; Farrell, R. The Role of Hyperglycemia in Endometrial Cancer Pathogenesis. Cancers 2020, 12, 1191. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2020, 8, 603837. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12. [Google Scholar] [CrossRef]
- Ramteke, P.; Deb, A.; Shepal, V.; Bhat, M.K. Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality. Cancers 2019, 11, 1402. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, F.; Xu, F.; Fang, K.; Fang, Z.; Shuai, X.; Cai, K.; Chen, J.; Hu, P.; Chen, D.; et al. A reciprocal feedback of Myc and lncRNA MTSS1-AS contributes to extracellular acidity-promoted metastasis of pancreatic cancer. Theranostics 2020, 10, 10120–10140. [Google Scholar] [CrossRef]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.F.; Ye, G.D.; Shen, D.Y.; Zhang, W.; Chen, M.L.; Chen, X.X.; Han, D.X.; Mi, Y.J.; Luo, Q.C.; Cai, W.Y.; et al. Chibby suppresses aerobic glycolysis and proliferation of nasopharyngeal carcinoma via the Wnt/beta-catenin-Lin28/let7-PDK1 cascade. J. Exp. Clin. Cancer Res. 2018, 37, 104. [Google Scholar] [CrossRef] [PubMed]
- Nagy, T.; Fisi, V.; Frank, D.; Katai, E.; Nagy, Z.; Miseta, A. Hyperglycemia-Induced Aberrant Cell Proliferation; A Metabolic Challenge Mediated by Protein O-GlcNAc Modification. Cells 2019, 8, 999. [Google Scholar] [CrossRef] [PubMed]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. 2019, 14, 50–59. [Google Scholar] [CrossRef]
- Daryabor, G.; Atashzar, M.R.; Kabelitz, D.; Meri, S.; Kalantar, K. The Effects of Type 2 Diabetes Mellitus on Organ Metabolism and the Immune System. Front. Immunol. 2020, 11, 1582. [Google Scholar] [CrossRef]
- Huang, L.Y.; Chiu, C.J.; Hsing, C.H.; Hsu, Y.H. Interferon Family Cytokines in Obesity and Insulin Sensitivity. Cells 2022, 11, 4041. [Google Scholar] [CrossRef]
- Kern, L.; Mittenbuhler, M.J.; Vesting, A.J.; Ostermann, A.L.; Wunderlich, C.M.; Wunderlich, F.T. Obesity-Induced TNFalpha and IL-6 Signaling: The Missing Link between Obesity and Inflammation-Driven Liver and Colorectal Cancers. Cancers 2018, 11, 24. [Google Scholar] [CrossRef]
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, J.W.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed]
- Mantuano, N.R.; Stanczak, M.A.; Oliveira, I.A.; Kirchhammer, N.; Filardy, A.A.; Monaco, G.; Santos, R.C.; Fonseca, A.C.; Fontes, M.; Bastos, C.S., Jr.; et al. Hyperglycemia Enhances Cancer Immune Evasion by Inducing Alternative Macrophage Polarization through Increased O-GlcNAcylation. Cancer Immunol. Res. 2020, 8, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Beatty, P.L.; McKolanis, J.; Brand, R.; Schoen, R.E.; Finn, O.J. Circulating Myeloid Derived Suppressor Cells (MDSC) That Accumulate in Premalignancy Share Phenotypic and Functional Characteristics With MDSC in Cancer. Front. Immunol. 2019, 10, 1401. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, Y.; Herrera, M.T.; Soldevila, G.; Garcia-Garcia, L.; Fabian, G.; Perez-Armendariz, E.M.; Bobadilla, K.; Guzman-Beltran, S.; Sada, E.; Torres, M. High glucose concentrations induce TNF-alpha production through the down-regulation of CD33 in primary human monocytes. BMC Immunol. 2012, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Edgar, L.; Akbar, N.; Braithwaite, A.T.; Krausgruber, T.; Gallart-Ayala, H.; Bailey, J.; Corbin, A.L.; Khoyratty, T.E.; Chai, J.T.; Alkhalil, M.; et al. Hyperglycemia Induces Trained Immunity in Macrophages and Their Precursors and Promotes Atherosclerosis. Circulation 2021, 144, 961–982. [Google Scholar] [CrossRef]
- Moganti, K.; Li, F.; Schmuttermaier, C.; Riemann, S.; Kluter, H.; Gratchev, A.; Harmsen, M.C.; Kzhyshkowska, J. Hyperglycemia induces mixed M1/M2 cytokine profile in primary human monocyte-derived macrophages. Immunobiology 2017, 222, 952–959. [Google Scholar] [CrossRef]
- Khodavirdipour, A.; Piri, M.; Jabbari, S.; Keshavarzi, S.; Safaralizadeh, R.; Alikhani, M.Y. Apoptosis Detection Methods in Diagnosis of Cancer and Their Potential Role in Treatment: Advantages and Disadvantages: A Review. J. Gastrointest. Cancer 2021, 52, 422–430. [Google Scholar] [CrossRef]
- Hernandez-Valencia, J.; Garcia-Villa, E.; Arenas-Hernandez, A.; Garcia-Mena, J.; Diaz-Chavez, J.; Gariglio, P. Induction of p53 Phosphorylation at Serine 20 by Resveratrol Is Required to Activate p53 Target Genes, Restoring Apoptosis in MCF-7 Cells Resistant to Cisplatin. Nutrients 2018, 10, 1148. [Google Scholar] [CrossRef]
- Cerychova, R.; Pavlinkova, G. HIF-1, Metabolism, and Diabetes in the Embryonic and Adult Heart. Front. Endocrinol. 2018, 9, 460. [Google Scholar] [CrossRef]
- Ramos, H.; Calheiros, J.; Almeida, J.; Barcherini, V.; Santos, S.; Carvalho, A.T.P.; Santos, M.M.M.; Saraiva, L. SLMP53-1 Inhibits Tumor Cell Growth through Regulation of Glucose Metabolism and Angiogenesis in a P53-Dependent Manner. Int. J. Mol. Sci. 2020, 21, 596. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. A review of the molecular mechanisms of hyperglycemia-induced free radical generation leading to oxidative stress. J. Cell Physiol. 2019, 234, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Rahn, S.; Zimmermann, V.; Viol, F.; Knaack, H.; Stemmer, K.; Peters, L.; Lenk, L.; Ungefroren, H.; Saur, D.; Schafer, H.; et al. Diabetes as risk factor for pancreatic cancer: Hyperglycemia promotes epithelial-mesenchymal-transition and stem cell properties in pancreatic ductal epithelial cells. Cancer Lett. 2018, 415, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, H.; Qian, W.; Cheng, L.; Yan, B.; Han, L.; Xu, Q.; Ma, Q.; Ma, J. Hyperglycemia aggravates microenvironment hypoxia and promotes the metastatic ability of pancreatic cancer. Comput. Struct. Biotechnol. J. 2018, 16, 479–487. [Google Scholar] [CrossRef]
- Fainsod-Levi, T.; Gershkovitz, M.; Vols, S.; Kumar, S.; Khawaled, S.; Sagiv, J.Y.; Sionov, R.V.; Grunewald, M.; Keshet, E.; Granot, Z. Hyperglycemia Impairs Neutrophil Mobilization Leading to Enhanced Metastatic Seeding. Cell Rep. 2017, 21, 2384–2392. [Google Scholar] [CrossRef]
- Jeong, O.; Kim, H.S. Dietary chokeberry and dried jujube fruit attenuates high-fat and high-fructose diet-induced dyslipidemia and insulin resistance via activation of the IRS-1/PI3K/Akt pathway in C57BL/6 J mice. Nutr. Metab. 2019, 16, 38. [Google Scholar] [CrossRef]
- Gartner, S.; Kruger, J.; Aghdassi, A.A.; Steveling, A.; Simon, P.; Lerch, M.M.; Mayerle, J. Nutrition in Pancreatic Cancer: A Review. Gastrointest. Tumors 2016, 2, 195–202. [Google Scholar] [CrossRef]
- Pannala, R.; Leirness, J.B.; Bamlet, W.R.; Basu, A.; Petersen, G.M.; Chari, S.T. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology 2008, 134, 981–987. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Inoue, M.; Iwasaki, M.; Otani, T.; Sasazuki, S.; Noda, M.; Tsugane, S. Diabetes mellitus and the risk of cancer: Results from a large-scale population-based cohort study in Japan. Arch. Intern. Med. 2006, 166, 1871–1877. [Google Scholar] [CrossRef]
- Saydah, S.H.; Platz, E.A.; Rifai, N.; Pollak, M.N.; Brancati, F.L.; Helzlsouer, K.J. Association of markers of insulin and glucose control with subsequent colorectal cancer risk. Cancer Epidemiol. Biomark. Prev. 2003, 12, 412–418. [Google Scholar]
- Stolzenberg-Solomon, R.Z.; Graubard, B.I.; Chari, S.; Limburg, P.; Taylor, P.R.; Virtamo, J.; Albanes, D. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA 2005, 294, 2872–2878. [Google Scholar] [CrossRef]
- Mizuno, S.; Nakai, Y.; Isayama, H.; Takahara, N.; Miyabayashi, K.; Yamamoto, K.; Kawakubo, K.; Mohri, D.; Kogure, H.; Sasaki, T.; et al. Diabetes is a useful diagnostic clue to improve the prognosis of pancreatic cancer. Pancreatology 2013, 13, 285–289. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36 (Suppl. S1), S67–S74. [Google Scholar] [CrossRef] [PubMed]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef]
- Korc, M. Pancreatic cancer-associated diabetes is an “exosomopathy”. Clin. Cancer Res. 2015, 21, 1508–1510. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef]
- Eibl, G.; Cruz-Monserrate, Z.; Korc, M.; Petrov, M.S.; Goodarzi, M.O.; Fisher, W.E.; Habtezion, A.; Lugea, A.; Pandol, S.J.; Hart, P.A.; et al. Diabetes Mellitus and Obesity as Risk Factors for Pancreatic Cancer. J. Acad. Nutr. Diet. 2018, 118, 555–567. [Google Scholar] [CrossRef]
- Hart, P.A.; Bellin, M.D.; Andersen, D.K.; Bradley, D.; Cruz-Monserrate, Z.; Forsmark, C.E.; Goodarzi, M.O.; Habtezion, A.; Korc, M.; Kudva, Y.C.; et al. Type 3c (pancreatogenic) diabetes mellitus secondary to chronic pancreatitis and pancreatic cancer. Lancet Gastroenterol. Hepatol. 2016, 1, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.P.; Nagpal, S.J.; Mukhopadhyay, D.; Chari, S.T. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Hu, C.M.; Hsu, Y.S.; Lee, W.H. Interplays of glucose metabolism and KRAS mutation in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 817. [Google Scholar] [CrossRef]
- Chikamoto, A.; Inoue, R.; Komohara, Y.; Sakamaki, K.; Hashimoto, D.; Shiraishi, S.; Takamori, H.; Yamashita, Y.I.; Yoshida, N.; Yamanaka, T.; et al. Preoperative High Maximum Standardized Uptake Value in Association with Glucose Transporter 1 Predicts Poor Prognosis in Pancreatic Cancer. Ann. Surg. Oncol. 2017, 24, 2040–2046. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Matsuda, S.; Sasaki, Y.; Masugi, Y.; Kitago, M.; Yagi, H.; Abe, Y.; Shinoda, M.; Tokino, T.; Sakamoto, M.; et al. Pathogenesis of multiple pancreatic cancers involves multicentric carcinogenesis and intrapancreatic metastasis. Cancer Sci. 2020, 111, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Bu, X.; Cai, B.; Liang, P.; Li, K.; Qu, X.; Shen, L. Biological role of metabolic reprogramming of cancer cells during epithelial-mesenchymal transition (Review). Oncol. Rep. 2019, 41, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, H.; Lee, S.; Youn, H.; Youn, B. Role of Metabolic Reprogramming in Epithelial(-)Mesenchymal Transition (EMT). Int. J. Mol. Sci. 2019, 20, 2042. [Google Scholar] [CrossRef]
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol. Cancer 2020, 19, 50. [Google Scholar] [CrossRef]
- Espiau-Romera, P.; Courtois, S.; Parejo-Alonso, B.; Sancho, P. Molecular and Metabolic Subtypes Correspondence for Pancreatic Ductal Adenocarcinoma Classification. J. Clin. Med. 2020, 9, 4128. [Google Scholar] [CrossRef]
- Hardie, R.A.; van Dam, E.; Cowley, M.; Han, T.L.; Balaban, S.; Pajic, M.; Pinese, M.; Iconomou, M.; Shearer, R.F.; McKenna, J.; et al. Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab. 2017, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Carmona, C.A.; Dalla Pozza, E.; Ambrosini, G.; Errico, A.; Dando, I. Divergent Roles of Mitochondria Dynamics in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 4128. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; He, X.; Gong, P.; Yang, Y.; Huang, S.; Zeng, Y.; Wei, L.; Zhang, J. Glycolysis-Related Gene Expression Profiling Screen for Prognostic Risk Signature of Pancreatic Ductal Adenocarcinoma. Front. Genet. 2021, 12, 639246. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q. Changes in mitochondrial function during EMT induced by TGFbeta-1 in pancreatic cancer. Oncol. Lett. 2017, 13, 1575–1580. [Google Scholar] [CrossRef]
- Guha, M.; Avadhani, N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013, 13, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef]
- Zaidieh, T.; Smith, J.R.; Ball, K.E.; An, Q. Mitochondrial DNA abnormalities provide mechanistic insight and predict reactive oxygen species-stimulating drug efficacy. BMC Cancer 2021, 21, 427. [Google Scholar] [CrossRef]
- Konate, M.M.; Antony, S.; Doroshow, J.H. Inhibiting the Activity of NADPH Oxidase in Cancer. Antioxid. Redox Signal 2020, 33, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Krapf, S.A.; Lund, J.; Lundkvist, M.; Dale, M.G.; Nyman, T.A.; Thoresen, G.H.; Kase, E.T. Pancreatic cancer cells show lower oleic acid oxidation and their conditioned medium inhibits oleic acid oxidation in human myotubes. Pancreatology 2020, 20, 676–682. [Google Scholar] [CrossRef]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., III; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Lafaro, K.J.; Melstrom, L.G. The Paradoxical Web of Pancreatic Cancer Tumor Microenvironment. Am. J. Pathol. 2019, 189, 44–57. [Google Scholar] [CrossRef]
- Veenstra, V.L.; Garcia-Garijo, A.; van Laarhoven, H.W.; Bijlsma, M.F. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers 2018, 10, 34. [Google Scholar] [CrossRef]
- Parente, P.; Parcesepe, P.; Covelli, C.; Olivieri, N.; Remo, A.; Pancione, M.; Latiano, T.P.; Graziano, P.; Maiello, E.; Giordano, G. Crosstalk between the Tumor Microenvironment and Immune System in Pancreatic Ductal Adenocarcinoma: Potential Targets for New Therapeutic Approaches. Gastroenterol. Res. Pr. 2018, 2018, 7530619. [Google Scholar] [CrossRef]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, M.; Imai, K.; Ishimoto, T.; Komohara, Y.; Yamashita, Y.I.; Nakagawa, S.; Umezaki, N.; Yamao, T.; Kitano, Y.; Miyata, T.; et al. PD-L1 expression enhancement by infiltrating macrophage-derived tumor necrosis factor-alpha leads to poor pancreatic cancer prognosis. Cancer Sci. 2019, 110, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef]
- Suri, R.; Zimmerman, J.W.; Burkhart, R.A. Modeling human pancreatic ductal adenocarcinoma for translational research: Current options, challenges, and prospective directions. Ann. Pancreat. Cancer 2020, 3, 17. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140. [Google Scholar] [CrossRef]
- Koikawa, K.; Kibe, S.; Suizu, F.; Sekino, N.; Kim, N.; Manz, T.D.; Pinch, B.J.; Akshinthala, D.; Verma, A.; Gaglia, G.; et al. Targeting Pin1 renders pancreatic cancer eradicable by synergizing with immunochemotherapy. Cell 2021, 184, 4753–4771. [Google Scholar] [CrossRef]
- Benny, S.; Mishra, R.; Manojkumar, M.K.; Aneesh, T.P. From Warburg effect to Reverse Warburg effect; the new horizons of anti-cancer therapy. Med. Hypotheses 2020, 144, 110216. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanita, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020, 10, 722. [Google Scholar] [CrossRef]
- Kang, C.; LeRoith, D.; Gallagher, E.J. Diabetes, Obesity, and Breast Cancer. Endocrinology 2018, 159, 3801–3812. [Google Scholar] [CrossRef]
- Schauer, D.P.; Feigelson, H.S.; Koebnick, C.; Caan, B.; Weinmann, S.; Leonard, A.C.; Powers, J.D.; Yenumula, P.R.; Arterburn, D.E. Bariatric Surgery and the Risk of Cancer in a Large Multisite Cohort. Ann. Surg. 2019, 269, 95–101. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pr. 2018, 143, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Qiu, S.; Wang, P.; Liang, X.; Huang, F.; Wu, H.; Zhang, B.; Zhang, W.; Tian, X.; Xu, R.; et al. Cardamonin inhibits breast cancer growth by repressing HIF-1alpha-dependent metabolic reprogramming. J. Exp. Clin. Cancer Res. 2019, 38, 377. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Tu, B.; Yao, J.; Gong, J.; Carugo, A.; Bristow, C.A.; Wang, Q.; Zhu, C.; Dai, B.; Kang, Y.; et al. Targeting Glucose Metabolism Sensitizes Pancreatic Cancer to MEK Inhibition. Cancer Res. 2021, 81, 4054–4065. [Google Scholar] [CrossRef] [PubMed]
- Klapdor, S.; Richter, E.; Klapdor, R. Vitamin D status and per-oral vitamin D supplementation in patients suffering from chronic pancreatitis and pancreatic cancer disease. Anticancer. Res. 2012, 32, 1991–1998. [Google Scholar] [PubMed]
- Ewald, N.; Bretzel, R.G. Diabetes mellitus secondary to pancreatic diseases (Type 3c)--are we neglecting an important disease? Eur. J. Intern. Med. 2013, 24, 203–206. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Therapeutic Agent | Therapeutic Strategy | Cancer type | Reference |
|---|---|---|---|
| Prevent risk factors | Weight loss and the adoption of a healthy diet, surgery | Most of malignant tumors | [109,110] |
| Metformin, sulfonylureas, thiazolidinediones (pioglitazone) | Control the blood glucose | Breast, Pancreatic, Colorectal, and Prostate cancers | [14,111] |
| Cardamonin | Inhibit HIF-1α, inhibition of the glycolytic process | Breast cancer | [112] |
| Trametinib, GDC-0623 | Inhibit the MEK pathway | PDAC | [113] |
| Vitamin D | Monitoring and supplementation of 25-hydroxyvitamin D levels is beneficial for treatment | Pancreatic cancer | [114] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Hayashi, H.; Matsumura, K.; Uemura, N.; Shiraishi, Y.; Sato, H.; Baba, H. Biological and Clinical Impacts of Glucose Metabolism in Pancreatic Ductal Adenocarcinoma. Cancers 2023, 15, 498. https://doi.org/10.3390/cancers15020498
Liu Z, Hayashi H, Matsumura K, Uemura N, Shiraishi Y, Sato H, Baba H. Biological and Clinical Impacts of Glucose Metabolism in Pancreatic Ductal Adenocarcinoma. Cancers. 2023; 15(2):498. https://doi.org/10.3390/cancers15020498
Chicago/Turabian StyleLiu, Zhao, Hiromitsu Hayashi, Kazuki Matsumura, Norio Uemura, Yuta Shiraishi, Hiroki Sato, and Hideo Baba. 2023. "Biological and Clinical Impacts of Glucose Metabolism in Pancreatic Ductal Adenocarcinoma" Cancers 15, no. 2: 498. https://doi.org/10.3390/cancers15020498
APA StyleLiu, Z., Hayashi, H., Matsumura, K., Uemura, N., Shiraishi, Y., Sato, H., & Baba, H. (2023). Biological and Clinical Impacts of Glucose Metabolism in Pancreatic Ductal Adenocarcinoma. Cancers, 15(2), 498. https://doi.org/10.3390/cancers15020498