
Perspectives of the Application of Non-Steroidal Anti-Inflammatory Drugs in Cancer Therapy: Attempts to Overcome Their Unfavorable Side Effects




Simple Summary
Abstract
1. Introduction
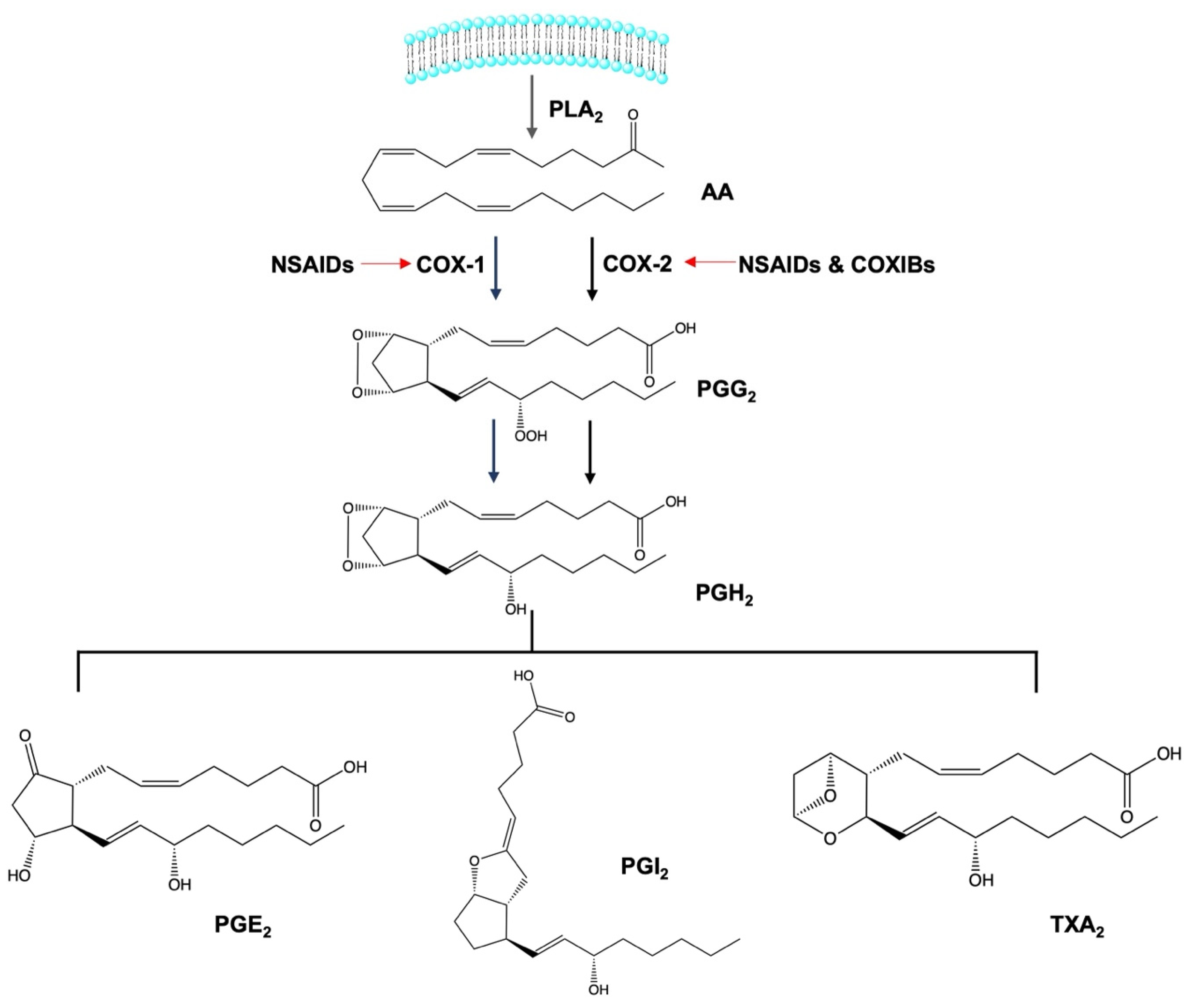
2. Anti-Inflammatory Activity of Non-Steroidal Anti-Inflammatory Drugs
3. Evidence of the Anti-Cancer Mechanism of Action of NSAIDs toward Tumoral Cell Lines
4. Evidence of the Anti-Cancer Mechanisms of NSAIDs in In Vivo Models
5. Evidence of the Anti-Cancer Mechanisms of Action of NSAIDs in Epidemiologic Studies
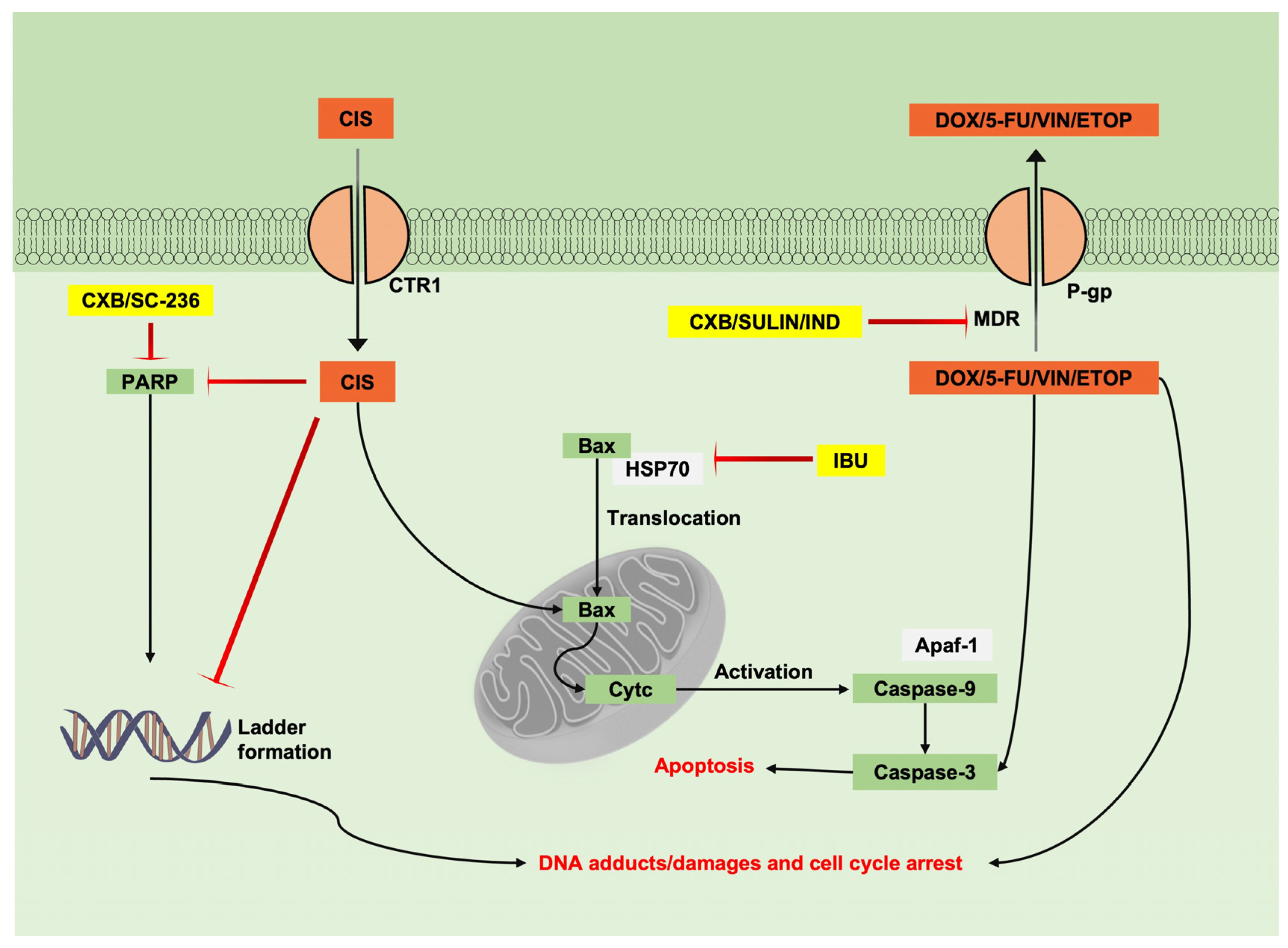
6. Combination of NSAIDs with Chemotherapeutic Drugs
6.1. Combination of NSAIDs with Chemotherapeutic Drugs Studied in Tumoural Cell Lines
6.2. Combination of NSAIDs with Chemotherapeutic Drugs in In Vivo Models
6.3. Combination of NSAIDs with Chemotherapeutic Drugs Studied in Clinical Trials
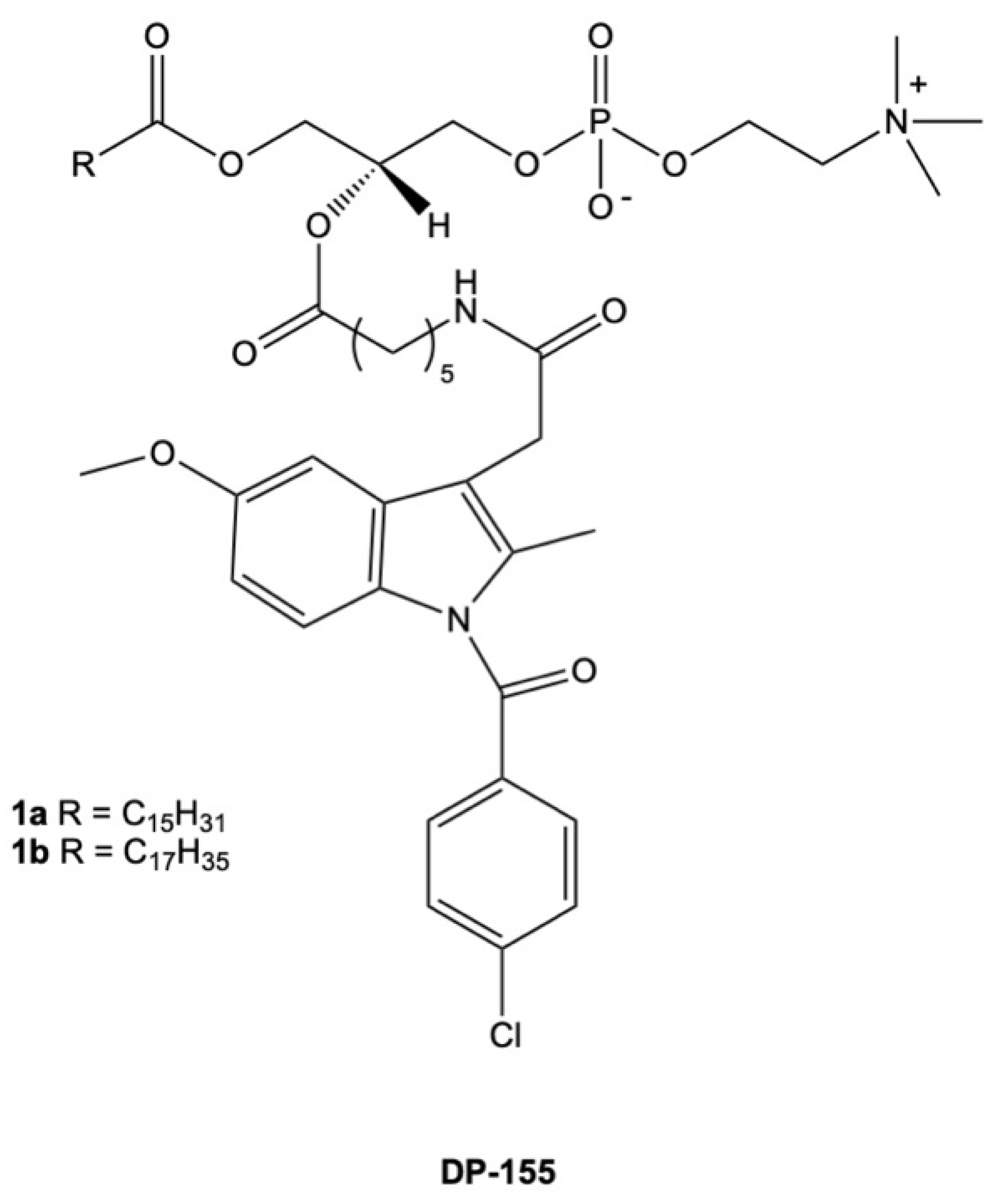
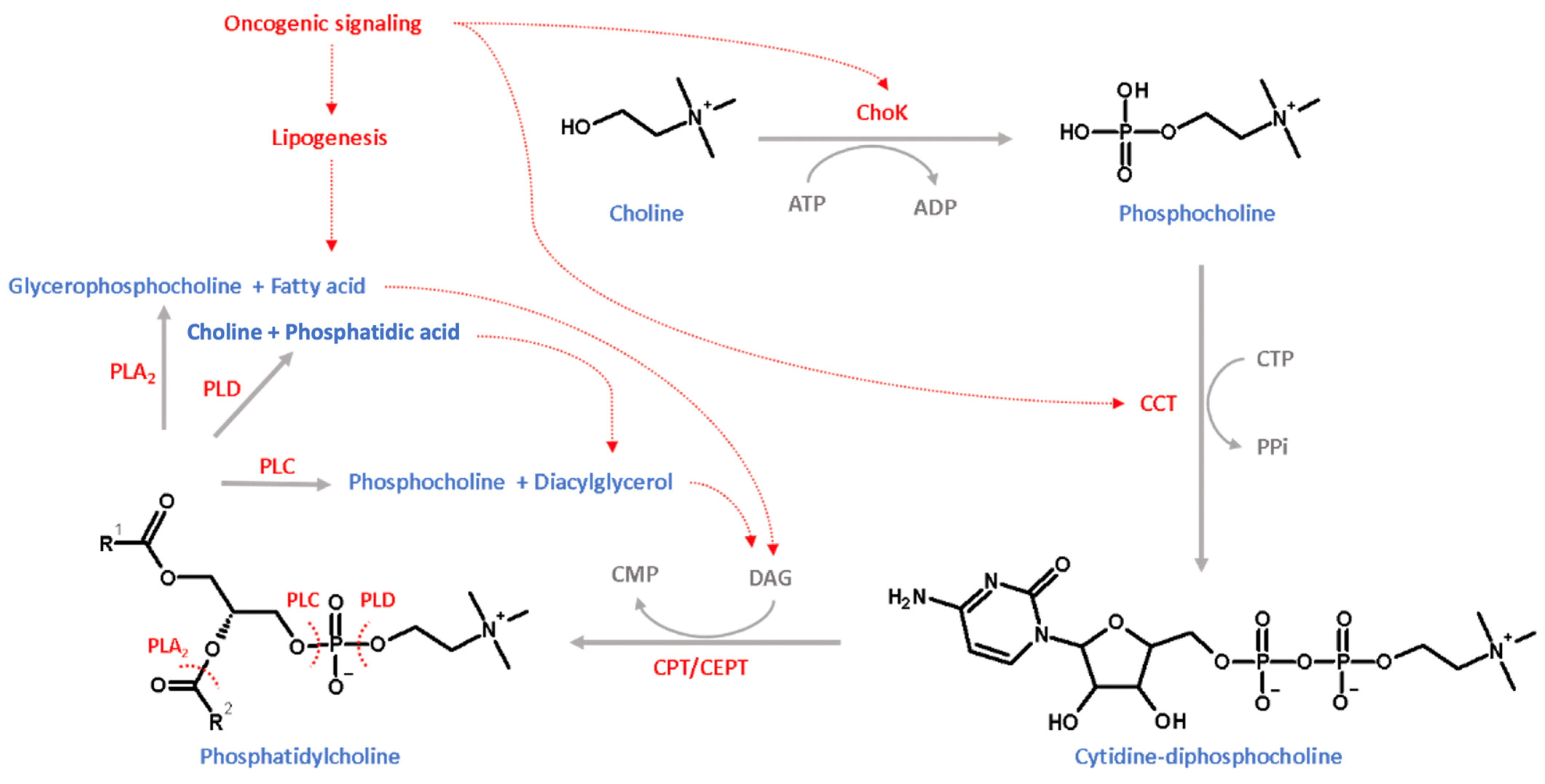
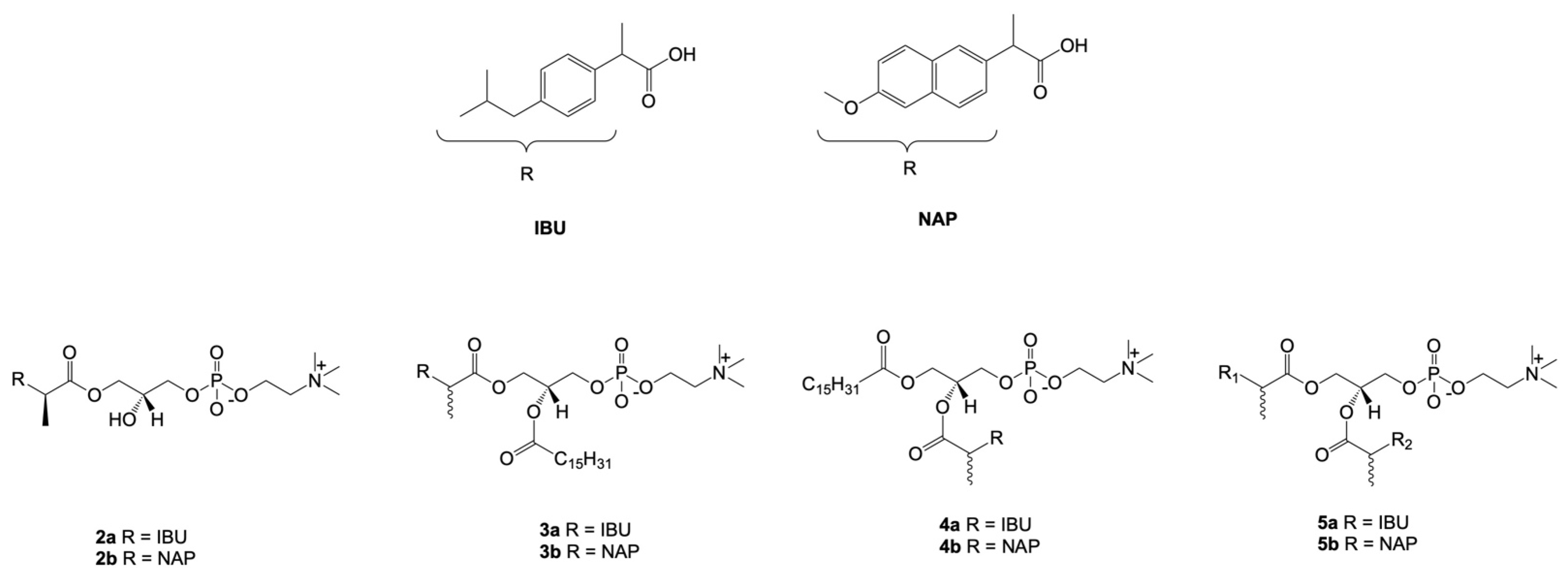
7. Combination of NSAIDs with Phosphatidylcholine
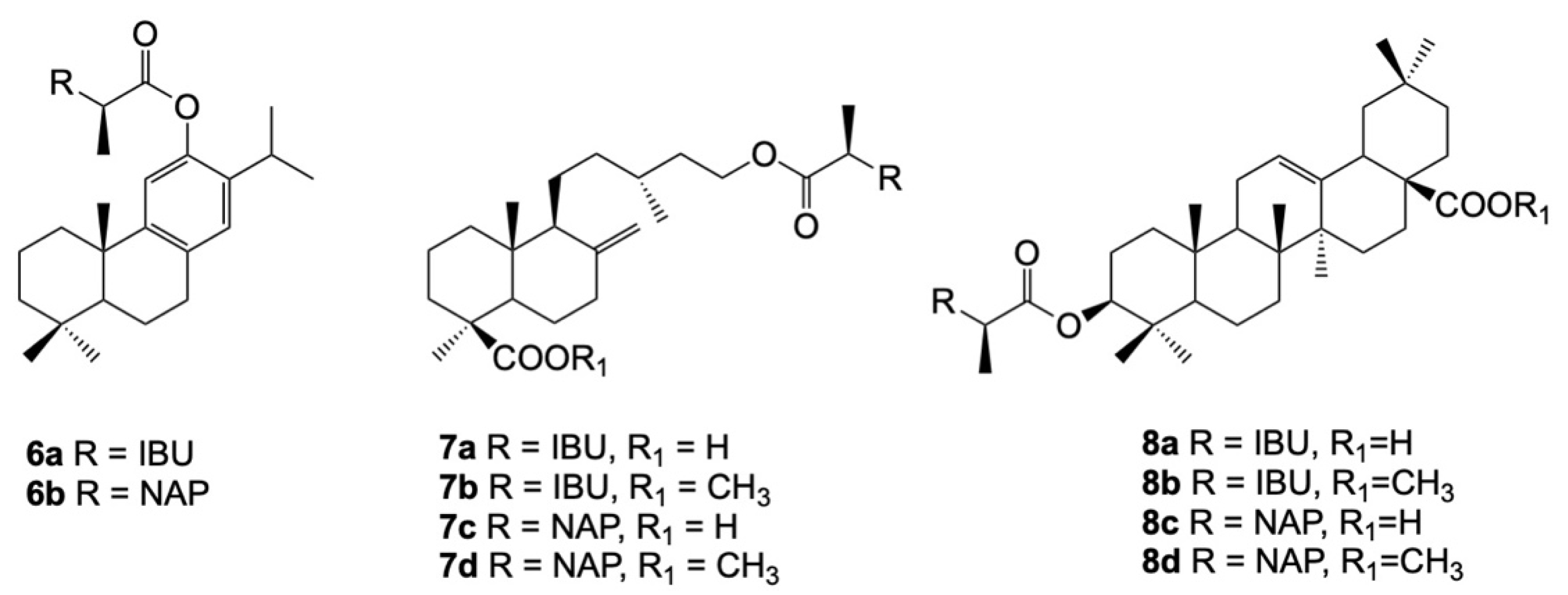
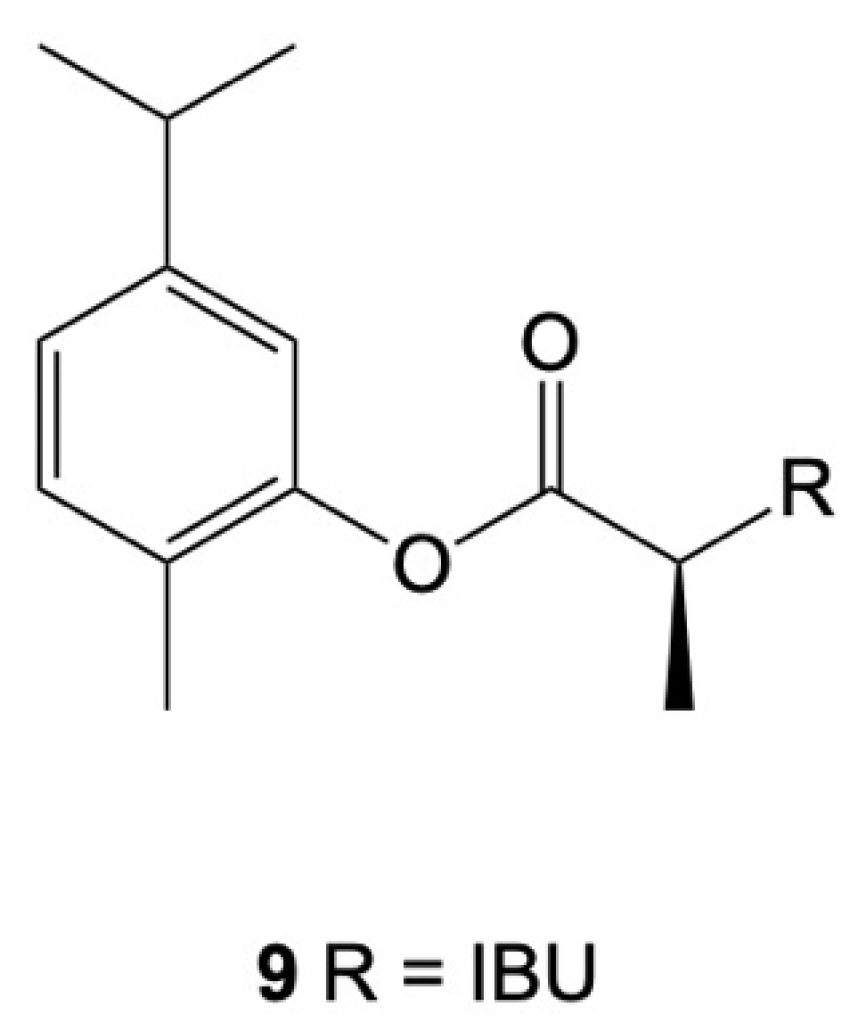
8. Combination of NSAIDs with Terpenoids
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PLA2 | Phospholipase A2 |
| Tr-NSAID | Traditional NSAIDs |
| TX | Thromboxane |
| EGFR | Epidermal growth factor receptor |
| Caspase | Cysteine-aspartic proteases- |
| DR5 | Death receptor 5 |
| CHOPS | CCAAT/enhancer binding protein homologous protein |
| TRAIL | Tumor necrosis factor related apoptosis inducing ligand |
| GLUT1 | Glucose transporter-1 |
| STAT-3 | Signal Transducer and Activator of Transcription 3 |
| ER | Endoplasmic reticulum |
| pEIF2α | Phosphorylated eukaryotic initiation factor 2 α |
| VEGFR-2 | Vascular endothelial growth factor receptor–2 |
| IL | Interleukin |
| PARP | Poly (ADP-ribose) polymerase |
| HSP70 | Heat shock protein 70 |
| Cyt-c | Cytochrome-c |
| Apaf-1 | Apoptotic protease activator 1 |
| CXB | Celecoxib |
| CIS | Cisplatin |
| IBU | Ibuprofen |
| SULIN | Sulindac |
| IND | Indomethacin |
| DOX | Doxorubicin |
| 5-FU | 5-Florouracil |
| VIN | Vincristine |
| ETOP | Etoposide |
| PLC | Phospholipase C |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI--Atheromatous affection of arteries. 1858. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Montinari, M.R.; Minelli, S.; De Caterina, R. The first 3500 years of aspirin history from its roots—A concise summary. Vascul. Pharmacol. 2019, 113, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Jane Henley, S.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Pollard, M.; Luckert, P.H.; Schmidt, M.A. The suppressive effect of piroxicam on autochthonous intestinal tumors in the rat. Cancer Lett. 1983, 21, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Desai, D.; Rivenson, A.; Simi, B.; Amin, S.; Reddy, B.S. Chemoprevention of Colon Carcinogenesis by Phenylethyl-3-methylcaffeate. Cancer Res. 1995, 55, 2310–2315. [Google Scholar]
- Craven, P.A.; Derubertis, F.R. Effects of aspirin on 1,2-dimethylhydrazine-induced colonic carcinogenesis. Carcinogenesis 1992, 13, 541–546. [Google Scholar] [CrossRef]
- Moorghen, M.; Ince, P.; Finney, K.J.; Sunter, J.P.; Appleton, D.R.; Watson, A.J. A protective effect of sulindac against chemically-induced primary colonic tumours in mice. J. Pathol. 1988, 156, 341–347. [Google Scholar] [CrossRef]
- Reddy, B.S.; Maruyama, H.; Kelloff, G. Dose-related Inhibition of Colon Carcinogenesis by Dietary Piroxicam, a Nonsteroidal Antiinflammatory Drug, during Different Stages of Rat Colon Tumor Development. Cancer Res. 1987, 47, 5401–5406. [Google Scholar]
- New Drugs at FDA: CDER’s New Molecular Entities and New Therapeutic Biological Products|FDA. Available online: https://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products (accessed on 9 September 2021).
- Human Medicines: Highlights of 2019|European Medicines Agency. Available online: https://www.ema.europa.eu/en/news/human-medicines-highlights-2019 (accessed on 9 September 2021).
- Human Medicines: Highlights of 2020|European Medicines Agency. Available online: https://www.ema.europa.eu/en/news/human-medicines-highlights-2020 (accessed on 9 September 2021).
- Warner, T.D.; Mitchell, J.A. Cyclooxygenase-3 (COX-3): Filling in the gaps toward a COX continuum? Proc. Natl. Acad. Sci. USA 2002, 99, 13371–13373. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Faki, Y.; Er, A. Different Chemical Structures and Physiological/Pathological Roles of Cyclooxygenases. Rambam Maimonides Med. J. 2021, 12, e0003. [Google Scholar] [CrossRef] [PubMed]
- Kirkby, N.S.; Sampaio, W.; Etelvino, G.; Alves, D.T.; Anders, K.L.; Temponi, R.; Shala, F.; Nair, A.S.; Ahmetaj-Shala, B.; Jiao, J.; et al. Cyclooxygenase-2 selectively controls renal blood flow through a novel PPARβ/δ-dependent vasodilator pathway. Hypertension 2018, 71, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Vara-Messler, M.; Buccellati, C.; Pustina, L.; Folco, G.; Rovati, G.E.; Hoxha, M. A potential role of PUFAs and COXIBs in cancer chemoprevention. Prostaglandins Other Lipid Mediat. 2015, 120, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Herschman, H.R. Regulation and Function of Prostaglandin Synthase 2/Cyclooxygenase II. In The Eicosanoids; Wiley: Hoboken, NJ, USA, 2004; pp. 43–52. [Google Scholar] [CrossRef]
- Mohale, D.S.; Tripathi, A.S.; Wahane, J.B.; Chandewar, A. V A Pharmacological Review on Cyclooxygenase Enzyme. Indian J. Pharm. Pharmacol. 2014, 1, 46–58. [Google Scholar]
- Smith, W.L.; Garavito, R.M.; Dewitt, D.L. Prostaglandin Endoperoxidase H Synthases (Cyclooxygenases)-1 and -2. J. Biol. Chem. 1991, 33157–33161. [Google Scholar]
- Bonnesen, K.; Schmidt, M. Recategorization of Non-Aspirin Nonsteroidal Anti-inflammatory Drugs According to Clinical Relevance: Abandoning the Traditional NSAID Terminology. Can. J. Cardiol. 2021, 37, 1705–1707. [Google Scholar] [CrossRef]
- Sun, L.; Chen, K.; Jiang, Z.; Chen, X.; Ma, J.; Ma, Q.; Duan, W. Indometacin inhibits the proliferation and activation of human pancreatic stellate cells through the downregulation of COX-2. Oncol. Rep. 2018, 39, 2243–2251. [Google Scholar] [CrossRef]
- Tai, Y.; Zhang, L.-H.; Gao, J.-H.; Zhao, C.; Tong, H.; Ye, C.; Huang, Z.-Y.; Liu, R.; Tang, C.-W. Suppressing growth and invasion of human hepatocellular carcinoma cells by celecoxib through inhibition of cyclooxygenase-2. Cancer Manag. Res. 2019, 11, 2831–2848. [Google Scholar] [CrossRef]
- Lampiasi, N.; Foderà, D.; D’Alessandro, N.; Cusimano, A.; Azzolina, A.; Tripodo, C.; Florena, A.M.; Minervini, M.I.; Notarbartolo, M.; Montalto, G.; et al. The selective cyclooxygenase-1 inhibitor SC-560 suppresses cell proliferation and induces apoptosis in human hepatocellular carcinoma cells. Int. J. Mol. Med. 2006, 17, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Hurst, E.A.; Pang, L.Y.; Argyle, D.J. The selective cyclooxygenase-2 inhibitor mavacoxib (Trocoxil) exerts anti-tumour effects in vitro independent of cyclooxygenase-2 expression levels. Vet. Comp. Oncol. 2019, 17, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Peng, J.; Liu, T.; Zhang, G. Effects of celecoxib on cell apoptosis and Fas, FasL and Bcl-2 expression in a BGC-823 human gastric cancer cell line. Exp. Ther. Med. 2017, 14, 1935–1940. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Akrami, H.; Aminzadeh, S.; Fallahi, H. Inhibitory effect of ibuprofen on tumor survival and angiogenesis in gastric cancer cell. Tumor Biol. 2015, 36, 3237–3243. [Google Scholar] [CrossRef]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, J.E.; Lim, D.Y.; Huang, Z.; Chen, H.; Langfald, A.; Lubet, R.A.; Grubbs, C.J.; Dong, Z.; Bode, A.M. Naproxen induces cell-cycle arrest and apoptosis in human urinary bladder cancer cell lines and chemically induced cancers by targeting PI3K. Cancer Prev. Res. 2014, 7, 236–245. [Google Scholar] [CrossRef]
- Abrahão, A.C.; Giudice, F.S.; Sperandio, F.F.; Pinto Junior, D. dos S. Effects of celecoxib treatment over the AKT pathway in head and neck squamous cell carcinoma. J. Oral Pathol. Med. 2013, 42, 793–798. [Google Scholar] [CrossRef]
- Arisan, E.D.; Ergül, Z.; Bozdağ, G.; Rencüzoğulları, Ö.; Çoker-Gürkan, A.; Obakan-Yerlikaya, P.; Coşkun, D.; Palavan-Ünsal, N. Diclofenac induced apoptosis via altering PI3K/Akt/MAPK signaling axis in HCT 116 more efficiently compared to SW480 colon cancer cells. Mol. Biol. Rep. 2018, 45, 2175–2184. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Jin, S.; Wu, X. Aspirin inhibits colon cancer cell line migration through regulating epithelial-mesenchymal transition via wnt signaling. Oncol. Lett. 2019, 17, 4675–4682. [Google Scholar] [CrossRef]
- Gottfried, E.; Lang, S.A.; Renner, K.; Bosserhoff, A.; Gronwald, W.; Rehli, M.; Einhell, S.; Gedig, I.; Singer, K.; Seilbeck, A.; et al. New Aspects of an Old Drug—Diclofenac Targets MYC and Glucose Metabolism in Tumor Cells. PLoS ONE 2013, 8, e66987. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wang, Y.; Feng, J.; Liu, Y.; Wang, T.; Zhao, M.; Ye, L.; Zhang, X. Aspirin suppresses the abnormal lipid metabolism in liver cancer cells via disrupting an NFκB-ACSL1 signaling. Biochem. Biophys. Res. Commun. 2017, 486, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, R.; Rodríguez-Enríquez, S.; Pacheco-Velázquez, S.C.; Bortnik, V.; Moreno-Sánchez, R.; Ralph, S. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem. Pharmacol. 2018, 154, 318–334. [Google Scholar] [CrossRef]
- Dai, X.; Yan, J.; Fu, X.; Pan, Q.; Sun, D.; Xu, Y.; Wang, J.; Nie, L.; Tong, L.; Shen, A.; et al. Aspirin inhibits cancer metastasis and angiogenesis via targeting heparanase. Clin. Cancer Res. 2017, 23, 6267–6279. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.I.J.; Kankipati, C.S.; Jones, S.; Newman, R.M.; Safrany, S.T.; Perry, C.J.; Nicholl, I.D. A novel mechanism for the anticancer activity of aspirin and salicylates. Int. J. Oncol. 2019, 54, 1256–1270. [Google Scholar] [CrossRef]
- Qin, S.; Xu, C.; Li, S.; Yang, C.; Sun, X.; Wang, X.; Tang, S.C.; Ren, H. Indomethacin induces apoptosis in the EC109 esophageal cancer cell line by releasing second mitochondria-derived activator of caspase and activating caspase-3. Mol. Med. Rep. 2015, 11, 4694–4700. [Google Scholar] [CrossRef] [PubMed]
- Tse, A.K.W.; Cao, H.H.; Cheng, C.Y.; Kwan, H.Y.; Yu, H.; Fong, W.F.; Yu, Z.L. Indomethacin sensitizes TRAIL-resistant melanoma cells to TRAIL-induced apoptosis through ROS-mediated upregulation of death receptor 5 and downregulation of survivin. J. Investig. Dermatol. 2014, 134, 1397–1407. [Google Scholar] [CrossRef]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150. [Google Scholar] [CrossRef]
- Yang, L.; Li, J.; Li, Y.; Zhou, Y.; Wang, Z.; Zhang, D.; Liu, J.; Zhang, X. Diclofenac impairs the proliferation and glucose metabolism of triple-negative breast cancer cells by targeting the c-Myc pathway. Exp. Ther. Med. 2021, 21, 584. [Google Scholar] [CrossRef]
- Leidgens, V.; Seliger, C.; Jachnik, B.; Welz, T.; Leukel, P.; Vollmann-Zwerenz, A.; Bogdahn, U.; Kreutz, M.; Grauer, O.M.; Hau, P. Ibuprofen and diclofenac restrict migration and proliferation of human glioma cells by distinct molecular mechanisms. PLoS ONE 2015, 10, e0140613. [Google Scholar] [CrossRef]
- Deb, J.; Majumder, J.; Bhattacharyya, S.; Jana, S.S. A novel naproxen derivative capable of displaying anti-cancer and anti-migratory properties against human breast cancer cells. BMC Cancer 2014, 14, 567. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, R.; Tong, J.; Risnik, D.; Leibowitz, B.J.; Wang, Y.J.; Concha-Benavente, F.; DeLiberty, J.M.; Stolz, D.B.; Pai, R.K.; Ferris, R.L.; et al. Non-steroidal anti-inflammatory drugs induce immunogenic cell death in suppressing colorectal tumorigenesis. Oncogene 2021, 40, 2035–2050. [Google Scholar] [CrossRef] [PubMed]
- Ağdaş, F.; Eryilmaz, A.; Gökmen Yilmaz, E.; Ergin, K. the Effects of Sulindac on Cell Viability, Cell Cycle andAnjiogenezi̇s in Pharyngeal Cancer Cell. ENT Updat. 2020, 10, 373–380. [Google Scholar] [CrossRef]
- Thabet, N.A.; El-Guendy, N.; Mohamed, M.M.; Shouman, S.A. Suppression of macrophages- Induced inflammation via targeting RAS and PAR-4 signaling in breast cancer cell lines. Toxicol. Appl. Pharmacol. 2019, 385, 114773. [Google Scholar] [CrossRef]
- Rai, N.; Sarkar, M.; Raha, S. Piroxicam, a traditional non-steroidal anti-inflammatory drug (NSAID) causes apoptosis by ROS mediated Akt activation. Pharmacol. Rep. 2015, 67, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Rahman, H.; Tyagi, E.; Liu, T.; Li, C.; Lu, R.; Lum, D.; Holmen, S.L.; Maschek, J.A.; Cox, J.E.; et al. Aspirin suppresses PGE2 and activates AMP kinase to inhibit melanoma cell motility, pigmentation, and selective tumor growth in vivo. Cancer Prev. Res. 2018, 11, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, J.; Zhang, L.; Huang, S.; Zhao, X.; Zhao, X. Celecoxib induced apoptosis against different breast cancer cell lines by down-regulated NF-κB pathway. Biochem. Biophys. Res. Commun. 2017, 490, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Eli, Y.; Przedecki, F.; Levin, G.; Kariv, N.; Raz, A. Comparative effects of indomethacin on cell proliferation and cell cycle progression in tumor cells grown in vitro and in vivo. Biochem. Pharmacol. 2001, 61, 565–571. [Google Scholar] [CrossRef]
- Johnson, S.D.; Young, M.R.I. Indomethacin treatment of mice with premalignant oral lesions sustains cytokine production and slows progression to cancer. Front. Immunol. 2016, 7, 379. [Google Scholar] [CrossRef]
- Quidville, V.; Segond, N.; Pidoux, E.; Cohen, R.; Jullienne, A.; Lausson, S. Tumor growth inhibition by indomethacin in a mouse model of human medullary thyroid cancer: Implication of cyclooxygenases and 15-hydroxyprostaglandin dehydrogenase. Endocrinology 2004, 145, 2561–2571. [Google Scholar] [CrossRef][Green Version]
- Will, O.M.; Purcz, N.; Chalaris, A.; Heneweer, C.; Boretius, S.; Purcz, L.; Nikkola, L.; Ashammakhi, N.; Kalthoff, H.; Glüer, C.C.; et al. Increased survival rate by local release of diclofenac in a murine model of recurrent oral carcinoma. Int. J. Nanomed. 2016, 11, 5311–5321. [Google Scholar] [CrossRef] [PubMed]
- Suri, A.; Sheng, X.; Schuler, K.M.; Zhong, Y.; Han, X.; Jones, H.M.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. The effect of celecoxib on tumor growth in ovarian cancer cells and a genetically engineered mouse model of serous ovarian cancer. Oncotarget 2016, 7, 39582–39594. [Google Scholar] [CrossRef] [PubMed]
- Esbona, K.; Inman, D.; Saha, S.; Jeffery, J.; Schedin, P.; Wilke, L.; Keely, P. COX-2 modulates mammary tumor progression in response to collagen density. Breast Cancer Res. 2016, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.A.; Skinner, S.A.; Malcontenti-Wilson, C.; Misajon, A.; Dejong, T.; Vogiagis, D.; O’Brien, P.E. Non-steroidal anti-inflammatory drugs with different cyclooxygenase inhibitory profiles that prevent aberrant crypt foci formation but vary in acute gastrotoxicity in a rat model. J. Gastroenterol. Hepatol. 2000, 15, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Abdeen, A.; Aboubakr, M.; Elgazzar, D.; Abdo, M.; Abdelkader, A.; Ibrahim, S.; Elkomy, A. Rosuvastatin attenuates piroxicam-mediated gastric ulceration and hepato-renal toxicity in rats. Biomed. Pharmacother. 2019, 110, 895–905. [Google Scholar] [CrossRef]
- Sukmawan, Y.P. Comparison and Impact of The Short-Term Use of Some Non-Aspirin NSAIDs on Bleeding Time of Mice. Cardiovasc. Hematol. Agents Med. Chem. 2018, 16, 120–122. [Google Scholar] [CrossRef]
- Paunescu, A.; Ponepal, C.M.; Zagardan, M.C.; Plesa, C.F.; Nemes, R.M.; Nicolae, C.; Bisoc, A.; Diaconu, M.; Fierascu, I.; Fierascu, R.C.; et al. Evaluation of histophysiological alterations associated with ketoprofen administration in albino NMRI mice. Naunyn. Schmiedebergs. Arch. Pharmacol. 2020, 393, 1033–1039. [Google Scholar] [CrossRef]
- Niranjan, R.; Manik, P.; Sivastav, A.K.; Palit, G.; Natu, S.M. Comparative Adverse Effects Of Cox-1 and Cox-2 Inhibitors in Rat Liver: An Experimental Study. J. Anat. Soc. India 2010, 59, 182–186. [Google Scholar] [CrossRef]
- Rosenberg, L.; Palmer, J.R.; Zauber, A.G.; Warshauer, M.E.; Stolley, P.D.; Shapiro, S. A hypothesis: Nonsteroidal anti-inflammatory drugs reduce the incidence of large-bowel cancer. J. Natl. Cancer Inst. 1991, 83, 355–358. [Google Scholar] [CrossRef]
- Coogan, P.F.; Rosenberg, L.; Palmer, J.R.; Strom, B.L.; Zauber, A.G.; Stolley, P.D.; Shapiro, S. Nonsteroidal anti-inflammatory drugs and risk of digestive cancers at sites other than the large bowel. Cancer Epidemiol. Biomarkers Prev. 2000, 9, 119–123. [Google Scholar]
- Wu, C.Y.; Wu, M.S.; Kuo, K.N.; Wang, C.B.; Chen, Y.J.; Lin, J.T. Effective reduction of gastric cancer risk with regular use of nonsteroidal anti-inflammatory drugs in Helicobacter pylori-infected patients. J. Clin. Oncol. 2010, 28, 2952–2957. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.N.; Theodoratou, E.; Farrington, S.M.; Tenesa, A.; Barnetson, R.A.; Cetnarskyj, R.; Stark, L.; Porteous, M.E.; Campbell, H.; Dunlop, M.G. Effect of aspirin and NSAIDs on risk and survival from colorectal cancer. Gut 2010, 59, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Friis, S.; Riis, A.H.; Erichsen, R.; Baron, J.A.; Sørensen, H.T. Low-Dose Aspirin or Nonsteroidal Anti-inflammatory Drug Use and Colorectal Cancer Risk. Ann. Intern. Med. 2015, 163, 347–355. [Google Scholar] [CrossRef]
- Zhao, X.; Xu, Z.; Li, H. NSAIDs Use and Reduced Metastasis in Cancer Patients: Results from a meta-analysis. Sci. Rep. 2017, 7, 1875. [Google Scholar] [CrossRef] [PubMed]
- Trelle, S.; Reichenbach, S.; Wandel, S.; Hildebrand, P.; Tschannen, B.; Villiger, P.M.; Egger, M.; Jüni, P. Cardiovascular safety of non-steroidal anti-inflammatory drugs: Network meta-analysis. BMJ 2011, 342, 154. [Google Scholar] [CrossRef] [PubMed]
- Bally, M.; Dendukuri, N.; Rich, B.; Nadeau, L.; Helin-Salmivaara, A.; Garbe, E.; Brophy, J.M. Risk of acute myocardial infarction with NSAIDs in real world use: Bayesian meta-analysis of individual patient data. BMJ 2017, 357, 1909. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.; Yakoob, J.; Jafri, W.; Islam, S.; Abid, S.; Islam, M. Frequency of NSAID induced peptic ulcer disease. J. Pakistan Med. Assoc. 2006, 56, 218. [Google Scholar]
- Bhala, N.; Emberson, J.; Merhi, A.; Abramson, S.; Arber, N.; Baron, J.A.; Bombardier, C.; Cannon, C.; Farkouh, M.E.; FitzGerald, G.A.; et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: Meta-analyses of individual participant data from randomised trials. Lancet 2013, 382, 769–779. [Google Scholar] [CrossRef]
- Balestracci, A.; Ezquer, M.; Elmo, M.E.; Molini, A.; Thorel, C.; Torrents, M.; Toledo, I. Ibuprofen-associated acute kidney injury in dehydrated children with acute gastroenteritis. Pediatr. Nephrol. 2015, 30, 1873–1878. [Google Scholar] [CrossRef]
- Duffy, C.P.; Elliott, C.J.; O’Connor, R.A.; Heenan, M.M.; Coyle, S.; Cleary, I.M.; Kavanagh, K.; Verhaegen, S.; O’Loughlin, C.M.; NicAmhlaoibh, R.; et al. Enhancement of chemotherapeutic drug toxicity to human tumour cells in vitro by a subset of non-steroidal anti-inflammatory drugs (NSAIDS). Eur. J. Cancer 1998, 34, 1250–1259. [Google Scholar] [CrossRef]
- Arunasree, K.M.; Roy, K.R.; Anilkumar, K.; Aparna, A.; Reddy, G.V.; Reddanna, P. Imatinib-resistant K562 cells are more sensitive to celecoxib, a selective COX-2 inhibitor: Role of COX-2 and MDR-1. Leuk. Res. 2008, 32, 855–864. [Google Scholar] [CrossRef]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Danø, K. Active outward transport of daunomycin in resistant ehrlich ascites tumor cells. Biochim. Biophys. Acta-Biomembr. 1973, 323, 466–483. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-K.; Lee, E.; Pyo, H.; Lim, S.-J. Cyclooxygenase-independent down-regulation of multidrug resistance–associated protein-1 expression by celecoxib in human lung cancer cells. Mol. Cancer Ther. 2005, 4, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Özvegy, C.; Varadi, A.; Sarkadi, B. Characterization of Drug Transport, ATP Hydrolysis, and Nucleotide Trapping by the Human ABCG2 Multidrug Transporter: Modulation of Substrate Specificity by a Point Mutation. J. Biol. Chem. 2002, 277, 47980–47990. [Google Scholar] [CrossRef] [PubMed]
- Elahian, F.; Kalalinia, F.; Behravan, J. Evaluation of indomethacin and dexamethasone effects on BCRP-mediated drug resistance in MCF-7 parental and resistant cell lines. Drug Chem. Toxicol. 2010, 33, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Zatelli, M.C.; Luchin, A.; Tagliati, F.; Leoni, S.; Piccin, D.; Bondanelli, M.; Rossi, R.; Degli Uberti, E.C. Cyclooxygenase-2 inhibitors prevent the development of chemoresistance phenotype in a breast cancer cell line by inhibiting glycoprotein p-170 expression. Endocr. Relat. Cancer 2007, 14, 1029–1038. [Google Scholar] [CrossRef]
- Xia, W.; Zhao, T.; Lv, J.; Xu, S.; Shi, J.; Wang, S.; Han, X.; Sun, Y. Celecoxib enhanced the sensitivity of cancer cells to anticancer drugs by inhibition of the expression of P-glycoprotein through a COX-2-independent manner. J. Cell. Biochem. 2009, 108, 181–194. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, R.A.; Saleh, E.M.; Ezz, M.; Elsayed, A.M. Interaction of celecoxib with different anti-cancer drugs is antagonistic in breast but not in other cancer cells. Toxicol. Appl. Pharmacol. 2011, 255, 271–286. [Google Scholar] [CrossRef]
- Endo, H.; Yano, M.; Okumura, Y.; Kido, H. Ibuprofen enhances the anticancer activity of cisplatin in lung cancer cells by inhibiting the heat shock protein 70. Cell Death Dis. 2014, 5, e1027. [Google Scholar] [CrossRef]
- Chen, M.; Yu, L.; Gu, C.; Zhong, D.; Wu, S.; Liu, S. Celecoxib antagonizes the cytotoxic effect of cisplatin in human gastric cancer cells by decreasing intracellular cisplatin accumulation. Cancer Lett. 2013, 329, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Ravera, M.; Zanellato, I.; Gabano, E.; Perin, E.; Rangone, B.; Coppola, M.; Osella, D. Antiproliferative activity of Pt(IV) conjugates containing the non-steroidal anti-inflammatory drugs (NSAIDs) Ketoprofen and Naproxen. Int. J. Mol. Sci. 2019, 20, 3074. [Google Scholar] [CrossRef]
- Awara, W.M.; El-Sisi, A.E.; El-Sayad, M.E.; Goda, A.E. The potential role of cyclooxygenase-2 inhibitors in the treatment of experimentally-induced mammary tumour: Does celecoxib enhance the anti-tumour activity of doxorubicin? Pharmacol. Res. 2004, 50, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Q.; Guo, Q.; Zhu, J.H.; Chen, W.C. Increase of cyclooxygenase-2 inhibition with celecoxib combined with 5-FU enhances tumor cell apoptosis and antitumor efficacy in a subcutaneous implantation tumor model of human colon cancer. World J. Surg. Oncol. 2013, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.; Heenan, M.; Connolly, L.; Larkin, A.; Clynes, M. Increased Anti-tumour Efficacy of Doxorubicin when Combined with Sulindac in a Xenograft Model of an MRP-1-positive Human Lung Cancer. Anticancer Res. 2004, 24, 457–464. [Google Scholar]
- Ponthan, F.; Wickström, M.; Gleissman, H.; Fuskevåg, O.M.; Segerström, L.; Sveinbjörnsson, B.; Redfern, C.P.F.; Eksborg, S.; Kogner, P.; Johnsen, J.I. Celecoxib prevents neuroblastoma tumor development and potentiates the effect of chemotherapeutic drugs in vitro and in vivo. Clin. Cancer Res. 2007, 13, 1036–1044. [Google Scholar] [CrossRef]
- Ong, S.M.; Saeki, K.; Kok, M.K.; Tanaka, Y.; Choisunirachon, N.; Yoshitake, R.; Nishimura, R.; Nakagawa, T. Anti-tumour efficacy of etoposide alone and in combination with piroxicam against canine osteosarcoma in a xenograft model. Res. Vet. Sci. 2017, 113, 130–135. [Google Scholar] [CrossRef]
- Gunjal, P.M.; Schneider, G.; Ismail, A.A.; Kakar, S.S.; Kucia, M.; Ratajczak, M.Z. Evidence for induction of a tumor metastasis-receptive microenvironment for ovarian cancer cells in bone marrow and other organs as an unwanted and underestimated side effect of chemotherapy/radiotherapy. J. Ovarian Res. 2015, 8, 20. [Google Scholar] [CrossRef]
- Young, S.D.; Whissell, M.; Noble, J.C.S.; Cano, P.O.; Lopez, P.G.; Germond, C.J. Phase II clinical trial results involving treatment with low-dose daily oral cyclophosphamide, weekly vinblastine, and rofecoxib in patients with advanced solid tumors. Clin. Cancer Res. 2006, 12, 3092–3098. [Google Scholar] [CrossRef]
- Reyners, A.K.L.; de Munck, L.; Erdkamp, F.L.G.; Smit, W.M.; Hoekman, K.; Lalisang, R.I.; de Graaf, H.; Wymenga, A.N.M.; Polee, M.; Hollema, H.; et al. A randomized phase ii study investigating the addition of the specific COX-2 inhibitor celecoxib to docetaxel plus carboplatin as first-line chemotherapy for stage IC to IV epithelial ovarian cancer, fallopian tube or primary peritoneal carcinomas: The Do. Ann. Oncol. 2012, 23, 2896–2902. [Google Scholar] [CrossRef]
- Perroud, H.A.; Alasino, C.M.; Rico, M.J.; Mainetti, L.E.; Queralt, F.; Pezzotto, S.M.; Rozados, V.R.; Graciela Scharovsky, O. Metastatic breast cancer patients treated with low-dose metronomic chemotherapy with cyclophosphamide and celecoxib: Clinical outcomes and biomarkers of response. Cancer Chemother. Pharmacol. 2016, 77, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Legge, F.; Paglia, A.; D’Asta, M.; Fuoco, G.; Scambia, G.; Ferrandina, G. Phase II study of the combination carboplatin plus celecoxib in heavily pre-treated recurrent ovarian cancer patients. BMC Cancer 2011, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Jüni, P.; Nartey, L.; Reichenbach, S.; Sterchi, R.; Dieppe, P.A.; Egger, P.M. Risk of cardiovascular events and rofecoxib: Cumulative meta-analysis. Lancet 2004, 364, 2021–2029. [Google Scholar] [CrossRef]
- Karai, E.; Szebényi, K.; Windt, T.; Fehér, S.; Szendi, E.; Dékay, V.; Vajdovich, P.; Szakács, G.; Füredi, A. Celecoxib Prevents Doxorubicin-Induced Multidrug Resistance in Canine and Mouse Lymphoma Cell Lines. Cancers 2020, 12, 1117. [Google Scholar] [CrossRef]
- Komaki, R.; Wei, X.; Allen, P.K.; Liao, Z.; Milas, L.; Cox, J.D.; O’Reilly, M.S.; Chang, J.Y.; McAleer, M.F.; Jeter, M.; et al. Phase I study of celecoxib with concurrent irinotecan, cisplatin, and radiation therapy for patients with unresectable locally advanced non-small cell lung cancer. Front. Oncol. 2011, 1, 52. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Mamon, H.J.; Szymonifka, J.; Bueno, R.; Choi, N.; Donahue, D.M.; Fidias, P.M.; Gaissert, H.A.; Jaklitsch, M.T.; Kulke, M.H.; et al. Neoadjuvant irinotecan, cisplatin, and concurrent radiation therapy with celecoxib for patients with locally advanced esophageal cancer. BMC Cancer 2016, 16, 468. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hills, B.A.; Butler, B.D.; Lichtenberger, L.M. Gastric mucosal barrier: Hydrophobic lining to the lumen of the stomach. Am. J. Physiol. Gastrointest. Liver Physiol. 1983, 244, G561–G568. [Google Scholar] [CrossRef]
- Goddard, P.J.; Hills, B.A.; Lichtenberger, L.M. Does aspirin damage canine gastric mucosa by reducing its surface hydrophobicity? Am. J. Physiol.-Gastrointest. Liver Physiol. 1987, 252, G421–G430. [Google Scholar] [CrossRef]
- Lichtenberger, L.M. The hydrophobic barrier properties of gastrointestinal mucus. Annu. Rev. Physiol. 1995, 57, 565–583. [Google Scholar] [CrossRef]
- Giraud, M.N.; Motta, C.; Romero, J.J.; Bommelaer, G.; Lichtenberger, L.M. Interaction of indomethacin and naproxen with gastric surface-active phospholipids: A possible mechanism for the gastric toxicity of nonsteroidal anti-inflammatory drugs (NSAIDs). Biochem. Pharmacol. 1999, 57, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.S.; Jim, J.J.; Sanduja, S.K.; Lichtenberger, L.M. Phospholipid association reduces the gastric mucosal toxicity of aspirin in human subjects. Am. J. Gastroenterol. 1999, 94, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Lanza, F.L.; Marathi, U.K.; Anand, B.S.; Lichtenberger, L.M. Clinical trial: Comparison of ibuprofen-phosphatidylcholine and ibuprofen on the gastrointestinal safety and analgesic efficacy in osteoarthritic patients. Aliment. Pharmacol. Ther. 2008, 28, 431–442. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Romero, J.J.; Dial, E.J. Gastrointestinal safety and therapeutic efficacy of parenterally administered phosphatidylcholine-associated indomethacin in rodent model systems. Br. J. Pharmacol. 2009, 157, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Cryer, B.; Bhatt, D.L.; Lanza, F.L.; Dong, J.F.; Lichtenberger, L.M.; Marathi, U.K. Low-dose aspirin-induced ulceration is attenuated by aspirin-phosphatidylcholine: A randomized clinical trial. Am. J. Gastroenterol. 2011, 106, 272–277. [Google Scholar] [CrossRef]
- Lichtenberger, L.M.; Phan, T.; Fang, D.; Dial, E.J. Chemoprevention with phosphatidylcholine non-steroidal anti-inflammatory drugs in vivo and in vitro. Oncol. Lett. 2018, 15, 6688–6694. [Google Scholar] [CrossRef]
- Davis, J.S.; Kanikarla-Marie, P.; Gagea, M.; Yu, P.L.; Fang, D.; Sebastian, M.; Yang, P.; Hawk, E.; Dashwood, R.; Lichtenberger, L.M.; et al. Sulindac plus a phospholipid is effective for polyp reduction and safer than sulindac alone in a mouse model of colorectal cancer development. BMC Cancer 2020, 20, 871. [Google Scholar] [CrossRef]
- Dvir, E.; Friedman, J.E.; Lee, J.Y.; Koh, J.Y.; Younis, F.; Raz, S.; Shapiro, I.; Hoffman, A.; Dahan, A.; Rosenberg, G.; et al. Erratum: A novel phospholipid derivative of indomethacin, DP-155 [mixture of 1-steroyl and 1-palmitoyl-2-{4-[1-(p-chlorobenzoyl)-5-methoxy-2-methyl-3- indolyl acetamido]butanoyl}-sn-glycero-3-phosophatidyl choline], shows superior safety and similar effic. J. Pharmacol. Exp. Ther. 2006, 319, 1492. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Ridgway, N.D. The role of phosphatidylcholine and choline metabolites to cell proliferation and survival. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 20–38. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Michel, V.; Yuan, Z.; Ramsubir, S.; Bakovic, M. Choline transport for phospholipid synthesis. Exp. Biol. Med. 2006, 231, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, E.; Nishiyama, R.; Iwao, B.; Kawai, Y.; Ishii, C.; Yamanaka, T.; Uchino, H.; Inazu, M. Molecular and Functional Characterization of Choline Transporter-Like Proteins in Esophageal Cancer Cells and Potential Therapeutic Targets. Biomol. Ther. 2018, 26, 399. [Google Scholar] [CrossRef] [PubMed]
- Kouji, H.; Inazu, M.; Yamada, T.; Tajima, H.; Aoki, T.; Matsumiya, T. Molecular and functional characterization of choline transporter in human colon carcinoma HT-29 cells. Arch. Biochem. Biophys. 2009, 483, 90–98. [Google Scholar] [CrossRef]
- Yuan, Z.; Tie, A.; Tarnopolsky, M.; Bakovic, M. Genomic organization, promoter activity, and expression of the human choline transporter-like protein 1. Physiol. Genom. 2006, 26, 76–90. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, L.; Fei, X.C.; Jiang, X.F.; Zheng, Z.; Zhao, Y.; Wang, C.F.; Li, B.; Chen, S.J.; Janin, A.; et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e582. [Google Scholar] [CrossRef]
- Hernández-Alcoceba, R.; Saniger, L.; Campos, J.; Núñez, M.C.; Khaless, F.; Gallo, M.A.; Espinosa, A.; Lacal, J.C. Choline kinase inhibitors as a novel approach for antiproliferative drug design. Oncogene 1997, 15, 2289–2301. [Google Scholar] [CrossRef]
- Rodríguez-González, A.; de Molina, A.R.; Fernández, F.; Ramos, M.A.; del Carmen Núñez, M.; Campos, J.; Lacal, J.C. Inhibition of choline kinase as a specific cytotoxic strategy in oncogene-transformed cells. Oncogene 2003, 22, 8803–8812. [Google Scholar] [CrossRef]
- García-Molina, P.; Sola-Leyva, A.; Luque-Navarro, P.M.; Laso, A.; Ríos-Marco, P.; Ríos, A.; Lanari, D.; Torretta, A.; Parisini, E.; López-Cara, L.C.; et al. Anticancer Activity of the Choline Kinase Inhibitor PL48 Is Due to Selective Disruption of Choline Metabolism and Transport Systems in Cancer Cell Lines. Pharmaceutics 2022, 14, 426. [Google Scholar] [CrossRef]
- Iorio, E.; Ricci, A.; Bagnoli, M.; Pisanu, M.E.; Castellano, G.; Di Vito, M.; Venturini, E.; Glunde, K.; Bhujwalla, Z.M.; Mezzanzanica, D.; et al. Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res. 2010, 70, 2126–2135. [Google Scholar] [CrossRef]
- Kłobucki, M.; Urbaniak, A.; Grudniewska, A.; Kocbach, B.; Maciejewska, G.; Kiełbowicz, G.; Ugorski, M.; Wawrzeńczyk, C. Syntheses and cytotoxicity of phosphatidylcholines containing ibuprofen or naproxen moieties. Sci. Rep. 2019, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Lima, P.S.S.; Lucchese, A.M.; Araújo-Filho, H.G.; Menezes, P.P.; Araújo, A.A.S.; Quintans-Júnior, L.J.; Quintans, J.S.S. Inclusion of terpenes in cyclodextrins: Preparation, characterization and pharmacological approaches. Carbohydr. Polym. 2016, 151, 965–987. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.V.; Silva, J.M.; Rodrigues, L.; Reis, R.L.; Paiva, A.; Duarte, A.R.C.; Matias, A. Unveil the Anticancer Potential of Limomene Based Therapeutic Deep Eutectic Solvents. Sci. Rep. 2019, 9, 14926. [Google Scholar] [CrossRef]
- Fujii, M.; Takeda, Y.; Yoshida, M.; Utoguchi, N.; Matsumoto, M.; Watanabe, Y. Comparison of skin permeation enhancement by 3-l-menthoxypropane-1,2-diol and l-menthol: The permeation of indomethacin and antipyrine through Yucatan micropig skin and changes in infrared spectra and X-ray diffraction patterns of stratum corneum. Int. J. Pharm. 2003, 258, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Okyar, A.; Nuriyev, M.; Yildiz, A.; Pala-Kara, Z.; Ozturk, N.; Kaptan, E. The effect of terpenes on percutaneous absorption of tiaprofenic acid gel. Arch. Pharmacal Res. 2010, 33, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.; Oliveira, F.; Silva, J.M.; Matias, A.; Reis, R.L.; Duarte, A.R.C. Optimal Design of THEDES Based on Perillyl Alcohol and Ibuprofen. Pharmaceutics 2020, 12, 1121. [Google Scholar] [CrossRef]
- Silva, E.; Oliveira, F.; Silva, J.M.; Reis, R.L.; Duarte, A.R.C. Untangling the bioactive properties of therapeutic deep eutectic solvents based on natural terpenes. Curr. Res. Chem. Biol. 2021, 1, 100003. [Google Scholar] [CrossRef]
- Theoduloz, C.; Delporte, C.; Valenzuela-Barra, G.; Silva, X.; Cádiz, S.; Bustamante, F.; Pertino, M.W.; Schmeda-Hirschmann, G. Topical Anti-inflammatory Activity of New Hybrid Molecules of Terpenes and Synthetic Drugs. Molecules 2015, 20, 11219–11235. [Google Scholar] [CrossRef]
- Theoduloz, C.; Alzate-Morales, J.; Jiménez-Aspee, F.; Isla, M.I.; Alberto, M.R.; Pertino, M.W.; Schmeda-Hirschmann, G. Inhibition of key enzymes in the inflammatory pathway by hybrid molecules of terpenes and synthetic drugs: In vitro and in silico studies. Chem. Biol. Drug Des. 2019, 93, 290–299. [Google Scholar] [CrossRef]
- de Oliveira Pedrosa Rolim, M.; de Almeida, A.R.; da Rocha Pitta, M.G.; de Melo Rêgo, M.J.B.; Quintans-Júnior, L.J.; de Souza Siqueira Quintans, J.; Heimfarth, L.; Scotti, L.; Scotti, M.T.; da Cruz, R.M.D.; et al. Design, synthesis and pharmacological evaluation of CVIB, a codrug of carvacrol and ibuprofen as a novel anti-inflammatory agent. Int. Immunopharmacol. 2019, 76, 105856. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NSAIDs | Therapeutic Concentration | Cancer Cell Line | Mechanism of Action | Reference |
|---|---|---|---|---|
| Acetylsalicylic acid | 0.5–4 mM for 2.5 h | MDA-MB-231, B16F10, CHO K1, and U87-MG | Dose-dependent direct binding on an oncogenic extracellular matrix enzyme called heparinase and the inhibition of cell migration and angiogenesis. | [38] |
| 0.1–1 mM for 12 h | SW480 | Dose-dependent relocation of EGF and the phosphorylation of EGFR. | [39] | |
| Indomethacin | 0.4 mM for 48 h | EC109 | Induction of mitochondria-derived caspase-3 in apoptosis. | [40] |
| 0.1–0.3 mM for 24 h | A375, MeWo, and SK-MEL-5 | Dose-dependent downregulation of survival via ROS induction and NFκB signaling. Induced ROS upregulation of DR5 and CHOPS in TRAIL-related cell death. | [41] | |
| Diclofenac | 0.1 and 0.2 mM for 24 h | 4T1 | Downregulation of lactate secretion in T-cell-mediated cell death. | [42] |
| 0.4 and 0.8 mM for 48 h | MDA-MB-231 and HCC1937 | Dose-dependent downregulation of GLUT1 and c-Myc expression, the inhibition of hexokinase activity, and the inhibition of cell proliferation. | [43] | |
| Ibuprofen | 0.5 mM for 48 h | AGS | Downregulation of VEGF-A, PCNA, Akt, CD44, and OCT3/4 gene transcription in apoptosis. | [28] |
| 2 mM for 24 h | HTZ-349 and A172 | Downregulation of c-Myc expression in the inhibition of cell growth and migration. | [44] | |
| Naproxen | 0.5–2 mM for 72 h | UM-UC-5 and UM-UC-14 | Dose-dependent downregulation of Bcl-2 and the upregulation of Bax expression during apoptosis. | [30] |
| 6 mM for 6 h | MDA-MB-231 | Increased activation of caspase-3 and caspase-9 in apoptosis. | [45] | |
| Sulindac | 0.03–0.12 mM for 24 h | HCT116 | Dose-dependent induction of ER stress makers, such as DR5, pPERK, and pEIF2α in ER-mediated apoptosis. | [46] |
| 0.03–1 mM for 48 h | FaDu | Induction of the production of VEGFR–2 and the arrest of cells at the G2/M phase. | [47] | |
| Piroxicam | 0.025–0.05 mM for 24 h and 48 h | MCF-7 and MDA-MB-231 | Time-dependent downregulation of IL-1β and IL-6 gene expression. | [48] |
| 0.03 mM for 3–48 h | MCF-7 | Time-dependent induction of ROS-activated PI3K/Akt signaling during apoptosis. | [49] | |
| Celecoxib | 0.02–0.04 mM for 24 h | A375 and Mel-STM | Inhibition of COX-2 expression and the downregulation of cell migration. | [50] |
| 0.04, 0.08 and 0.1 mM for 24–48 h | MDA-MB-231 and SK-BR-3 | Dose-dependent and time-dependent upregulation of caspase-3 induced cell cycle arrest at the G1 and G2 phases. | [51] |
| Compound/THEDES | Molar Ratio | EC50 Values (mM) | Selectivity Index | |
|---|---|---|---|---|
| Colon Adenocarcinoma Cell Line (HT29) | Cytotoxicity Assay | |||
| IBU | - | 2.346 ± 0.09 | 2.89 ± 0.06 | 1.23 |
| LIM | - | 0.67 ± 0.03 | 2.64 ± 0.11 | - |
| POH | - | 2.37 ± 0.20 | 4.86 ± 1.58 | 2.06 |
| THY | - | 5.22 ± 1.16 | 6.73 ± 1.69 | 1.29 |
| ME | - | 4.31 ± 0.63 | 5.09 ± 0.73 | 1.18 |
| POH + IBU | 3:1 | 4.51 ± 0.26 | - | - |
| LIM:IBU | 4:1 | 2.390 ± 2.919 | 10.5 ± 0.883 | - |
| POH:IBU | 3:1 | 1.316 ± 0.07 | 8.46 ± 1.13 | 5.89 |
| THY:IBU | 3:1 | 0.30 ± 0.04 | 1.07 ± 0.37 | 3.5 |
| ME:IBU | 3:1 | 4.3 ± 0.71 | 8.92 ± 1.39 | 2.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thiruchenthooran, V.; Sánchez-López, E.; Gliszczyńska, A. Perspectives of the Application of Non-Steroidal Anti-Inflammatory Drugs in Cancer Therapy: Attempts to Overcome Their Unfavorable Side Effects. Cancers 2023, 15, 475. https://doi.org/10.3390/cancers15020475
Thiruchenthooran V, Sánchez-López E, Gliszczyńska A. Perspectives of the Application of Non-Steroidal Anti-Inflammatory Drugs in Cancer Therapy: Attempts to Overcome Their Unfavorable Side Effects. Cancers. 2023; 15(2):475. https://doi.org/10.3390/cancers15020475
Chicago/Turabian StyleThiruchenthooran, Vaikunthavasan, Elena Sánchez-López, and Anna Gliszczyńska. 2023. "Perspectives of the Application of Non-Steroidal Anti-Inflammatory Drugs in Cancer Therapy: Attempts to Overcome Their Unfavorable Side Effects" Cancers 15, no. 2: 475. https://doi.org/10.3390/cancers15020475
APA StyleThiruchenthooran, V., Sánchez-López, E., & Gliszczyńska, A. (2023). Perspectives of the Application of Non-Steroidal Anti-Inflammatory Drugs in Cancer Therapy: Attempts to Overcome Their Unfavorable Side Effects. Cancers, 15(2), 475. https://doi.org/10.3390/cancers15020475