

The Contribution of Germline Pathogenic Variants in Breast Cancer Genes to Contralateral Breast Cancer Risk in BRCA1/BRCA2/PALB2-Negative Women


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population, Library Preparation and Sequencing of Samples
2.2. Alignment, Variant Calling and Annotation
2.3. Selecting Genes and Pathogenic Variants
- Variants annotated as Pathogenic/Likely pathogenic by ClinVar or
- Loss of Function variants as predicted by VEP (stop/start gain/loss, frameshift, essential splice sites) or
- Missense variants that were simultaneously called Deleterious by PolyPhen, Probably Damaging by SIFT and had CADD Phred score > 25.
2.4. Statistical Analysis
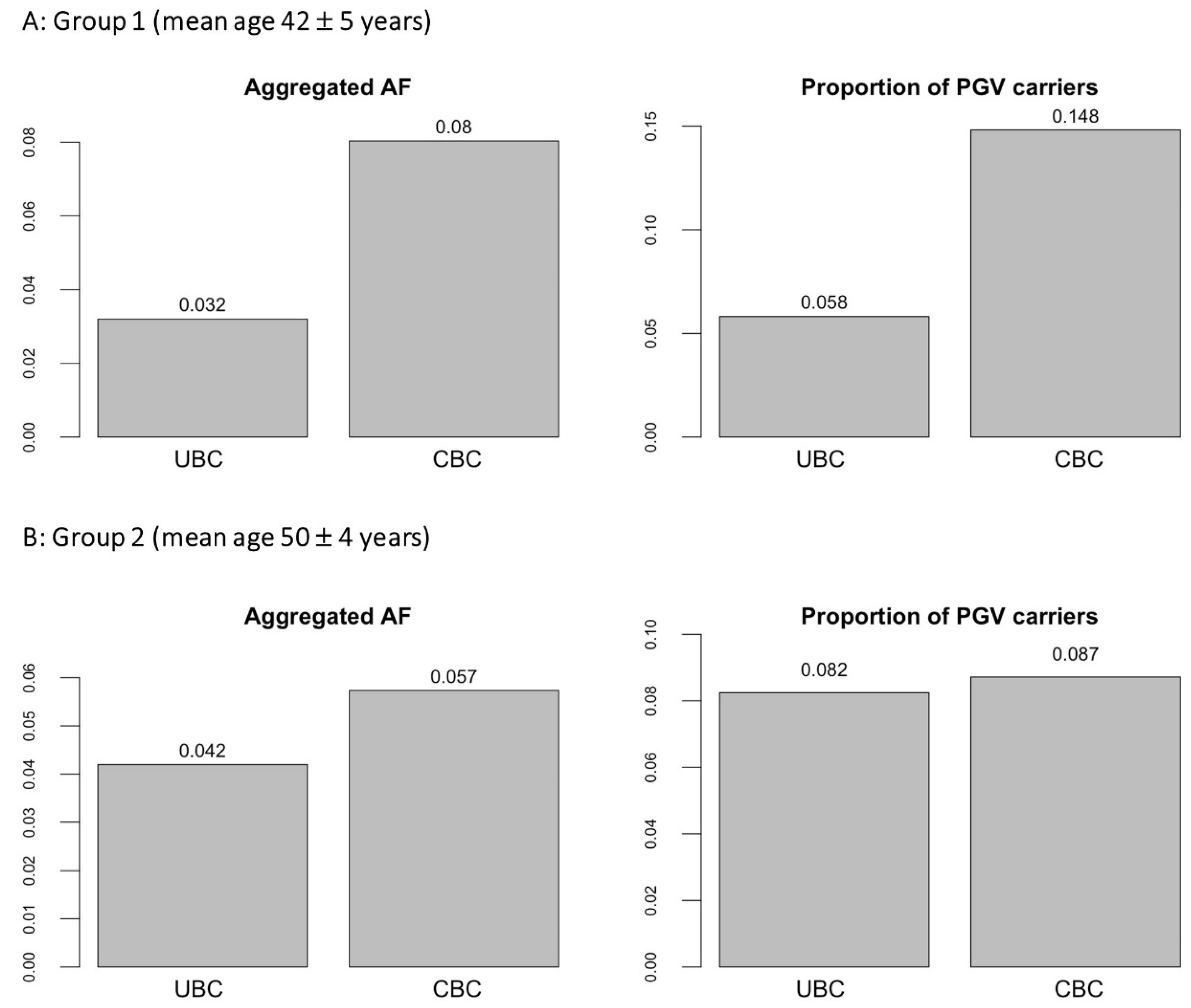
3. Results
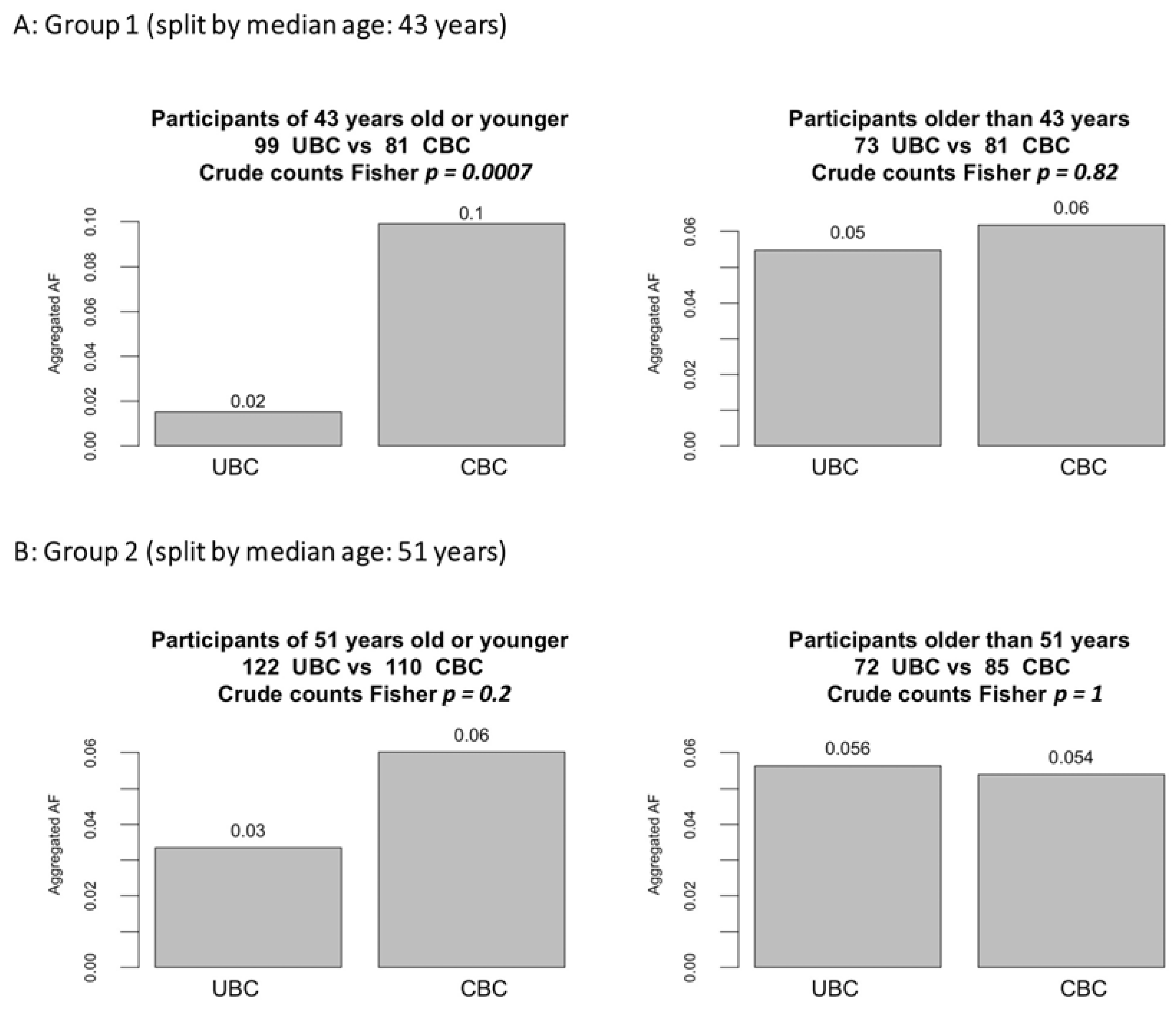
3.1. Subgroup Analyses by Age and Latency
3.2. Metanalysis and Comparison of PGVs Burdens with NFFE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bernstein, J.L.; Thompson, W.D.; Risch, N.; Holford, T.R. Risk factors predicting the incidence of second primary breast cancer among women diagnosed with a first primary breast cancer. Am. J. Epidemiol. 1992, 136, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A. Bilateral breast cancers. Nat. Rev. Clin. Oncol. 2014, 11, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Giannakeas, V.; Lim, D.W.; Narod, S.A. The risk of contralateral breast cancer: A SEER-based analysis. Br. J. Cancer 2021, 125, 601–610. [Google Scholar] [CrossRef]
- Reiner, A.S.; John, E.M.; Brooks, J.D.; Lynch, C.F.; Bernstein, L.; Mellemkjaer, L.; Malone, K.E.; Knight, J.A.; Capanu, M.; Teraoka, S.N.; et al. Risk of asynchronous contralateral breast cancer in noncarriers of BRCA1 and BRCA2 mutations with a family history of breast cancer: A report from the Women’s Environmental Cancer and Radiation Epidemiology Study. J. Clin. Oncol. 2013, 31, 433–439. [Google Scholar] [CrossRef]
- Hartman, M.; Czene, K.; Reilly, M.; Adolfsson, J.; Bergh, J.; Adami, H.O.; Dickman, P.W.; Hall, P. Incidence and prognosis of synchronous and metachronous bilateral breast cancer. J. Clin. Oncol. 2007, 25, 4210–4216. [Google Scholar] [CrossRef] [PubMed]
- Schaapveld, M.; Visser, O.; Louwman, W.J.; Willemse, P.H.; de Vries, E.G.; van der Graaf, W.T.; Otter, R.; Coebergh, J.W.; van Leeuwen, F.E. The impact of adjuvant therapy on contralateral breast cancer risk and the prognostic significance of contralateral breast cancer: A population based study in the Netherlands. Breast Cancer Res. Treat. 2008, 110, 189–197. [Google Scholar] [CrossRef]
- Nash, R.; Goodman, M.; Lin, C.C.; Freedman, R.A.; Dominici, L.S.; Ward, K.; Jemal, A. State Variation in the Receipt of a Contralateral Prophylactic Mastectomy Among Women Who Received a Diagnosis of Invasive Unilateral Early-Stage Breast Cancer in the United States, 2004–2012. JAMA Surg. 2017, 152, 648–657. [Google Scholar] [CrossRef]
- Chiesa, F.; Sacchini, V.S. Risk-reducing mastectomy. Minerva Ginecol. 2016, 68, 544–547. [Google Scholar]
- Liede, A.; Cai, M.; Crouter, T.F.; Niepel, D.; Callaghan, F.; Evans, D.G. Risk-reducing mastectomy rates in the US: A closer examination of the Angelina Jolie effect. Breast Cancer Res. Treat. 2018, 171, 435–442. [Google Scholar] [CrossRef]
- Basu, N.N.; Ross, G.L.; Evans, D.G.; Barr, L. The Manchester guidelines for contralateral risk-reducing mastectomy. World J. Surg. Oncol. 2015, 13, 237. [Google Scholar] [CrossRef]
- Chowdhury, M.; Euhus, D.; Onega, T.; Biswas, S.; Choudhary, P.K. A model for individualized risk prediction of contralateral breast cancer. Breast Cancer Res. Treat. 2017, 161, 153–160. [Google Scholar] [CrossRef]
- O’Donnell, M. Estimating contralateral breast cancer risk. Curr. Breast Cancer Rep. 2018, 10, 91–97. [Google Scholar] [CrossRef]
- Giardiello, D.; Hooning, M.J.; Hauptmann, M.; Keeman, R.; Heemskerk-Gerritsen, B.A.M.; Becher, H.; Blomqvist, C.; Bojesen, S.E.; Bolla, M.K.; Camp, N.J.; et al. PredictCBC-2.0: A contralateral breast cancer risk prediction model developed and validated in ~200,000 patients. Breast Cancer Res. 2022, 24, 69. [Google Scholar] [CrossRef]
- Giardiello, D.; Hauptmann, M.; Steyerberg, E.W.; Adank, M.A.; Akdeniz, D.; Blom, J.C.; Blomqvist, C.; Bojesen, S.E.; Bolla, M.K.; Brinkhuis, M.; et al. Prediction of contralateral breast cancer: External validation of risk calculators in 20 international cohorts. Breast Cancer Res. Treat. 2020, 181, 423–434. [Google Scholar] [CrossRef]
- Reiner, A.S.; Sisti, J.; John, E.M.; Lynch, C.F.; Brooks, J.D.; Mellemkjaer, L.; Boice, J.D.; Knight, J.A.; Concannon, P.; Capanu, M.; et al. Breast Cancer Family History and Contralateral Breast Cancer Risk in Young Women: An Update From the Women’s Environmental Cancer and Radiation Epidemiology Study. J. Clin. Oncol. 2018, 36, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Kramer, I.; Hooning, M.J.; Mavaddat, N.; Hauptmann, M.; Keeman, R.; Steyerberg, E.W.; Giardiello, D.; Antoniou, A.C.; Pharoah, P.D.P.; Canisius, S.; et al. Breast Cancer Polygenic Risk Score and Contralateral Breast Cancer Risk. Am. J. Hum. Genet. 2020, 107, 837–848. [Google Scholar] [CrossRef]
- Gronwald, J.; Tung, N.; Foulkes, W.D.; Offit, K.; Gershoni, R.; Daly, M.; Kim-Sing, C.; Olsson, H.; Ainsworth, P.; Eisen, A.; et al. Tamoxifen and contralateral breast cancer in BRCA1 and BRCA2 carriers: An update. Int. J. Cancer 2006, 118, 2281–2284. [Google Scholar] [CrossRef]
- Reding, K.W.; Bernstein, J.L.; Langholz, B.M.; Bernstein, L.; Haile, R.W.; Begg, C.B.; Lynch, C.F.; Concannon, P.; Borg, A.; Teraoka, S.N.; et al. Adjuvant systemic therapy for breast cancer in BRCA1/BRCA2 mutation carriers in a population-based study of risk of contralateral breast cancer. Breast Cancer Res. Treat. 2010, 123, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.D.; Teraoka, S.N.; Malone, K.E.; Haile, R.W.; Bernstein, L.; Lynch, C.F.; Mellemkjaer, L.; Duggan, D.J.; Reiner, A.S.; Concannon, P.; et al. Variants in tamoxifen metabolizing genes: A case-control study of contralateral breast cancer risk in the WECARE study. Int. J. Mol. Epidemiol. Genet. 2013, 4, 35–48. [Google Scholar]
- Bernstein, J.L.; Haile, R.W.; Stovall, M.; Boice, J.D., Jr.; Shore, R.E.; Langholz, B.; Thomas, D.C.; Bernstein, L.; Lynch, C.F.; Olsen, J.H.; et al. Radiation exposure, the ATM Gene, and contralateral breast cancer in the women’s environmental cancer and radiation epidemiology study. J. Natl. Cancer Inst. 2010, 102, 475–483. [Google Scholar] [CrossRef]
- Bertelsen, L.; Bernstein, L.; Olsen, J.H.; Mellemkjaer, L.; Haile, R.W.; Lynch, C.F.; Malone, K.E.; Anton-Culver, H.; Christensen, J.; Langholz, B.; et al. Effect of systemic adjuvant treatment on risk for contralateral breast cancer in the Women’s Environment, Cancer and Radiation Epidemiology Study. J. Natl. Cancer Inst. 2008, 100, 32–40. [Google Scholar] [CrossRef]
- Langballe, R.; Mellemkjaer, L.; Malone, K.E.; Lynch, C.F.; John, E.M.; Knight, J.A.; Bernstein, L.; Brooks, J.; Andersson, M.; Reiner, A.S.; et al. Systemic therapy for breast cancer and risk of subsequent contralateral breast cancer in the WECARE Study. Breast Cancer Res. 2016, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, O.; Johnson, N.; Dos Santos Silva, I.; Kilpivaara, O.; Aittomaki, K.; Blomqvist, C.; Nevanlinna, H.; Wasielewski, M.; Meijers-Heijerboer, H.; Broeks, A.; et al. Family history, genetic testing, and clinical risk prediction: Pooled analysis of CHEK2 1100delC in 1,828 bilateral breast cancers and 7030 controls. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, K.; Gershman, S.; Lynch, H.T.; Ghadirian, P.; Tung, N.; Kim-Sing, C.; Olopade, O.I.; Domchek, S.; McLennan, J.; Eisen, A.; et al. Predictors of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2011, 104, 1384–1392. [Google Scholar] [CrossRef]
- Malone, K.E.; Begg, C.B.; Haile, R.W.; Borg, A.; Concannon, P.; Tellhed, L.; Xue, S.; Teraoka, S.; Bernstein, L.; Capanu, M.; et al. Population-based study of the risk of second primary contralateral breast cancer associated with carrying a mutation in BRCA1 or BRCA2. J. Clin. Oncol. 2010, 28, 2404–2410. [Google Scholar] [CrossRef] [PubMed]
- Tischkowitz, M.; Capanu, M.; Sabbaghian, N.; Li, L.; Liang, X.; Vallee, M.P.; Tavtigian, S.V.; Concannon, P.; Foulkes, W.D.; Bernstein, L.; et al. Rare germline mutations in PALB2 and breast cancer risk: A population-based study. Hum. Mutat. 2012, 33, 674–680. [Google Scholar] [CrossRef]
- Reiner, A.S.; Robson, M.E.; Mellemkjaer, L.; Tischkowitz, M.; John, E.M.; Lynch, C.F.; Brooks, J.D.; Boice, J.D.; Knight, J.A.; Teraoka, S.N.; et al. Radiation Treatment, ATM, BRCA1/2, and CHEK2*1100delC Pathogenic Variants and Risk of Contralateral Breast Cancer. J. Natl. Cancer Inst. 2020, 112, 1275–1279. [Google Scholar] [CrossRef]
- Mellemkjaer, L.; Dahl, C.; Olsen, J.H.; Bertelsen, L.; Guldberg, P.; Christensen, J.; Borresen-Dale, A.L.; Stovall, M.; Langholz, B.; Bernstein, L.; et al. Risk for contralateral breast cancer among carriers of the CHEK2*1100delC mutation in the WECARE Study. Br. J. Cancer 2008, 98, 728–733. [Google Scholar] [CrossRef]
- Bernstein, J.L.; Langholz, B.; Haile, R.W.; Bernstein, L.; Thomas, D.C.; Stovall, M.; Malone, K.E.; Lynch, C.F.; Olsen, J.H.; Anton-Culver, H.; et al. Study design: Evaluating gene-environment interactions in the etiology of breast cancer—The WECARE study. Breast Cancer Res. 2004, 6, R199–R214. [Google Scholar] [CrossRef]
- Borg, A.; Haile, R.W.; Malone, K.E.; Capanu, M.; Diep, A.; Torngren, T.; Teraoka, S.; Begg, C.B.; Thomas, D.C.; Concannon, P.; et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: The WECARE study. Hum. Mutat. 2010, 31, E1200–E1240. [Google Scholar] [CrossRef]
- Robson, M.E.; Reiner, A.S.; Brooks, J.D.; Concannon, P.J.; John, E.M.; Mellemkjaer, L.; Bernstein, L.; Malone, K.E.; Knight, J.A.; Lynch, C.F.; et al. Association of Common Genetic Variants With Contralateral Breast Cancer Risk in the WECARE Study. J. Natl. Cancer Inst. 2017, 109, djx051. [Google Scholar] [CrossRef] [PubMed]
- Sisti, J.S.; Bernstein, J.L.; Lynch, C.F.; Reiner, A.S.; Mellemkjaer, L.; Brooks, J.D.; Knight, J.A.; Bernstein, L.; Malone, K.E.; Woods, M.; et al. Reproductive factors, tumor estrogen receptor status and contralateral breast cancer risk: Results from the WECARE study. Springerplus 2015, 4, 825. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.C.; Brooks, J.D.; Conti, D.V.; Poynter, J.N.; Teraoka, S.N.; Malone, K.E.; Bernstein, L.; Lee, W.D.; Duggan, D.J.; Siniard, A.; et al. Risk of contralateral breast cancer associated with common variants in BRCA1 and BRCA2: Potential modifying effect of BRCA1/BRCA2 mutation carrier status. Breast Cancer Res. Treat. 2011, 127, 819–829. [Google Scholar] [CrossRef]
- Poynter, J.N.; Langholz, B.; Largent, J.; Mellemkjaer, L.; Bernstein, L.; Malone, K.E.; Lynch, C.F.; Borg, A.; Concannon, P.; Teraoka, S.N.; et al. Reproductive factors and risk of contralateral breast cancer by BRCA1 and BRCA2 mutation status: Results from the WECARE study. Cancer Causes Control 2010, 21, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Konig, K.; Peifer, M.; Fassunke, J.; Ihle, M.A.; Kunstlinger, H.; Heydt, C.; Stamm, K.; Ueckeroth, F.; Vollbrecht, C.; Bos, M.; et al. Implementation of Amplicon Parallel Sequencing Leads to Improvement of Diagnosis and Therapy of Lung Cancer Patients. J. Thorac. Oncol. 2015, 10, 1049–1057. [Google Scholar] [CrossRef]
- Clarke, L.; Fairley, S.; Zheng-Bradley, X.; Streeter, I.; Perry, E.; Lowy, E.; Tasse, A.M.; Flicek, P. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017, 45, D854–D859. [Google Scholar] [CrossRef]
- Hadfield, J.; Eldridge, M.D. Multi-genome alignment for quality control and contamination screening of next-generation sequencing data. Front. Genet. 2014, 5, 31. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v1302. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Breast Cancer Association, C.; Dorling, L.; Carvalho, S.; Allen, J.; Gonzalez-Neira, A.; Luccarini, C.; Wahlstrom, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; et al. Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Wu, M.C.; Lee, S.; Cai, T.; Li, Y.; Boehnke, M.; Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011, 89, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Prive, F.; Luu, K.; Blum, M.G.B.; McGrath, J.J.; Vilhjalmsson, B.J. Efficient toolkit implementing best practices for principal component analysis of population genetic data. Bioinformatics 2020, 36, 4449–4457. [Google Scholar] [CrossRef] [PubMed]
- Venables, W.N.; Ripley, B.D. Modern applied statistics with S-PLUS, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.K.; Clifford, J.; Li, S.; LaDuca, H.; Hulick, P.; Gutierrez, S.; Black, M.H. Prevalence of Germline Pathogenic and Likely Pathogenic Variants in Patients With Second Breast Cancers. JNCI Cancer Spectr. 2020, 4, pkaa094. [Google Scholar] [CrossRef]
- Fanale, D.; Incorvaia, L.; Filorizzo, C.; Bono, M.; Fiorino, A.; Calo, V.; Brando, C.; Corsini, L.R.; Barraco, N.; Badalamenti, G.; et al. Detection of Germline Mutations in a Cohort of 139 Patients with Bilateral Breast Cancer by Multi-Gene Panel Testing: Impact of Pathogenic Variants in Other Genes beyond BRCA1/2. Cancers 2020, 12, 2415. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Parameters | Group 1 | Group 2 | |||||
|---|---|---|---|---|---|---|---|
| UBC | CBC | p | UBC | CBC | p | ||
| Samples Count | 172 | 162 | - | 194 | 195 | - | |
| Age at the 1st cancer | 42 ± 5.5 | 42 ± 5.4 | 0.29 | 50 ± 4.1 | 50 ± 3.9 | 0.19 | |
| Time to the 2nd cancer or lack of it | 6.2 ± 3.5 | 6.2 ± 3.5 | 0.88 | 4.6 ± 2.8 | 4.6 ± 2.8 | 0.87 | |
| Stage | 1 | 118 | 119 | 0.34 | 143 | 144 | 1.00 |
| 2 | 54 | 43 | 51 | 51 | |||
| Estrogen receptors | Neg | 47 | 34 | 0.31 | 47 | 53 | 0.68 |
| Pos | 92 | 89 | 116 | 108 | |||
| Unknown | 33 | 39 | 31 | 34 | |||
| Cytotoxic chemotherapy | No | 75 | 83 | 0.19 | 99 | 113 | 0.19 |
| Yes | 97 | 79 | 95 | 82 | |||
| Hormonal therapy | No | 115 | 113 | 0.55 | 95 | 113 | 0.08 |
| Yes | 56 | 49 | 99 | 82 | |||
| Unknown | 1 | 0 | - | - | |||
| Breast irradiation | No | 72 | 89 | 0.02 | 63 | 130 | <0.001 |
| Yes | 100 | 73 | 131 | 65 | |||
| Number of pregnancies | None | 26 | 38 | 0.04 | 32 | 36 | 0.78 |
| 1–2 | 123 | 113 | 86 | 80 | |||
| 3 or more | 23 | 11 | 76 | 79 | |||
| Family history | No | 117 | 97 | 0.14 | 133 | 132 | 0.98 |
| Yes | 55 | 65 | 54 | 56 | |||
| Unknown | - | - | 7 | 7 | |||
| Gene | Variant Count | UBC | CBC | ||||
|---|---|---|---|---|---|---|---|
| Aggregated AC | Mean AN | Aggregated AF | Aggregated AC | Mean AN | Aggregated AF | ||
| ATM | 11 | 7 | 344.0 | 0.020 | 10 | 323.5 | 0.031 |
| CHEK2 | 5 | 2 | 344.0 | 0.006 | 7 | 324.0 | 0.022 |
| NF1 | 2 | 0 | 344.0 | 0.000 | 2 | 324.0 | 0.006 |
| RAD51D | 2 | 2 | 343.0 | 0.006 | 2 | 324.0 | 0.006 |
| NBN | 1 | 0 | 344 | 0.0000 | 2 | 324 | 0.0062 |
| CDH1 | 1 | 0 | 344 | 0.0000 | 1 | 324 | 0.0031 |
| TP53 | 1 | 0 | 336 | 0.0000 | 1 | 320 | 0.0031 |
| RAD51C | 1 | 0 | 344 | 0.0000 | 1 | 324 | 0.0031 |
| Total | 24 | 11 | 343.6 | 0.032 | 26 | 323.6 | 0.080 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larionov, A.; Fewings, E.; Redman, J.; Goldgraben, M.; Clark, G.; Boice, J.; Concannon, P.; Bernstein, J.; Conti, D.V.; the WECARE Study Collaborative Group; et al. The Contribution of Germline Pathogenic Variants in Breast Cancer Genes to Contralateral Breast Cancer Risk in BRCA1/BRCA2/PALB2-Negative Women. Cancers 2023, 15, 415. https://doi.org/10.3390/cancers15020415
Larionov A, Fewings E, Redman J, Goldgraben M, Clark G, Boice J, Concannon P, Bernstein J, Conti DV, the WECARE Study Collaborative Group, et al. The Contribution of Germline Pathogenic Variants in Breast Cancer Genes to Contralateral Breast Cancer Risk in BRCA1/BRCA2/PALB2-Negative Women. Cancers. 2023; 15(2):415. https://doi.org/10.3390/cancers15020415
Chicago/Turabian StyleLarionov, Alexey, Eleanor Fewings, James Redman, Mae Goldgraben, Graeme Clark, John Boice, Patrick Concannon, Jonine Bernstein, David V. Conti, the WECARE Study Collaborative Group, and et al. 2023. "The Contribution of Germline Pathogenic Variants in Breast Cancer Genes to Contralateral Breast Cancer Risk in BRCA1/BRCA2/PALB2-Negative Women" Cancers 15, no. 2: 415. https://doi.org/10.3390/cancers15020415
APA StyleLarionov, A., Fewings, E., Redman, J., Goldgraben, M., Clark, G., Boice, J., Concannon, P., Bernstein, J., Conti, D. V., the WECARE Study Collaborative Group, & Tischkowitz, M. (2023). The Contribution of Germline Pathogenic Variants in Breast Cancer Genes to Contralateral Breast Cancer Risk in BRCA1/BRCA2/PALB2-Negative Women. Cancers, 15(2), 415. https://doi.org/10.3390/cancers15020415