
Unraveling the Synergy between Atezolizumab and Bevacizumab for the Treatment of Hepatocellular Carcinoma

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. The Current Position of Systemic Agents in the Treatment of HCC
3. Antiangiogenics in the Treatment of HCC
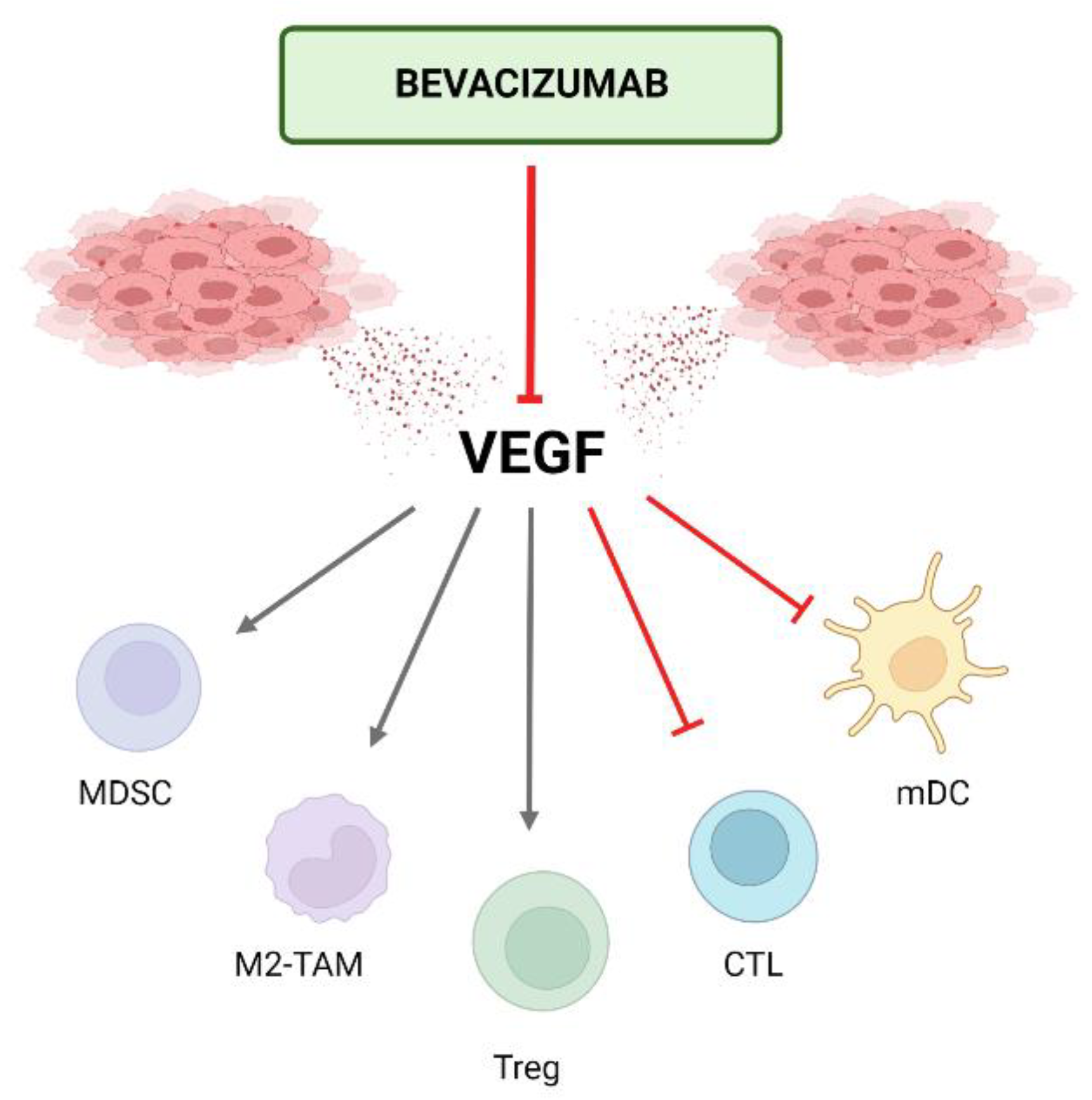
3.1. Mechanism of Action of Antiangiogenics
3.2. Clinical Efficacy of Antiangiogenics in HCC
3.3. Potential Biomarkers for Antiangiogenics in HCC
4. Immune Checkpoint Inhibition in the Treatment of HCC
4.1. Mechanism of Action of Immune Checkpoint Inhibitors
4.2. Clinical Efficacy of Immune Checkpoint Inhibitors in HCC
4.2.1. PD-1/PD-L1 Inhibitors
4.2.2. CTLA-4 Inhibitors
4.2.3. Other Immune Checkpoint Inhibitors
4.2.4. ICI-Combination Therapies
4.3. Potential Biomarkers for Immune Checkpoint Inhibitors in HCC
5. Atezolizumab with Bevacizumab for the Treatment of HCC
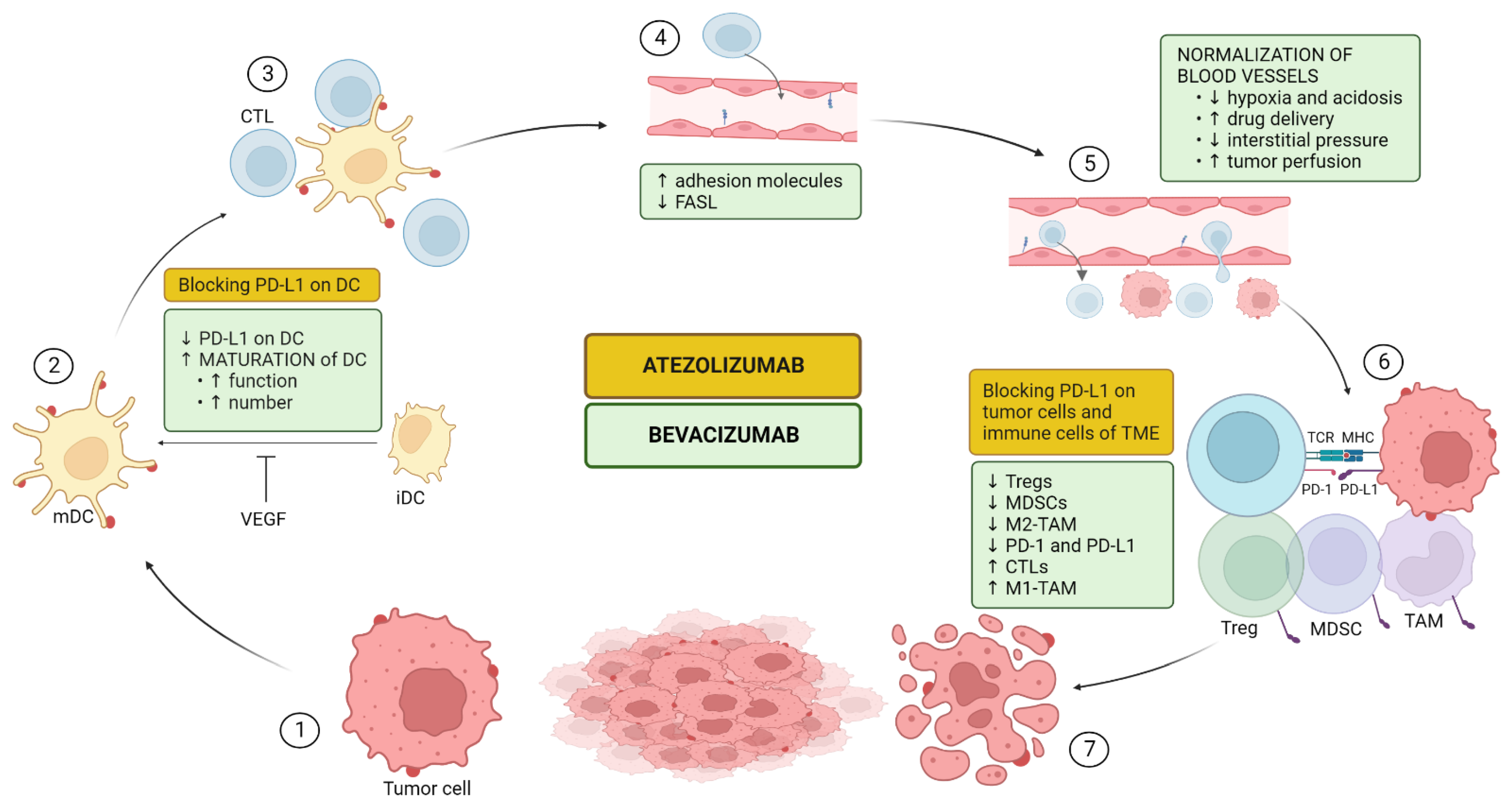
5.1. Mechanism of Action of the Combination
5.1.1. Upregulation of Antigen Presentation Via Dendritic Cell (DC) Maturation and Functioning (CIC Steps 1–3)
5.1.2. Upregulation of T-Cell Proliferation, Trafficking, and Infiltration (CIC Steps 4–5)
5.1.3. Impairing Recruitment and Proliferation of Immunosuppressive Cells (CIC Steps 6–7)
5.2. Clinical Efficacy of Atezolizumab with Bevacizumab
5.3. Potential Biomarkers for Combination Therapy of Atezolizumab with Bevacizumab

{kind=link}
{kind=link}
| Antiangiogenics | Immune Checkpoint Inhibitors | Atezolizumab/Bevacizumab | |||
|---|---|---|---|---|---|
| HCC | RCC | HCC | RCC | HCC | RCC |
| AFP (in particular for ramucirumab; [16,39] | Soluble VEGF [50,51,52] | Expression of PD-L1 e.g., TPS, CPS [9,10] | NLR-ratio [96,97] | Immune cell signature (genes linked to the adaptive and innate immune system) corresponding to upregulated PD-L1 expression and effector T cells [116] | Gene expression signatures reflecting the high expression of effector T cells and high myeloid infiltration in tumor tissue [58] |
| Soluble VEGF-A [45] | SNP in VEGFR1 [53,54] | Downregulated Wnt/β-catenin signaling e.g., CTNNB1-wt [80,86] | Transcriptomic immune-related gene signatures [59] | High CD8+ T cell infiltration and PD-L1 expression on immunohistochemistry [116] | |
| Ang2 [47,48] | Transcriptomic angiogenesis-related gene signatures including e.g., KDR [57,58,59] | High TMB [89,90] | Single-cell TCR-sequencing (in particular maintenance of TCR-clonality) [103] | Low levels of GPC3 and AFP [116] | |
| IFN-y gene signaling [91] | CTNNB1-wt or TERT-mutation [116] | ||||
| Single-cell TCR-sequencing: TCR-clonality and TCR-sharing between tumor and blood [92] | |||||
| CXCR + CD8+ effector memory T cells in blood [93] | |||||
| CD103+ tissue-resident memory T-cells [78] | |||||
6. Conclusions and Further Directions
Author Contributions
Funding
Conflicts of Interest
References
- Qin, S.; Bai, Y.; Lim, H.Y.; Thongprasert, S.; Chao, Y.; Fan, J.; Yang, T.S.; Bhudhisawasdi, V.; Kang, W.K.; Zhou, Y.; et al. Randomized, multicenter, open-label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J. Clin. Oncol. 2013, 31, 3501–3508. [Google Scholar] [CrossRef] [PubMed]
- El Dika, I.; Capanu, M.; Chou, J.F.; Harding, J.J.; Ly, M.; Hrabovsky, A.D.; Do, R.K.G.; Shia, J.; Millang, B.; Ma, J.; et al. Phase II trial of sorafenib and doxorubicin in patients with advanced hepatocellular carcinoma after disease progression on sorafenib. Cancer Med. 2020, 9, 7453–7459. [Google Scholar] [CrossRef] [PubMed]
- Lohitesh, K.; Chowdhury, R.; Mukherjee, S. Resistance a major hindrance to chemotherapy in hepatocellular carcinoma: An insight. Cancer Cell Int. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Sandhu, S.; Lai, J.P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Trojan, J.; Sarrazin, C. Complete Response of Hepatocellular Carcinoma in a Patient With End-stage Liver Disease treated With Nivolumab: Whishful thinking or Possible? Am. J. Gastroenterol. 2016, 111, 1208–1209. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.; Chan, S.; Kudo, M.; Lau, G.; Kelley, R.; Furuse, J. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. In Proceedings of the 2022 Gastrointestinal Cancers Symposium, San Francisco, CA, USA, 20–22 January 2022. [Google Scholar]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC strategy for prognosis prediction and treatment recommendation: The 2022 update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef]
- Şenbabaoğlu, Y.; Gejman, R.S.; Winer, A.G.; Liu, M.; Van Allen, E.M.; de Velasco, G.; Miao, D.; Ostrovnaya, I.; Drill, E.; Luna, A.; et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016, 17, 231. [Google Scholar] [CrossRef]
- Powles, T.; Albiges, L.; Bex, A.; Grünwald, V.; Porta, C.; Procopio, G.; Schmidinger, M.; Suárez, C.; de Velasco, G.; on behalf of the ESMO Guidelines Committee. ESMO Clinical Practice Guideline update on the use of immunotherapy in early stage and advanced renal cell carcinoma. Ann. Oncol. 2021, 32, 1511–1519. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Hanna, D.L.; Llovet, J.; Kelley, R.K. Cabozantinib: An evolving therapy for hepatocellular carcinoma. Cancer Treat. Rev. 2021, 98, 102221. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Galle, R.; Forner, A.; Llovet, J.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Llovet, J.M.; De Baere, T.; Kulik, L.; Haber, P.K.; Greten, T.F.; Meyer, T.; Lencioni, R. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 293–313. [Google Scholar] [CrossRef]
- Katariya, N.N.; Lizaola-Mayo, B.C.; Chascsa, D.M.; Giorgakis, E.; Aqel, B.A.; Moss, A.A.; Uson Junior, P.L.S.; Borad, M.J.; Mathur, A.K. Immune Checkpoint Inhibitors as Therapy to Down-Stage Hepatocellular Carcinoma Prior to Liver Transplantation. Cancers 2022, 14, 2056. [Google Scholar] [CrossRef] [PubMed]
- Berretta, M.; Rinaldi, L.; Di Benedetto, F.; Lleshi, A.; De Re, V.; Facchini, G.; De Paoli, P.; Di Francia, R. Angiogenesis Inhibitors for the Treatment of Hepatocellular Carcinoma. Front. Pharmacol. 2016, 7, 428. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A.; Adamek, A. Role of Endoglin (CD105) in the Progression of Hepatocellular Carcinoma and Anti-Angiogenic Therapy. Int. J. Mol. Sci. 2018, 19, 3887. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Tozer, G.M.; Kanthou, C.; Baguley, B.C. Disrupting tumour blood vessels. Nat. Rev. Cancer 2005, 5, 423–435. [Google Scholar] [CrossRef]
- Al-Abd, A.M.; Alamoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Augustin, H.G.; Koh, G.Y. Antiangiogenesis: Vessel Regression, Vessel Normalization, or Both? Cancer Res. 2022, 82, 15–17. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, L.G.; Reig, M.; Bruix, J. Tyrosine Kinase Inhibitors and Hepatocellular Carcinoma. Clin. Liver Dis. 2020, 24, 719–737. [Google Scholar] [CrossRef]
- Fang, P.; Hu, J.H.; Cheng, Z.G.; Liu, Z.F.; Wang, J.L.; Jiao, S.C. Efficacy and safety of bevacizumab for the treatment of advanced hepatocellular carcinoma: A systematic review of phase II trials. PLoS ONE 2012, 7, e49717. [Google Scholar] [CrossRef]
- Van Wynsberghe, M.; Flejeo, J.; Sakhi, H.; Ollero, M.; Sahali, D.; Izzedine, H.; Henique, C. Nephrotoxicity of Anti-Angiogenic Therapies. Diagnostics 2021, 11, 640. [Google Scholar] [CrossRef]
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The Role of Angiogenesis in Hepatocellular Carcinoma. Clin. Cancer Res. 2019, 25, 912–920. [Google Scholar] [CrossRef]
- Singal, A.G.; Hoshida, Y.; Pinato, D.J.; Marrero, J.; Nault, J.C.; Paradis, V.; Tayob, N.; Sherman, M.; Lim, Y.S.; Feng, Z.; et al. International Liver Cancer Association (ILCA) White Paper on Biomarker Development for Hepatocellular Carcinoma. Gastroenterology 2021, 160, 2572–2584. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Montal, R.; Andreu-Oller, C.; Bassaganyas, L.; Esteban-Fabró, R.; Moran, S.; Montironi, C.; Moeini, A.; Pinyol, R.; Peix, J.; Cabellos, L.; et al. Molecular portrait of high alpha-fetoprotein in hepatocellular carcinoma: Implications for biomarker-driven clinical trials. Br. J. Cancer 2019, 121, 340–343. [Google Scholar] [CrossRef]
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.X.; et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008, 68, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Sala, M.; Forner, A.; Varela, M.; Bruix, J. Prognostic prediction in patients with hepatocellular carcinoma. Semin. Liver Dis. 2005, 25, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.Y.; Hsu, C.H.; Cheng, A.L. Predictive biomarkers of sorafenib efficacy in advanced hepatocellular carcinoma: Are we getting there? World J. Gastroenterol. 2015, 21, 10336–10347. [Google Scholar] [CrossRef]
- Sánchez, A.I.P.; Roces, L.V.; García, I.Z.; López, E.L.; Hernandez, M.A.C.; Parejo, M.I.B.; Peña-Díaz, J. Value of α-fetoprotein as an early biomarker for treatment response to sorafenib therapy in advanced hepatocellular carcinoma. Oncol. Lett. 2018, 15, 8863–8870. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Peña, C.E.; Lathia, C.D.; Shan, M.; Meinhardt, G.; Bruix, J.; Group, S.I.S. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 2012, 18, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Pircher, A.; Hilbe, W.; Heidegger, I.; Drevs, J.; Tichelli, A.; Medinger, M. Biomarkers in tumor angiogenesis and anti-angiogenic therapy. Int. J. Mol. Sci. 2011, 12, 7077–7099. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, K.; Nouso, K.; Tomoda, T.; Kobayashi, S.; Hagihara, H.; Kuwaki, K.; Toshimori, J.; Onishi, H.; Ikeda, F.; Miyake, Y.; et al. Predicting the treatment effect of sorafenib using serum angiogenesis markers in patients with hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 1604–1611. [Google Scholar] [CrossRef]
- Sun, H.C.; Tang, Z.Y. Angiogenesis in hepatocellular carcinoma: The retrospectives and perspectives. J. Cancer Res. Clin. Oncol. 2004, 130, 307–319. [Google Scholar] [CrossRef]
- Verbiest, A.; Renders, I.; Caruso, S.; Couchy, G.; Job, S.; Laenen, A.; Verkarre, V.; Rioux-Leclercq, N.; Schöffski, P.; Vano, Y.; et al. Clear-cell Renal Cell Carcinoma: Molecular Characterization of IMDC Risk Groups and Sarcomatoid Tumors. Clin. Genitourin. Cancer 2019, 17, e981–e994. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A.A.; Rolland, F.; et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J. Clin. Oncol. 2009, 27, 3312–3318. [Google Scholar] [CrossRef]
- Deprimo, S.E.; Bello, C.L.; Smeraglia, J.; Baum, C.M.; Spinella, D.; Rini, B.I.; Michaelson, M.D.; Motzer, R.J. Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: Modulation of VEGF and VEGF-related proteins. J. Transl. Med. 2007, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Michaelson, M.D.; Rosenberg, J.E.; Bukowski, R.M.; Sosman, J.A.; Stadler, W.M.; Hutson, T.E.; Margolin, K.; Harmon, C.S.; DePrimo, S.E.; et al. Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J. Clin. Oncol. 2008, 26, 3743–3748. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Donas, J.; Esteban, E.; Leandro-García, L.J.; Castellano, D.E.; González del Alba, A.; Climent, M.A.; Arranz, J.A.; Gallardo, E.; Puente, J.; Bellmunt, J.; et al. Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: A multicentre, observational, prospective study. Lancet Oncol. 2011, 12, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Beuselinck, B.; Karadimou, A.; Lambrechts, D.; Claes, B.; Wolter, P.; Couchy, G.; Berkers, J.; Paridaens, R.; Schöffski, P.; Méjean, A.; et al. Single-nucleotide polymorphisms associated with outcome in metastatic renal cell carcinoma treated with sunitinib. Br. J. Cancer 2013, 108, 887–900. [Google Scholar] [CrossRef][Green Version]
- Lambrechts, D.; Claes, B.; Delmar, P.; Reumers, J.; Mazzone, M.; Yesilyurt, B.T.; Devlieger, R.; Verslype, C.; Tejpar, S.; Wildiers, H.; et al. VEGF pathway genetic variants as biomarkers of treatment outcome with bevacizumab: An analysis of data from the AViTA and AVOREN randomised trials. Lancet Oncol. 2012, 13, 724–733. [Google Scholar] [CrossRef]
- D’Aniello, C.; Berretta, M.; Cavaliere, C.; Rossetti, S.; Facchini, B.A.; Iovane, G.; Mollo, G.; Capasso, M.; Pepa, C.D.; Pesce, L.; et al. Biomarkers of Prognosis and Efficacy of Anti-angiogenic Therapy in Metastatic Clear Cell Renal Cancer. Front. Oncol. 2019, 9, 1400. [Google Scholar] [CrossRef] [PubMed]
- Beuselinck, B.; Verbiest, A.; Couchy, G.; Job, S.; de Reynies, A.; Meiller, C.; Albersen, M.; Verkarre, V.; Lerut, E.; Méjean, A.; et al. Pro-angiogenic gene expression is associated with better outcome on sunitinib in metastatic clear-cell renal cell carcinoma. Acta Oncol. 2018, 57, 498–508. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef]
- Beuselinck, B.; Job, S.; Becht, E.; Karadimou, A.; Verkarre, V.; Couchy, G.; Giraldo, N.; Rioux-Leclercq, N.; Molinié, V.; Sibony, M.; et al. Molecular subtypes of clear cell renal cell carcinoma are associated with sunitinib response in the metastatic setting. Clin. Cancer Res. 2015, 21, 1329–1339. [Google Scholar] [CrossRef]
- Motzer, R.J.; Banchereau, R.; Hamidi, H.; Powles, T.; McDermott, D.; Atkins, M.B.; Escudier, B.; Liu, L.F.; Leng, N.; Abbas, A.R.; et al. Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell 2020, 38, 803–817.e804. [Google Scholar] [CrossRef]
- Singla, N.; Xie, Z.; Zhang, Z.; Gao, M.; Yousuf, Q.; Onabolu, O.; McKenzie, T.; Tcheuyap, V.T.; Ma, Y.; Choi, J.; et al. Pancreatic tropism of metastatic renal cell carcinoma. JCI Insight 2020, 5, e134564. [Google Scholar] [CrossRef]
- Roussel, E.; Kinget, L.; Verbiest, A.; Boeckx, B.; Zucman-Rossi, J.; Couchy, G.; Caruso, S.; Baldewijns, M.; Joniau, S.; Van Poppel, H.; et al. Molecular underpinnings of glandular tropism in metastatic clear cell renal cell carcinoma: Therapeutic implications. Acta Oncol. 2021, 60, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Vano, Y.A.; Elaidi, R.; Bennamoun, M.; Chevreau, C.; Borchiellini, D.; Pannier, D.; Maillet, D.; Gross-Goupil, M.; Tournigand, C.; Laguerre, B.; et al. Nivolumab, nivolumab-ipilimumab, and VEGFR-tyrosine kinase inhibitors as first-line treatment for metastatic clear-cell renal cell carcinoma (BIONIKK): A biomarker-driven, open-label, non-comparative, randomised, phase 2 trial. Lancet Oncol. 2022, 23, 612–624. [Google Scholar] [CrossRef]
- Tran, J.; Ornstein, M.C. Clinical Review on the Management of Metastatic Renal Cell Carcinoma. JCO Oncol. Pract. 2022, 18, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Grünwald, V.; Gillessen, S.; Horwich, A.; ESMO Guidelines Committee. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Herzberg, B.; Campo, M.J.; Gainor, J.F. Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer. Oncologist 2017, 22, 81–88. [Google Scholar] [CrossRef]
- Schizas, D.; Charalampakis, N.; Kole, C.; Mylonas, K.S.; Katsaros, I.; Zhao, M.; Ajani, J.A.; Psyrri, A.; Karamouzis, M.V.; Liakakos, T. Immunotherapy for esophageal cancer: A 2019 update. Immunotherapy 2020, 12, 203–218. [Google Scholar] [CrossRef]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef]
- Kudo, M. Scientific Rationale for Combination Immunotherapy of Hepatocellular Carcinoma with Anti-PD-1/PD-L1 and Anti-CTLA-4 Antibodies. Liver Cancer 2019, 8, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, M.; Xiang, R. Clonal replacement of novel T cells: A new phenomenon in the tumor microenvironment following PD-1 blockade. Signal Transduct. Target. Ther. 2019, 4, 43. [Google Scholar] [CrossRef]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, B.G.; Pallett, L.J.; Li, X.; Davies, S.P.; Amin, O.E.; Gill, U.S.; Kucykowicz, S.; Patel, A.M.; Aliazis, K.; Liu, Y.S.; et al. The human liver microenvironment shapes the homing and function of CD4. Gut 2022, 71, 1399–1411. [Google Scholar] [CrossRef]
- Barsch, M.; Salié, H.; Schlaak, A.E.; Zhang, Z.; Hess, M.; Mayer, L.S.; Tauber, C.; Otto-Mora, P.; Ohtani, T.; Nilsson, T.; et al. T-cell exhaustion and residency dynamics inform clinical outcomes in hepatocellular carcinoma. J. Hepatol. 2022, 77, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sánchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chen, Z.; Fang, W.; Ren, Z.; Xu, R.; Ryoo, B.Y.; Hao, C. Pembrolizumab (pembro) plus best supportive care (BSC) versus placebo plus BSC as second-line therapy in patients in Asia with advanced hepatocellular carcinoma (HCC): Phase 3 KEYNOTE-394 study. In Proceedings of the American Society of Clinical Oncology Gastrointestinal Cancers Symposium (ASCO GI), San Francisco, CA, USA, 20–22 January 2022. [Google Scholar]
- Qin, S.; Kudo, M.; Meyer, T.; Finn, R.; Vogel, A.; Bai, Y.; Guo, Y.; Meng, Z.; Zhang, T.; Satoh, T.; et al. Final analysis of RATIONALE-301: Randomized, phase III study of tislelizumab versus sorafenib as first-line treatment for unresectable hepatocellular carcinoma. Ann. Oncol. 2022, 33 (Suppl. S7), S808–S869. [Google Scholar] [CrossRef]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- Fulgenzi, C.A.M.; Talbot, T.; Murray, S.M.; Silletta, M.; Vincenzi, B.; Cortellini, A.; Pinato, D.J. Immunotherapy in Hepatocellular Carcinoma. Curr. Treat. Options Oncol. 2021, 22, 87. [Google Scholar] [CrossRef] [PubMed]
- Ganjalikhani Hakemi, M.; Jafarinia, M.; Azizi, M.; Rezaeepoor, M.; Isayev, O.; Bazhin, A.V. The Role of TIM-3 in Hepatocellular Carcinoma: A Promising Target for Immunotherapy? Front. Oncol. 2020, 10, 601661. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. LAG3 inhibition improves outcomes. Nat. Rev. Clin. Oncol. 2022, 19, 149. [Google Scholar] [CrossRef]
- Saung, M.T.; Pelosof, L.; Casak, S.; Donoghue, M.; Lemery, S.; Yuan, M.; Rodriguez, L.; Schotland, P.; Chuk, M.; Davis, G.; et al. FDA Approval Summary: Nivolumab Plus Ipilimumab for the Treatment of Patients with Hepatocellular Carcinoma Previously Treated with Sorafenib. Oncologist 2021, 26, 797–806. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K.; Duda, D.G. The Current Landscape of Immune Checkpoint Blockade in Hepatocellular Carcinoma: A Review. JAMA Oncol. 2021, 7, 113–123. [Google Scholar] [CrossRef]
- Jung, H.I.; Jeong, D.; Ji, S.; Ahn, T.S.; Bae, S.H.; Chin, S.; Chung, J.C.; Kim, H.C.; Lee, M.S.; Baek, M.J. Overexpression of PD-L1 and PD-L2 Is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma. Cancer Res. Treat. 2017, 49, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Carretero-González, A.; Lora, D.; Martín Sobrino, I.; Sáez Sanz, I.; Bourlon, M.T.; Anido Herranz, U.; Martínez Chanzá, N.; Castellano, D.; de Velasco, G. The Value of PD-L1 Expression as Predictive Biomarker in Metastatic Renal Cell Carcinoma Patients: A Meta-Analysis of Randomized Clinical Trials. Cancers 2020, 12, 1945. [Google Scholar] [CrossRef]
- Ang, C.; Klempner, S.J.; Ali, S.M.; Madison, R.; Ross, J.S.; Severson, E.A.; Fabrizio, D.; Goodman, A.; Kurzrock, R.; Suh, J.; et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget 2019, 10, 4018–4025. [Google Scholar] [CrossRef]
- Wong, C.N.; Fessas, P.; Dominy, K.; Mauri, F.A.; Kaneko, T.; Parcq, P.D.; Khorashad, J.; Toniutto, P.; Goldin, R.D.; Avellini, C.; et al. Qualification of tumour mutational burden by targeted next-generation sequencing as a biomarker in hepatocellular carcinoma. Liver Int. 2021, 41, 192–203. [Google Scholar] [CrossRef]
- Haber, P.K.; Torres-Martin, M.; Dufour, J.F.; Verslype, C.; Marquardt, J.; Galle, P.R.; Vogel, A.; Meyer, T.; Labgaa, I.; Roberts, L.R.; et al. Molecular markers of response to anti-PD1 therapy in advanced hepatocellular carcinoma. J. Clin. Oncol. 2021, 39 (Suppl. S15), 4100. [Google Scholar] [CrossRef]
- Cappuyns, S.; Philips, G.; Vandecaveye, V.; Verslype, C.; Van Cutsem, E.; Lambrechts, D.; Dekervel, J. Pre-treatment cross-talk between the tumoural and peripheral immune system predicts response to checkpoint inhibition in advanced HCC: A single-cell study. J. Hepatol. 2022, 77, S385. [Google Scholar] [CrossRef]
- Chuah, S.; Lee, J.; Song, Y.; Kim, H.D.; Wasser, M.; Kaya, N.A.; Bang, K.; Lee, Y.J.; Jeon, S.H.; Suthen, S.; et al. Uncoupling immune trajectories of response and adverse events from anti-PD-1 immunotherapy in hepatocellular carcinoma. J. Hepatol. 2022, 77, 683–694. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.J.; McNamara, M.G.; Šeruga, B.; Vera-Badillo, F.E.; Aneja, P.; Ocaña, A.; Leibowitz-Amit, R.; Sonpavde, G.; Knox, J.J.; Tran, B.; et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2014, 106, dju124. [Google Scholar] [CrossRef] [PubMed]
- Lalani, A.A.; Xie, W.; Martini, D.J.; Steinharter, J.A.; Norton, C.K.; Krajewski, K.M.; Duquette, A.; Bossé, D.; Bellmunt, J.; Van Allen, E.M.; et al. Change in Neutrophil-to-lymphocyte ratio (NLR) in response to immune checkpoint blockade for metastatic renal cell carcinoma. J. Immunother. Cancer 2018, 6, 5. [Google Scholar] [CrossRef]
- Simonaggio, A.; Elaidi, R.; Fournier, L.; Fabre, E.; Ferrari, V.; Borchiellini, D.; Thouvenin, J.; Barthelemy, P.; Thibault, C.; Tartour, E.; et al. Variation in neutrophil to lymphocyte ratio (NLR) as predictor of outcomes in metastatic renal cell carcinoma (mRCC) and non-small cell lung cancer (mNSCLC) patients treated with nivolumab. Cancer Immunol. Immunother. 2020, 69, 2513–2522. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Mori, K.; Abufaraj, M.; Mostafaei, H.; Quhal, F.; Fajkovic, H.; Remzi, M.; Karakiewicz, P.I.; Egawa, S.; Schmidinger, M.; Shariat, S.F.; et al. The Predictive Value of Programmed Death Ligand 1 in Patients with Metastatic Renal Cell Carcinoma Treated with Immune-checkpoint Inhibitors: A Systematic Review and Meta-analysis. Eur. Urol. 2021, 79, 783–792. [Google Scholar] [CrossRef]
- Motzer, R.J.; Choueiri, T.K.; McDermott, D.F.; Powles, T.; Vano, Y.A.; Gupta, S.; Yao, J.; Han, C.; Ammar, R.; Papillon-Cavanagh, S.; et al. Biomarker analysis from CheckMate 214: Nivolumab plus ipilimumab versus sunitinib in renal cell carcinoma. J. Immunother. Cancer 2022, 10, e004316. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Motzer, R.J.; Rini, B.I.; Haanen, J.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Gravis-Mescam, G.; Uemura, M.; Lee, J.L.; et al. Updated efficacy results from the JAVELIN Renal 101 trial: First-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann. Oncol. 2020, 31, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Au, L.; Hatipoglu, E.; Robert de Massy, M.; Litchfield, K.; Beattie, G.; Rowan, A.; Schnidrig, D.; Thompson, R.; Byrne, F.; Horswell, S.; et al. Determinants of anti-PD-1 response and resistance in clear cell renal cell carcinoma. Cancer Cell 2021, 39, 1497–1518.e1411. [Google Scholar] [CrossRef] [PubMed]
- Hack, S.P.; Zhu, A.X.; Wang, Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front. Immunol. 2020, 11, 598877. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Wallin, J.J.; Mancao, C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin. Cancer Biol. 2018, 52(Pt2), 117–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Dikov, M.M.; Ohm, J.E.; Ray, N.; Tchekneva, E.E.; Burlison, J.; Moghanaki, D.; Nadaf, S.; Carbone, D.P. Differential roles of vascular endothelial growth factor receptors 1 and 2 in dendritic cell differentiation. J. Immunol. 2005, 174, 215–222. [Google Scholar] [CrossRef]
- Osada, T.; Chong, G.; Tansik, R.; Hong, T.; Spector, N.; Kumar, R.; Hurwitz, H.I.; Dev, I.; Nixon, A.B.; Lyerly, H.K.; et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol. Immunother. 2008, 57, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaav7431. [Google Scholar] [CrossRef]
- Bouzin, C.; Brouet, A.; De Vriese, J.; Dewever, J.; Feron, O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: Identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Ryoo, B.Y.; Hsu, C.H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef]
- Zhu, A.X.; Abbas, A.R.; de Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.H.; He, A.R.; Ryoo, B.Y.; et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef]
- Palmer, A.C.; Izar, B.; Hwangbo, H.; Sorger, P.K. Predictable Clinical Benefits without Evidence of Synergy in Trials of Combination Therapies with Immune-Checkpoint Inhibitors. Clin. Cancer Res. 2022, 28, 368–377. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brackenier, C.; Kinget, L.; Cappuyns, S.; Verslype, C.; Beuselinck, B.; Dekervel, J. Unraveling the Synergy between Atezolizumab and Bevacizumab for the Treatment of Hepatocellular Carcinoma. Cancers 2023, 15, 348. https://doi.org/10.3390/cancers15020348
Brackenier C, Kinget L, Cappuyns S, Verslype C, Beuselinck B, Dekervel J. Unraveling the Synergy between Atezolizumab and Bevacizumab for the Treatment of Hepatocellular Carcinoma. Cancers. 2023; 15(2):348. https://doi.org/10.3390/cancers15020348
Chicago/Turabian StyleBrackenier, Cedric, Lisa Kinget, Sarah Cappuyns, Chris Verslype, Benoit Beuselinck, and Jeroen Dekervel. 2023. "Unraveling the Synergy between Atezolizumab and Bevacizumab for the Treatment of Hepatocellular Carcinoma" Cancers 15, no. 2: 348. https://doi.org/10.3390/cancers15020348
APA StyleBrackenier, C., Kinget, L., Cappuyns, S., Verslype, C., Beuselinck, B., & Dekervel, J. (2023). Unraveling the Synergy between Atezolizumab and Bevacizumab for the Treatment of Hepatocellular Carcinoma. Cancers, 15(2), 348. https://doi.org/10.3390/cancers15020348