
Resistance to Antiangiogenic Therapy in Hepatocellular Carcinoma: From Molecular Mechanisms to Clinical Impact

 ,
,  ,
,  ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Angiogenesis in Hepatocellular Carcinoma: From the Anatomical Point of View to Clinical Relevance
3. Resistance to Antiangiogenic Drugs
3.1. Sorafenib
- (a)
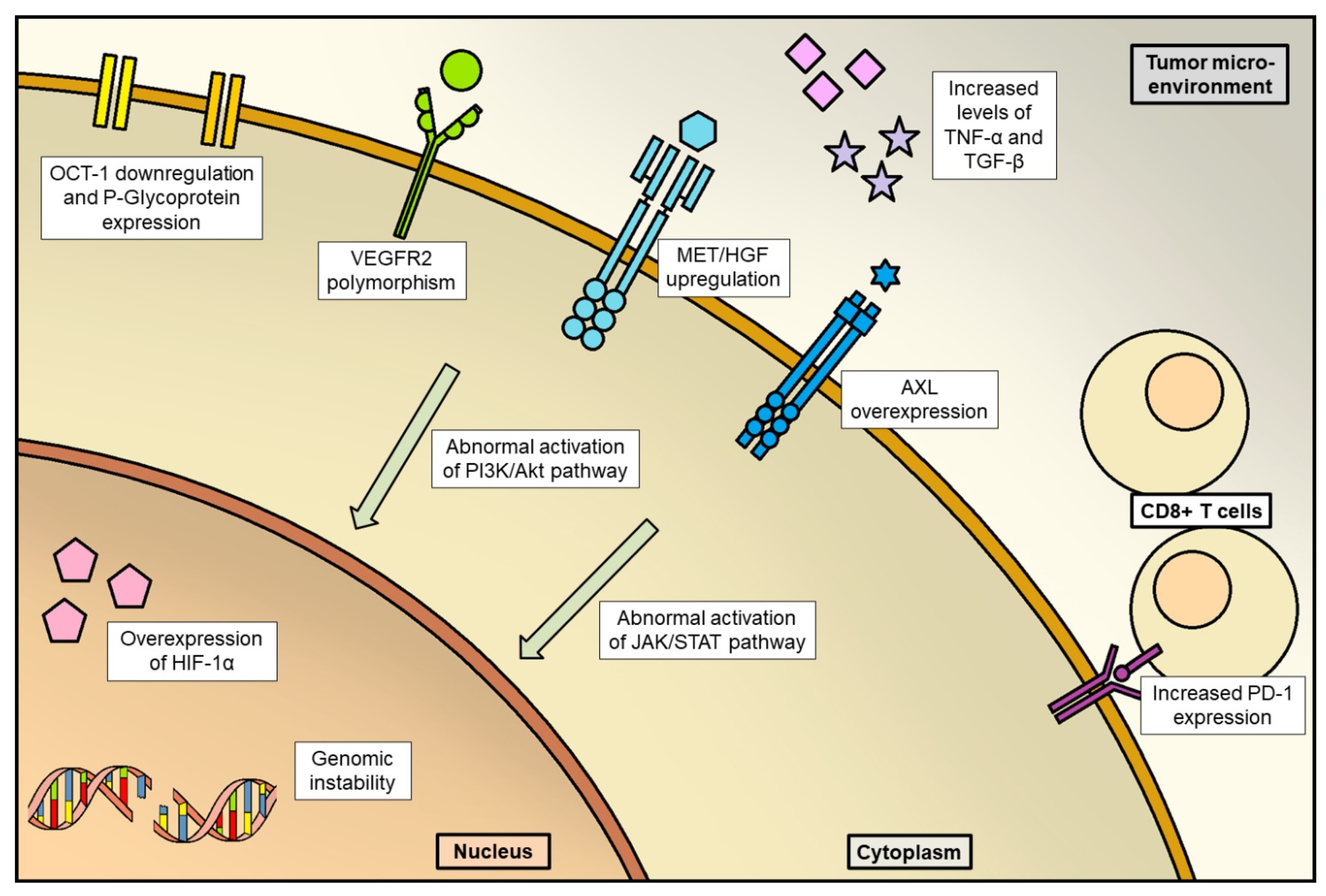
- Disruption of tumor vessels could select resistant cells overexpressing HIF-1α, which is a transcription factor regulating angiogenesis and, definitely, tumor progression [40];
- (b)
- Other mechanisms of acquired resistance could be the “co-option of liver vessels”, that is recruitment of pre-existing liver vessels by HCC without resorting to neoangiogenesis [41]; abnormal activation of PI3K/Akt and JAK-STAT pathways and fibroblast growth factor (FGF) signaling pathways [42]; elevated expression of MET [43]; genome instability and epigenetic regulation [43].
3.2. Lenvatinib
3.3. Regorafenib and Cabozantinib
3.4. Anti-VEGF/VEGFR Monoclonal Antibodies
4. Overcoming Resistance: From the Concept of “Starve the Tumor” to the “Vessel Normalization”
5. Overcoming Resistance: Depicting the Future of HCC Treatment
5.1. Combination Strategies with Approved Antiangiogenetic Drugs
5.1.1. Sorafenib
5.1.2. Lenvatinib
5.1.3. Regorafenib and Cabozantinib
5.2. Experimental Single-Agent Drugs
5.2.1. Antiangiogenetic Drugs
5.2.2. Other Molecules
5.3. Combination of Immunotherapy and Antiangiogenic Drugs
5.3.1. IMbrave 150 Trial: Pioneer of Successful Combination Therapy in HCC
5.3.2. Immunotherapy and Heterogeneous Responses: Role of the Immune Landscape in the Tumor Microenvironment
5.3.3. Combining Immunotherapy in First-Line Setting: Recent Clinical Trials
6. Changing the Algorithm to Optimize the “Continuum of Care” and Future Perspectives
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mohammadian, M.; Mahdavifar, N.; Mohammadian-Hafshejani, A.; Salehiniya, H. Liver cancer in the world: Epidemiology, incidence, mortality and risk factors. WCRJ 2018, 5, e1082. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Burroughs, A.; Bruix, J. Hepatocellular carcinoma. Lancet 2003, 362, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66, Erratum in Lancet 2017, 389, 36. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121, Erratum in Physiol. Rev. 2014, 94, 707. [Google Scholar] [CrossRef]
- Magnussen, A.L.; Mills, I.G. Vascular normalisation as the stepping stone into tumour microenvironment transformation. Br. J. Cancer 2021, 125, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.; Ducreux, M.; Zhu, A.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; et al. IMbrave150: Efficacy and safety results from a ph III study evaluating atezolizumab (atezo) + bevacizumab (bev) vs sorafenib (Sor) as first treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). Ann. Oncol. 2019, 30, ix195–ix196. [Google Scholar] [CrossRef]
- Kudo, M. Scientific Rationale for Combined Immunotherapy with PD-1/PD-L1 Antibodies and VEGF Inhibitors in Advanced Hepatocellular Carcinoma. Cancers 2020, 12, 1089. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, S.; Tovoli, F.; Trevisani, F. Mechanisms of Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Patients with Hepatocellular Carcinoma. Cancers 2022, 14, 4616. [Google Scholar] [CrossRef]
- Saxena, R.; Theise, N.D.; Crawford, J.M. Microanatomy of the human liver-exploring the hidden interfaces. Hepatology 1999, 30, 1339–1346. [Google Scholar] [CrossRef]
- Si-Tayeb, K.; Lemaigre, F.P.; Duncan, S.A. Organogenesis and development of the liver. Dev. Cell 2010, 18, 175–189. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Semela, D.; Dufour, J.F. Angiogenesis and hepatocellular carcinoma. J. Hepatol. 2004, 41, 864–880. [Google Scholar] [CrossRef]
- Xiong, X.X.; Qiu, X.Y.; Hu, D.X.; Chen, X.Q. Advances in Hypoxia-Mediated Mechanisms in Hepatocellular Carcinoma. Mol. Pharmacol. 2017, 92, 246–255. [Google Scholar] [CrossRef]
- Yang, Z.F.; Poon, R.T. Vascular changes in hepatocellular carcinoma. Anat. Rec. 2008, 291, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Elpek, G.O. Molecular pathways in viral hepatitis-associated liver carcinogenesis: An update. World J. Clin. Cases 2021, 9, 4890–4917. [Google Scholar] [CrossRef] [PubMed]
- Daud, M.; Rana, M.A.; Husnain, T.; Ijaz, B. Modulation of Wnt signaling pathway by hepatitis B virus. Arch. Virol. 2017, 162, 2937–2947. [Google Scholar] [CrossRef]
- Tang, T.C.; Poon, R.T.; Lau, C.P.; Xie, D.; Fan, S.T. Tumor cyclooxygenase-2 levels correlate with tumor invasiveness in human hepatocellular carcinoma. World J. Gastroenterol. 2005, 11, 1896–1902. [Google Scholar] [CrossRef]
- Liu, J.; Ding, X.; Tang, J.; Cao, Y.; Hu, P.; Zhou, F.; Shan, X.; Cai, X.; Chen, Q.; Ling, N.; et al. Enhancement of canonical Wnt/β-catenin signaling activity by HCV core protein promotes cell growth of hepatocellular carcinoma cells. PLoS ONE 2011, 6, e27496. [Google Scholar] [CrossRef]
- Shao, Y.Y.; Hsieh, M.S.; Wang, H.Y.; Li, Y.S.; Lin, H.; Hsu, H.W.; Huang, C.Y.; Hsu, C.H.; Cheng, A.L. Hepatitis C virus core protein potentiates proangiogenic activity of hepatocellular carcinoma cells. Oncotarget 2017, 8, 86681–86692. [Google Scholar] [CrossRef][Green Version]
- Poon, R.T.; Ng, I.O.; Lau, C.; Zhu, L.X.; Yu, W.C.; Lo, C.M.; Fan, S.T.; Wong, J. Serum vascular endothelial growth factor predicts venous invasion in hepatocellular carcinoma: A prospective study. Ann. Surg. 2001, 233, 227–235. [Google Scholar] [CrossRef]
- Al-Abd, A.M.; Alamoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Asić, K. Dominant mechanisms of primary resistance differ from dominant mechanisms of secondary resistance to targeted therapies. Crit. Rev. Oncol. Hematol. 2015, 97, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Lang, L. FDA approves sorafenib for patients with inoperable liver cancer. Gastroenterology 2008, 134, 379. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2008, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Macias, R.I.R.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1663. [Google Scholar] [CrossRef]
- Geier, A.; Macias, R.I.; Bettinger, D.; Weiss, J.; Bantel, H.; Jahn, D.; Al-Abdulla, R.; Marin, J.J. The lack of the organic cation transporter OCT1 at the plasma membrane of tumor cells precludes a positive response to sorafenib in patients with hepatocellular carcinoma. Oncotarget 2017, 8, 15846–15857. [Google Scholar] [CrossRef]
- Hu, S.; Chen, Z.; Franke, R.; Orwick, S.; Zhao, M.; Rudek, M.A.; Sparreboom, A.; Baker, S.D. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin. Cancer Res. 2009, 15, 6062–6069. [Google Scholar] [CrossRef]
- Zheng, Y.B.; Zhan, M.X.; Zhao, W.; Liu, B.; Huang, J.W.; He, X.; Fu, S.R.; Zhao, Y.; Li, Y.; Hu, B.S.; et al. The relationship of kinase insert domain receptor gene polymorphisms and clinical outcome in advanced hepatocellular carcinoma pa-tients treated with sorafenib. Med. Oncol. 2014, 31, 209. [Google Scholar] [CrossRef]
- Méndez-Blanco, C.; Fondevila, F.; García-Palomo, A.; González-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Yin, M.; Bar-Zion, A.; Lee, C.R.; Butz, H.; Man, S.; Daley, F.; Vermeulen, P.B.; Yousef, G.M.; Foster, F.S.; et al. Co-option of Liver Vessels and Not Sprouting Angiogenesis Drives Acquired Sorafenib Resistance in Hepatocellular Carcinoma. JNCI J. Natl. Cancer Inst. 2016, 108, djw030. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.S.; Kim, B.J.; Jang, H.J.; Lee, J. Prognostic value of c-Met overexpression in hepatocellular carcinoma: A meta-analysis and review. Oncotarget 2017, 8, 90351–90357. [Google Scholar] [CrossRef]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-α is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. Ebiomedicine 2018, 40, 446–456, Erratum in EBioMedicine 2022, 80, 104074. [Google Scholar] [CrossRef]
- Ungerleider, N.; Han, C.; Zhang, J.; Yao, L.; Wu, T. TGFβ signaling confers sorafenib resistance via induction of multiple RTKs in hepatocellular carcinoma cells. Mol. Carcinog. 2016, 56, 1302–1311. [Google Scholar] [CrossRef]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef]
- Chen, J.; Ji, T.; Zhao, J.; Li, G.; Zhang, J.; Jin, R.; Liu, J.; Liu, X.; Liang, X.; Huang, D.; et al. Sorafenib-resistant hepatocellular carcinoma stratified by phosphorylated ERK activates PD-1 immune checkpoint. Oncotarget 2016, 7, 41274–41284. [Google Scholar] [CrossRef]
- Cheng, Z.; Wei-Qi, J.; Jin, D. New insights on sorafenib resistance in liver cancer with correlation of individualized therapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188382. [Google Scholar] [CrossRef]
- Finn, R.S.; Kudo, M.; Cheng, A.L.; Wyrwicz, L.; Ngan, R.; Blanc, J.F.; Baron, A.D.; Vogel, A.; Ikeda, M.; Piscaglia, F.; et al. Final analysis of serum biomarkers in patients (pts) from the phase III study of lenvatinib (LEN) vs sorafenib (SOR) in unresectable hepatocellular carcinoma (uHCC) [REFLECT]. Ann. Oncol. 2018, 29, viii17–viii18. [Google Scholar] [CrossRef]
- Tahara, M.; Schlumberger, M.; Elisei, R.; Habra, M.A.; Kiyota, N.; Paschke, R.; Dutcus, C.E.; Hihara, T.; McGrath, S.; Matijevic, M.; et al. Exploratory analysis of biomarkers associated with clinical outcomes from the study of lenvatinib in differentiated cancer of the thyroid. Eur. J. Cancer 2017, 75, 213–221. [Google Scholar] [CrossRef]
- Fu, R.; Jiang, S.; Li, J.; Chen, H.; Zhang, X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med. Oncol. 2020, 37, 24. [Google Scholar] [CrossRef]
- Venepalli, N.K.; Goff, L. Targeting the HGF-cMET Axis in Hepatocellular Carcinoma. Int. J. Hepatol. 2013, 2013, 341636. [Google Scholar] [CrossRef]
- Ye, D.Z.; Field, J. PAK signaling in cancer. Cell. Logist. 2012, 2, 105–116. [Google Scholar] [CrossRef]
- Shrestha, Y.; Schafer, E.J.; Boehm, J.S.; Thomas, S.R.; He, F.; Du, J.; Wang, S.; Barretina, J.; Weir, B.A.; Zhao, J.J.; et al. PAK1 is a breast cancer oncogene that coordinately activates MAPK and MET signaling. Oncogene 2011, 31, 3397–3408. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, C.; Hou, B. Genome-wide CRISPR/Cas9 library screening to identify PAK1 as a critical driver for lenvatinib resistance in HCC. J. Clin. Oncol. 2020, 38, e16687. [Google Scholar] [CrossRef]
- Teufel, M.; Seidel, H.; Köchert, K.; Meinhardt, G.; Finn, R.S.; Llovet, J.M.; Bruix, J. Biomarkers Associated with Response to Regorafenib in Patients with Hepatocellular Carcinoma. Gastroenterology 2019, 156, 1731–1741. [Google Scholar] [CrossRef]
- Juengpanich, S.; Topatana, W.; Lu, C.; Staiculescu, D.; Li, S.; Cao, J.; Lin, J.; Hu, J.; Chen, M.; Chen, J.; et al. Role of cellular, molecular and tumor microenvironment in hepatocellular carcinoma: Possible targets and future directions in the regorafenib era. Int. J. Cancer 2020, 147, 1778–1792. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, S.; Ma, D.; Yan, D.; Zhang, G.; Cao, Y.; Wang, Z.; Wu, J.; Jiang, C. Targeting SphK2 Reverses Acquired Resistance of Regorafenib in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 694. [Google Scholar] [CrossRef]
- Arechederra, M.; Bazai, S.K.; Abdouni, A.; Sequera, C.; Mead, T.J.; Richelme, S.; Daian, F.; Audebert, S.; Dono, R.; Lozano, A.; et al. ADAMTSL5 is an epigenetically activated gene underlying tumorigenesis and drug resistance in hepatocellular carcinoma. J. Hepatol. 2020, 74, 893–906. [Google Scholar] [CrossRef]
- Tutusaus, A.; Stefanovic, M.; Boix, L.; Cucarull, B.; Zamora, A.; Blasco, L.; de Frutos, P.G.; Reig, M.; Fernandez-Checa, J.C.; Marí, M.; et al. Antiapoptotic BCL-2 proteins determine sorafenib/regorafenib resistance and BH3-mimetic efficacy in hepatocellular carcinoma. Oncotarget 2018, 9, 16701–16717. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, Y.; Tian, L.; Shi, H.; Wang, J.; Liang, Y.; Sun, B.; Wang, S.; Zhou, M.; Wu, L.; et al. Gankyrin drives metabolic reprogramming to promote tumorigenesis, metastasis and drug resistance through activating beta-catenin/c-Myc signaling in human hepatocellular carcinoma. Cancer Lett. 2019, 443, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Grüllich, C. Cabozantinib: Multi-kinase Inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. 2018, 211, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.F.; Huang, Y.L.; Xie, Y.K.; Tan, Y.H.; Chen, B.C.; Zhou, M.T.; Shi, H.Q.; Yu, Z.P.; Song, Q.T.; Zhang, Q.Y. Angiogenesis and clinicopathologic characteristics in different hepatocellular carcinoma subtypes defined by EpCAM and α-fetoprotein expression status. Med. Oncol. 2010, 28, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Galle, P.R.; Foerster, F.; Kudo, M.; Chan, S.L.; Llovet, J.M.; Qin, S.; Schelman, W.R.; Chintharlapalli, S.; Abada, P.B.; Sherman, M.; et al. Biology and significance of alpha-fetoprotein in hepatocellular carcinoma. Liver Int. 2019, 39, 2214–2229. [Google Scholar] [CrossRef]
- Kelley, R.K.; Meyer, T.; Rimassa, L.; Merle, P.; Park, J.W.; Yau, T.; Chan, S.L.; Blanc, J.F.; Tam, V.C.; Tran, A.; et al. Serum Alpha-fetoprotein Levels and Clinical Outcomes in the Phase III CELESTIAL Study of Cabozantinib versus Placebo in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2020, 26, 4795–4804. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, X.; Xu, W.; Chen, J.; Yu, J.; Gamble, J.R.; McCaughan, G.W. Targeting the vasculature in hepatocellular carcinoma treatment: Starving versus normalizing blood supply. Clin. Transl. Gastroenterol. 2017, 8, e98. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Heishi, T.; Khan, O.F.; Kowalski, P.S.; Incio, J.; Rahbari, N.N.; Chung, E.; Clark, J.W.; Willett, C.G.; Luster, A.D.; et al. Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J. Clin. Investig. 2017, 127, 3039–3051. [Google Scholar] [CrossRef]
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Connelly, C.; Connolly, E.M.; Li, J.; Manos, M.P.; Lawrence, D.; McDermott, D.; Severgnini, M.; et al. Angiopoietin-2 as a Biomarker and Target for Immune Checkpoint Therapy. Cancer Immunol. Res. 2017, 5, 17–28. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Mi, Y.J.; Liang, Y.J.; Huang, H.B.; Zhao, H.Y.; Wu, C.P.; Wang, F.; Tao, L.Y.; Zhang, C.Z.; Dai, C.L.; Tiwari, A.K.; et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010, 70, 7981–7991. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, G.; Miao, H.; Song, Z.; Zhang, X.; Fan, W.; Wang, Y.; Li, J.; Chen, Y. Apatinib treatment may improve survival outcomes of patients with hepatitis B virus-related sorafenib-resistant hepatocellular carcinoma. Ther. Adv. Med. Oncol. 2020, 12, 1758835920937422. [Google Scholar] [CrossRef]
- Shen, G.; Zheng, F.; Ren, D.; Du, F.; Dong, Q.; Wang, Z.; Zhao, F.; Ahma, R.; Zhao, J. Anlotinib: A novel multi-targeting tyrosine kinase inhibitor in clinical development. J. Hematol. Oncol. 2018, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.; Qin, S.; Gu, S.; Bai, Y.; Chen, Z.; Wang, Z.; Ying, J.; Lu, Y.; Meng, Z.; Pan, H.; et al. Donafenib versus sorafenib as first-line therapy in advanced hepatocellular carcinoma: An open-label, randomized, multicenter phase II/III trial. J. Clin. Oncol. 2020, 38 (Suppl. 15), 4506. [Google Scholar] [CrossRef]
- Dolan, M.; Mastri, M.; Tracz, A.; Christensen, J.G.; Chatta, G.; Ebos, J.M.L. Enhanced efficacy of sitravatinib in metastatic models of antiangiogenic therapy resistance. PLoS ONE 2019, 14, e0220101. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Lee, L.Y.; Goh, K.Y.; Ong, R.; Hao, H.X.; Huang, A.; Wang, Y.; Graus Porta, D.; Chow, P.; Chung, A. Infigratinib Mediates Vascular Normalization, Impairs Metastasis, and Improves Chemotherapy in Hepatocellular Carcinoma. Hepatology 2018, 69, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Rezende Miranda, R.; Fu, Y.; Chen, X.; Perino, J.; Cao, P.; Carpten, J.; Chen, Y.; Zhang, C. Development of a Potent and Specific FGFR4 Inhibitor for the Treatment of Hepatocellular Carcinoma. J. Med. Chem. 2020, 63, 11484–11497. [Google Scholar] [CrossRef]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Rota Caremoli, E.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- Kudo, M.; Morimoto, M.; Moriguchi, M.; Izumi, N.; Takayama, T.; Yoshiji, H.; Hino, K.; Oikawa, T.; Chiba, T.; Motomura, K.; et al. A randomized, double-blind, placebo-controlled, phase 3 study of tivantinib in Japanese patients with MET-high hepatocellular carcinoma. Cancer Sci. 2020, 111, 3759–3769. [Google Scholar] [CrossRef]
- Ito, Y.; Takeda, T.; Sakon, M.; Tsujimoto, M.; Higashiyama, S.; Noda, K.; Miyoshi, E.; Monden, M.; Matsuura, N. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br. J. Cancer 2001, 84, 1377–1383. [Google Scholar] [CrossRef]
- Hsieh, C.-Y.; Ooi, L.; Ong, R.W.; Lindmark, B.; McHale, M.; Huynh, H.T. Varlitinib to demonstrate anti-tumour efficacy in patient-derived hepatocellular carcinoma xenograft models. J. Clin. Oncol. 2016, 34 (Suppl. 15), e15598. [Google Scholar] [CrossRef]
- Covington, M.; He, X.; Scuron, M.; Li, J.; Collins, R.; Juvekar, A.; Shin, N.; Favata, M.; Gallagher, K.; Sarah, S.; et al. Preclinical characterization of itacitinib (INCB039110), a novel selective inhibitor of JAK1, for the treatment of inflammatory diseases. Eur. J. Pharmacol. 2020, 885, 173505. [Google Scholar] [CrossRef]
- Fan, Y.; Li, S.; Ding, X.; Yue, J.; Jiang, J.; Zhao, H.; Hao, R.; Qiu, W.; Liu, K.; Li, Y.; et al. First-in-class immune-modulating small molecule Icaritin in advanced hepatocellular carcinoma: Preliminary results of safety, durable survival and immune biomarkers. BMC Cancer 2019, 19, 279. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, Q.; Xu, J.M.; Liang, J.; Cheng, Y.; Li, S.; Zheng, L.; Ye, B.; Meng, K.; Qin, S. A multicenter, single arm phase II trial of a small molecule immune-modulator icaritin: Safety, overall survival, immune dynamics, and PD-L1 expression in advanced hepatocellular carcinoma. J. Clin. Oncol. 2018, 36 (Suppl. 15), 4077. [Google Scholar] [CrossRef]
- Tong, X.; Wang, Q.; Wu, D.; Bao, L.; Yin, T.; Chen, H. MEK inhibition by cobimetinib suppresses hepatocellular carcinoma and angiogenesis in vitro and in vivo. Biochem. Biophys. Res. Commun. 2019, 523, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Tan, E.; Wang, E.; Mahipal, A.; Chen, D.T.; Cao, B.; Masawi, F.; Machado, C.; Yu, J.; Kim, D.W. A Phase I Trial of Trametinib in Combination with Sorafenib in Patients with Advanced Hepatocellular Cancer. Oncologist 2020, 25, e1893–e1899. [Google Scholar] [CrossRef]
- Mazzocca, A.; Fransvea, E.; Lavezzari, G.; Antonaci, S.; Giannelli, G. Inhibition of transforming growth factor beta receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology 2009, 50, 1140–1151. [Google Scholar] [CrossRef]
- Kelley, R.K.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.R.; Merle, P.; Hourmand, I.O.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Su, Z.; Fei, H.; Liu, X.; Pan, Q. The novel mTOR inhibitor Torin-2 induces autophagy and downregulates the expression of UHRF1 to suppress hepatocarcinoma cell growth. Oncol. Rep. 2015, 34, 1708–1716. [Google Scholar] [CrossRef]
- Yang, X.; Liu, J.; Liang, Q.; Sun, G. Valproic acid reverses sorafenib resistance through inhibiting activated Notch/Akt signaling pathway in hepatocellular carcinoma. Fundam. Clin. Pharmacol. 2020, 35, 690–699. [Google Scholar] [CrossRef]
- Liu, J.; Yang, X.; Liang, Q.; Yu, Y.; Shen, X.; Sun, G. Valproic acid overcomes sorafenib resistance by reducing the migration of Jagged2-mediated Notch1 signaling pathway in hepatocellular carcinoma cells. Int. J. Biochem. Cell Biol. 2020, 126, 105820. [Google Scholar] [CrossRef]
- Jilkova, Z.M.; Kuyucu, A.Z.; Kurma, K.; Pour, S.T.A.; Roth, G.S.; Abbadessa, G.; Yu, Y.; Schwartz, B.; Sturm, N.; Marche, P.N.; et al. Combination of AKT inhibitor ARQ 092 and sorafenib potentiates inhibition of tumor progression in cirrhotic rat model of hepatocellular carcinoma. Oncotarget 2018, 9, 11145–11158. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Lee, Y.B.; Cho, E.J.; Lee, J.H.; Yu, S.J.; Kim, Y.J.; Yoon, J.H. CKD-5, a novel pan-histone deacetylase inhibitor, synergistically enhances the efficacy of sorafenib for hepatocellular carcinoma. BMC Cancer 2020, 20, 1001. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Elkholy, K.H.; Wani, N.A.; Li, D.; Hu, P.; Barajas, J.M.; Yu, L.; Zhang, X.; Jacob, S.T.; Khan, W.N.; et al. Ibrutinib Potentiates Antihepatocarcinogenic Efficacy of Sorafenib by Targeting EGFR in Tumor Cells and BTK in Immune Cells in the Stroma. Mol. Cancer Ther. 2020, 19, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.J.; Guo, L.X.; Zheng, K.Y.; Song, J.N.; Wang, Q.; Zheng, Y.G. Chelidonine enhances the antitumor effect of lenvatinib on hepatocellular carcinoma cells. OncoTargets Ther. 2019, 12, 6685–6697. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, D.; Wu, F.; Tu, J.; Song, J.; Xu, M.; Ji, J. Sophoridine suppresses lenvatinib-resistant hepatocellular carcinoma growth by inhibiting RAS/MEK/ERK axis via decreasing VEGFR2 expression. J. Cell. Mol. Med. 2020, 25, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Song, J.; Zhang, D.; Wu, F.; Tu, J.; Ji, J. Oxysophocarpine suppresses FGFR1-overexpressed hepatocellular carcinoma growth and sensitizes the therapeutic effect of lenvatinib. Life Sci. 2020, 264, 118642. [Google Scholar] [CrossRef]
- Finn, R.; Kudo, M.; Merle, P.; Meyer, T.; Qin, S.; Ikeda, M.; Xu, R.; Edeline, J.; Ryoo, B.-Y.; Ren, Z.; et al. Primary results from the phase III LEAP-002 study: Lenvatinib plus pembrolizumab versus lenvatinib as first- line (1L) therapy for advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2022, 33 (Suppl. 7), S808–S869. [Google Scholar] [CrossRef]
- Shang, R.; Song, X.; Wang, P.; Zhou, Y.; Lu, X.; Wang, J.; Xu, M.; Chen, X.; Utpatel, K.; Che, L.; et al. Cabozantinib-based combination therapy for the treatment of hepatocellular carcinoma. Gut 2020, 70, 1746–1757. [Google Scholar] [CrossRef]
- Le, T.B.U.; Vu, T.C.; Ho, R.Z.W.; Prawira, A.; Wang, L.; Goh, B.C.; Huynh, H. Bevacizumab Augments the Antitumor Efficacy of Infigratinib in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 9405. [Google Scholar] [CrossRef]
- Prawira, A.; Le, T.B.U.; Vu, T.C.; Huynh, H. Ribociclib enhances infigratinib-induced cancer cell differentiation and delays resistance in FGFR-driven hepatocellular carcinoma. Liver Int. 2020, 41, 608–620. [Google Scholar] [CrossRef]
- Casak, S.J.; Donoghue, M.; Fashoyin-Aje, L.; Jiang, X.; Rodriguez, L.; Shen, Y.L.; Xu, Y.; Jiang, X.; Liu, J.; Zhao, H.; et al. FDA Approval Summary: Atezolizumab Plus Bevacizumab for the Treatment of Patients with Advanced Unresectable or Metastatic Hepatocellular Carcinoma. Clin. Cancer Res. 2020, 27, 1836–1841. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.V.; Merle, P.; et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J. Clin. Oncol. 2021, 39 (Suppl. 33), 267. [Google Scholar] [CrossRef]
- Toh, H.C.; Galle, P.R.; Zhu, A.X.; Nicholas, A.; Gaillard, V.; Ducreux, M.; Cheng, A.L.; Finn, R.S. IMbrave150: Exploratory efficacy and safety in patients with unresectable hepatocellular carcinoma (HCC) treated with atezolizumab beyond radiological progression until loss of clinical benefit in a global phase III study. Meeting Abstract|2022 ASCO Gastrointestinal Cancers Symposium. J. Clin. Oncol. 2022, 40 (Suppl. 4), 470. [Google Scholar] [CrossRef]
- D’Alessio, A.; Weinmann, A.; Galle, P.R.; Fulgenzi, C.A.M.; Bettinger, D.; Bengsch, B.; Vogel, A.; Balcar, L.; Scheiner, B.; Navaid, M.; et al. Real-world use of atezolizumab plus bevacizumab in patients with hepatocellular carcinoma and Child-Pugh A and B cirrhosis. J. Clin. Oncol. 2022, 40 (Suppl. 4), 393. [Google Scholar] [CrossRef]
- NIH. Clinical Trials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 30 September 2022).
- Foerster, F.; Kloeckner, R.; Reig, M.; Chan, S.L.; Chung, J.W.; Merle, P.; Park, J.W.; Piscaglia, F.; Vogel, A.; Gaillard, V.; et al. ABC-HCC: A phase IIIb, randomized, multicenter, open-label trial of atezolizumab plus bevacizumab versus transarterial chemoembolization (TACE) in intermediate-stage hepatocellular carcinoma. J. Clin. Oncol. 2022, 40 (Suppl. 4), TPS498. [Google Scholar] [CrossRef]
- Kudo, M.; Guo, Y.; Hua, Y.; Zhao, M.; Xing, W.; Zhang, Y.; Liu, R.; Ren, Z.; Gu, S.; Lin, Z.; et al. TALENTACE: A phase III, open-label, randomized study of on-demand transarterial chemoembolization combined with atezolizumab + bevacizumab or on-demand transarterial chemoembolization alone in patients with untreated hepatocellular carcinoma. 2022 ASCO Gastrointestinal Cancers Symposium. J. Clin. Oncol. 2022, 40 (Suppl. 4), TPS487. [Google Scholar] [CrossRef]
- Harding, J.J. Immune checkpoint blockade in advanced hepatocellular carcinoma: An update and critical review of ongoing clinical trials. Futur. Oncol. 2018, 14, 2293–2302. [Google Scholar] [CrossRef]
- Lin, Z.; Lu, D.; Wei, X.; Wang, J.; Xu, X. Heterogeneous responses in hepatocellular carcinoma: The achilles heel of immune checkpoint inhibitors. Am. J. Cancer Res. 2020, 10, 1085–1102. [Google Scholar]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, S.N.; Anjum, K.M.; Gui, L.X.; Zhu, S.S.; Liu, J.; Chen, J.K.; Liu, Q.F.; Ye, G.D.; Wang, W.J.; et al. A double-negative feedback loop between Wnt-β-catenin signaling and HNF4α regulates epithelial-mesenchymal transition in hepatocellular carcinoma. J. Cell Sci. 2013, 126, 5692–5703. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhao, J.; Ge, H.; Li, G.; Wu, J. Clinicopathological and prognostic significance of FoxM1 in hepatocellular carcinoma patients: A meta-analysis. OncoTargets Ther. 2018, 11, 3561–3571. [Google Scholar] [CrossRef] [PubMed]
- Vercher, E.; Tamayo, I.; Mancheño, U.; Elizalde, E.; Conde, E.; Reparaz, D.; Lasarte, J.J.; Villar, V.; Iñarrairaegui, M.; Sangro, B.; et al. Identification of neoantigen-reactive T cells in hepatocellular carcinoma: Implication in adoptive T cell therapy. J. Hepatol. 2020, 73, S39–S40. [Google Scholar] [CrossRef]
- Zhu, A.X.; Abbas, A.R.; de Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.H.; He, A.R.; Ryoo, B.Y.; et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.K.; Dao, T.V.; De Toni, E.N.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab and durvalumab as first-line therapy in patients with unresectable hepatocellular carcinoma: HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Qin, S.; Chan, L.; Gu, S.; Bai, Y.; Ren, Z.; Lin, X.; Chen, Z.; Jia, W.; Jin, Y.; Guo, Y.; et al. Camrelizumab (C) plus rivoceranib (R) vs. sorafenib (S) as first-line therapy for unresectable hepatocellular carcinoma (uHCC): A randomized, phase III trial. Ann. Oncol. 2022, 33 (Suppl. 7), S1401–S1402. [Google Scholar] [CrossRef]
- Piñero, F.; Silva, M.; Iavarone, M. Sequencing of systemic treatment for hepatocellular carcinoma: Second line competitors. World J. Gastroenterol. 2020, 26, 1888–1900. [Google Scholar] [CrossRef] [PubMed]
- Busato, D.; Mossenta, M.; Baboci, L.; Di Cintio, F.; Toffoli, G.; Dal Bo, M. Novel immunotherapeutic approaches for hepatocellular carcinoma treatment. Expert Rev. Clin. Pharmacol. 2019, 12, 453–470. [Google Scholar] [CrossRef]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix Tudó, J.; Qin, S.; Thuluvath, P.J.; Llovet i Bayer, J.M.; et al. A phase III, randomized, double-blind, placebo controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef]
- Koeberle, D.; Dufour, J.F.; Demeter, G.; Li, Q.; Ribi, K.; Samaras, P.; Saletti, P.; Roth, A.D.; Horber, D.; Bühlmann, M.; et al. Sorafenib with or without everolimus in patients with advanced hepatocellular carcinoma (HCC): A randomized multicenter, multinational phase II trial (SAKK 77/08 and SASL 29). Ann. Oncol. 2016, 27, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-S.; Chang, C.-C.; Wu, C.-H.; Dinh, T.K.; Jan, J.-J.; Huang, K.-W.; Chou, M.-C.; Shiue, T.-Y.; Yeh, K.-C.; Ke, Y.-Y.; et al. A highly selective and potent CXCR4 antagonist for hepatocellular carcinoma treatment. Proc. Natl. Acad. Sci. USA 2021, 118, e2015433118. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.S.; Kamoun, W.S.; Farrar, C.T.; Kirkpatrick, N.D.; Niemeyer, E.; de Graaf, A.M.; Sorensen, A.G.; Munn, L.L.; Jain, R.K.; Fukumura, D. Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalization and survival benefit in mice bearing gliomas. Clin. Cancer Res. 2010, 16, 3618–3627. [Google Scholar] [CrossRef] [PubMed]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9, eaak9670. [Google Scholar] [CrossRef]
- Wada, H.; Nagano, H.; Yamamoto, H.; Yang, Y.; Kondo, M.; Ota, H.; Nakamura, M.; Yoshioka, S.; Kato, H.; Damdinsuren, B.; et al. Expression pattern of angiogenic factors and prognosis after hepatic resection in hepatocellular carcinoma: Importance of angiopoietin-2 and hypoxia-induced factor-1 alpha. Liver Int. 2006, 26, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Nishida, N.; Kudo, M. Clinical Significance of the Duality of Wnt/β-Catenin Signaling in Human Hepatocellular Carcinoma. Cancers 2022, 14, 444. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Mechanism of Action | Evidence | Reference |
|---|---|---|---|
| Antiangiogenetic drugs | |||
| Apatinib | VEGFR-2, RET, cKIT and cSRC inhibitor | Phase II/III trial (post- sorafenib patients): improved OS | Mi et al. [79] Zhang et al. [80] |
| Anlotinib | VEGFR(1,2,3), FGFR(1,2,3,4), PDGFR(α,β) and cKIT inhibitor | Phase II trial (post-lenvatinib patients): ongoing | Shen et al. [81] |
| Donafenib | RAF and VEGFR inhibitor | Phase II/III trial (versus sorafenib): improved OS | Bi et al. [82] |
| Sitravatinib | VEGFR, PDGFR(α,β), cKIT, MET, AXL and EphA1 inhibitor | Preclinical studies | Dolan et al. [83] |
| Infigratinib | Pan-FGFR inhibitor | Preclinical studies (alone or in combination) | Huynh et al. [84] Rezende et al. [85] |
| Other molecules | |||
| Tivantinib | MET inhibitor | Phase III trials (post-sorafenib MET-high patients): negative in OS | Rimassa et al. [86] Kudo [87] |
| Varlitinib | Pan-HER inhibitor | Phase Ib trial (post-sorafenib or post-lenvatinib patients): ongoing | Ito et al. [88] Hsieh et al. [89] |
| Itacitinib | JAK1 inhibitor | Phase Ib trial (post-sorafenib or post-lenvatinib patients): ongoing | Covington et al. [90] |
| Icaritin | JAK2/STAT3 inhibitor, immune modulator | Phase III trial (versus sorafenib in PD-L1+ patients): ongoing | Fan et al. [91] Sun et al. [92] |
| Median OS, mo (95% CI) | Median PFS, mo (95% CI) | ORR | |
|---|---|---|---|
| Himalaya (NCT03298451) | |||
| Primary objective: OS for STRIDE vs. S Secondary objective: non-inferiority for OS for D vs. S | |||
| STRIDE (n = 393) Durvalumab (n = 389) Sorafenib (n = 389) | 16.4 (14.2–19.6) 16.6 (14.1–19.1) 13.8 (12.3–16.1) | 3.8 (3.7–5.3) 3.7 (3.2–3.8) 4.1 (3.8–5.5) | 20.1% 17.0% 5.1% |
| SHR-1210-III-310 (NCT03764293) | |||
| Primary objective: OS and PFS for C + R vs. S Secondary objective: ORR | |||
| Camrelizumab + Apatinib Sorafenib | 22.1 (19.1–27.2) 15.2 (13.0–18.5) | 5.6 (5.5–6.3) 3.7 (2.8–3.7) | 25.4% 5.9% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Federico, P.; Giunta, E.F.; Tufo, A.; Tovoli, F.; Petrillo, A.; Daniele, B. Resistance to Antiangiogenic Therapy in Hepatocellular Carcinoma: From Molecular Mechanisms to Clinical Impact. Cancers 2022, 14, 6245. https://doi.org/10.3390/cancers14246245
Federico P, Giunta EF, Tufo A, Tovoli F, Petrillo A, Daniele B. Resistance to Antiangiogenic Therapy in Hepatocellular Carcinoma: From Molecular Mechanisms to Clinical Impact. Cancers. 2022; 14(24):6245. https://doi.org/10.3390/cancers14246245
Chicago/Turabian StyleFederico, Piera, Emilio Francesco Giunta, Andrea Tufo, Francesco Tovoli, Angelica Petrillo, and Bruno Daniele. 2022. "Resistance to Antiangiogenic Therapy in Hepatocellular Carcinoma: From Molecular Mechanisms to Clinical Impact" Cancers 14, no. 24: 6245. https://doi.org/10.3390/cancers14246245
APA StyleFederico, P., Giunta, E. F., Tufo, A., Tovoli, F., Petrillo, A., & Daniele, B. (2022). Resistance to Antiangiogenic Therapy in Hepatocellular Carcinoma: From Molecular Mechanisms to Clinical Impact. Cancers, 14(24), 6245. https://doi.org/10.3390/cancers14246245