

Cell-of-Origin Targeted Drug Repurposing for Triple-Negative and Inflammatory Breast Carcinoma with HDAC and HSP90 Inhibitors Combined with Niclosamide
 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction

2. Materials and Methods
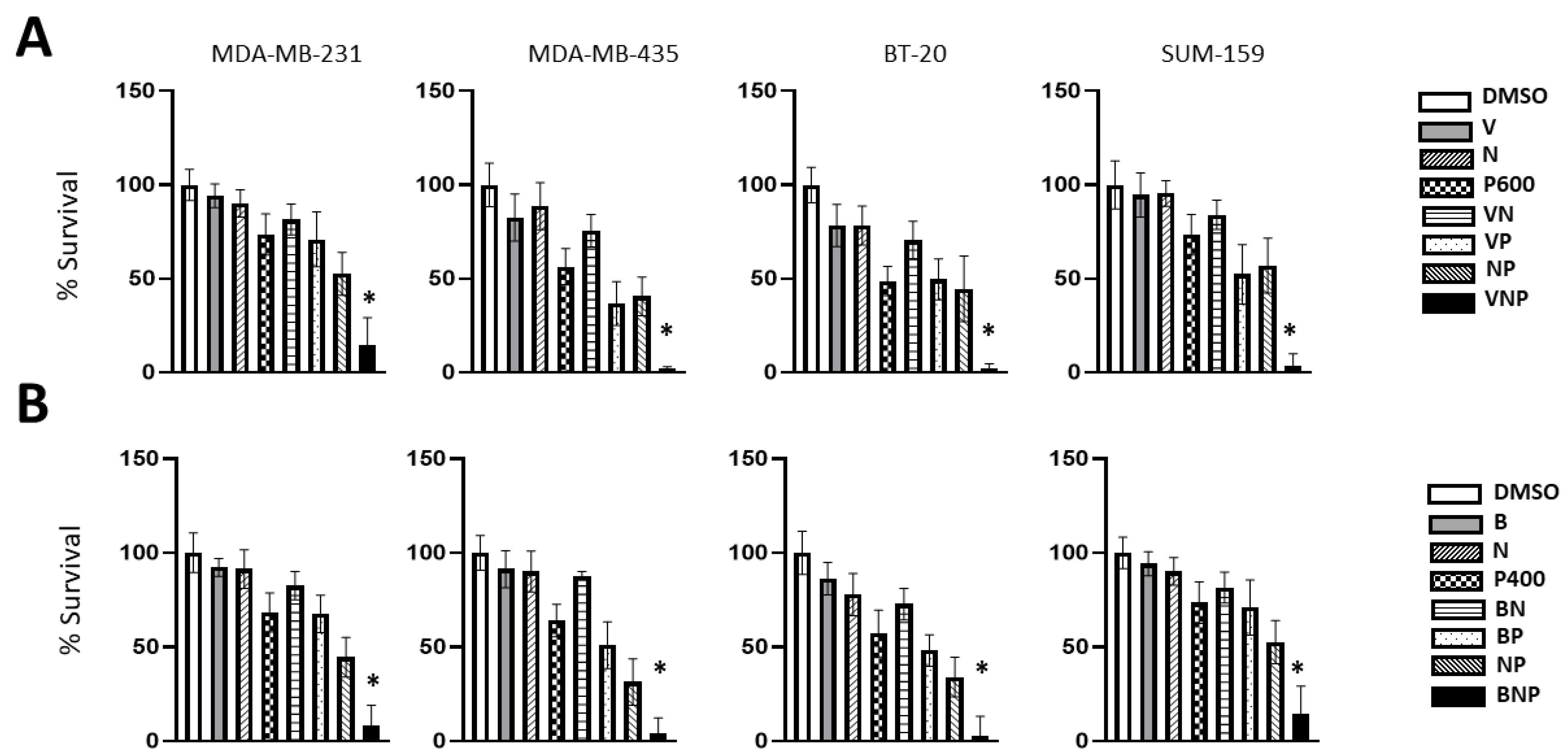
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, W.F.; Schairer, C.; Chen, B.E.; Hance, K.W.; Levine, P.H. Epidemiology of Inflammatory Breast Cancer (IBC). Breast Dis. 2006, 22, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Ince, T.A.; Richardson, A.L.; Bell, G.W.; Saitoh, M.; Godar, S.; Karnoub, A.E.; Iglehart, J.D.; Weinberg, R.A. Transformation of Different Human Breast Epithelial Cell Types Leads to Distinct Tumor Phenotypes. Cancer Cell 2007, 12, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Bhagirath, D.; Zhao, X.; West, W.W.; Qiu, F.; Band, H.; Band, V. Cell type of origin as well as genetic alterations contribute to breast cancer phenotypes. Oncotarget 2015, 6, 9018–9030. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Kim, S.S.; Nebeck, H.E.; Ahn, E.H. Immortalization of Different Breast Epithelial Cell Types Results in Distinct Mitochondrial Mutagenesis. Int. J. Mol. Sci. 2019, 20, 2813. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Liu, Z.; Jiang, W.; Nagi, C.; Huang, S.; Edwards, D.P.; Jo, E.; Mo, Q.; Creighton, C.J.; Hilsenbeck, S.G.; et al. Mammary Precancerous Stem and Non-Stem Cells Evolve into Cancers of Distinct Subtypes. Cancer Res. 2019, 79, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Waldum, H.; Mjones, P. Time to Classify Tumours of the Stomach and the Kidneys According to Cell of Origin. Int. J. Mol. Sci. 2021, 22, 13386–13399. [Google Scholar] [CrossRef]
- Weiss, J.M.; Hunter, M.V.; Cruz, N.M.; Baggiolini, A.; Tagore, M.; Ma, Y.; Misale, S.; Marasco, M.; Simon-Vermot, T.; Campbell, N.R.; et al. Anatomic position determines oncogenic specificity in melanoma. Nature 2022, 604, 354–361. [Google Scholar] [CrossRef]
- Abdulla, M.; Hollander, P.; Pandzic, T.; Mansouri, L.; Ednersson, S.B.; Andersson, P.; Hultdin, M.; Fors, M.; Erlanson, M.; Degerman, S.; et al. Cell-of-origin determined by both gene expression profiling and immunohistochemistry is the strongest predictor of survival in patients with diffuse large B-cell lymphoma. Am. J. Hematol. 2019, 95, 57–67. [Google Scholar] [CrossRef]
- Tabbò, F.; Nottegar, A.; Guerrera, F.; Migliore, E.; Luchini, C.; Maletta, F.; Veronese, N.; Montagna, L.; Gaudiano, M.; Di Giacomo, F.; et al. Cell of origin markers identify different prognostic subgroups of lung adenocarcinoma. Hum. Pathol. 2018, 75, 167–178. [Google Scholar] [CrossRef]
- Flowers, B.M.; Xu, H.; Mulligan, A.S.; Hanson, K.J.; Seoane, J.A.; Vogel, H.; Curtis, C.; Wood, L.D.; Attardi, L.D. Cell of Origin Influences Pancreatic Cancer Subtype. Cancer Discov. 2021, 11, 660–677. [Google Scholar] [CrossRef]
- Santagata, S.; Thakkar, A.; Ergonul, A.; Wang, B.; Woo, T.; Hu, R.; Harrell, J.C.; McNamara, G.; Schwede, M.; Culhane, A.; et al. Taxonomy of breast cancer based on normal cell phenotype predicts outcome. J. Clin. Investig. 2014, 124, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Ince, T.A. Normal cell phenotypes of breast epithelial cells provide the foundation of a breast cancer taxonomy. Expert Rev. Anticancer Ther. 2014, 14, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Merritt, M.A.; Bentink, S.; Schwede, M.; Iwanicki, M.P.; Quackenbush, J.; Woo, T.; Agoston, E.S.; Reinhardt, F.; Crum, C.P.; Berkowitz, R.S.; et al. Gene Expression Signature of Normal Cell-of-Origin Predicts Ovarian Tumor Outcomes. PLoS ONE 2013, 8, e80314. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Livne-Bar, I.; Vanderluit, J.L.; Slack, R.; Agochiya, M.; Bremner, R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell 2004, 5, 539–551. [Google Scholar] [CrossRef]
- Shimizu, D.; Taniue, K.; Matsui, Y.; Haeno, H.; Araki, H.; Miura, F.; Fukunaga, M.; Shiraishi, K.; Miyamoto, Y.; Tsukamoto, S.; et al. Pan-cancer methylome analysis for cancer diagnosis and classification of cancer cell of origin. Cancer Gene Ther. 2021, 29, 428–436. [Google Scholar] [CrossRef]
- Mancarella, D.; Plass, C. Epigenetic signatures in cancer: Proper controls, current challenges and the potential for clinical translation. Genome Med. 2021, 13, 23. [Google Scholar] [CrossRef]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct Epigenomic Landscapes of Pluripotent and Lineage-Committed Human Cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef]
- Xin, L. Cells of Origin for Prostate Cancer. Adv. Exp. Med. Biol. 2019, 1210, 67–86. [Google Scholar] [CrossRef]
- Gombar, S.; MacCarthy, T.; Bergman, A. Epigenetics Decouples Mutational from Environmental Robustness. Did It Also Facilitate Multicellularity? PLoS Comput. Biol. 2014, 10, e1003450. [Google Scholar] [CrossRef][Green Version]
- Godar, S.; Ince, T.A.; Bell, G.W.; Feldser, D.; Donaher, J.L.; Bergh, J.; Liu, A.; Miu, K.; Watnick, R.S.; Reinhardt, F.; et al. Growth-Inhibitory and Tumor- Suppressive Functions of p53 Depend on Its Repression of CD44 Expression. Cell 2008, 134, 62–73. [Google Scholar] [CrossRef]
- McAllister, S.S.; Gifford, A.M.; Greiner, A.L.; Kelleher, S.P.; Saelzler, M.P.; Ince, T.A.; Reinhardt, F.; Harris, L.N.; Hylander, B.L.; Repasky, E.A.; et al. Systemic Endocrine Instigation of Indolent Tumor Growth Requires Osteopontin. Cell 2008, 133, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 Drives a Transcriptional Program Distinct from Heat Shock to Support Highly Malignant Human Cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Petrocca, F.; Altschuler, G.; Tan, S.M.; Mendillo, M.L.; Yan, H.; Jerry, D.J.; Kung, A.L.; Hide, W.; Ince, T.A.; Lieberman, J. A Genome-wide siRNA Screen Identifies Proteasome Addiction as a Vulnerability of Basal-like Triple-Negative Breast Cancer Cells. Cancer Cell 2013, 24, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Hu, R.; Lin, N.U.; Mendillo, M.L.; Collins, L.C.; Hankinson, S.E.; Schnitt, S.J.; Whitesell, L.; Tamimi, R.M.; Lindquist, S.; et al. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 18378–18383. [Google Scholar] [CrossRef]
- Witt, A.E.; Lee, C.-W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2017, 36, 1707–1720. [Google Scholar] [CrossRef]
- Kamran, M.; Bhattacharya, U.; Omar, M.; Marchionni, L.; Ince, T.A. ZNF92, an unexplored transcription factor with remarkably distinct breast cancer over-expression associated with prognosis and cell-of-origin. npj Breast Cancer 2022, 8, 99. [Google Scholar] [CrossRef]
- Caslini, C.; Hong, S.; Ban, Y.J.; Chen, X.S.; Ince, T.A. HDAC7 regulates histone 3 lysine 27 acetylation and transcriptional activity at super-enhancer-associated genes in breast cancer stem cells. Oncogene 2019, 38, 6599–6614. [Google Scholar] [CrossRef]
- Stormo, C.; Kringen, M.K.; Lyle, R.; Olstad, O.K.; Sachse, D.; Berg, J.P.; Piehler, A.P. RNA-Sequencing Analysis of HepG2 Cells Treated with Atorvastatin. PLoS ONE 2014, 9, e105836. [Google Scholar] [CrossRef]
- Thakkar, A.; Wang, B.; Picon-Ruiz, M.; Buchwald, P.; Ince, T.A. Vitamin D and androgen receptor-targeted therapy for triple-negative breast cancer. Breast Cancer Res. Treat. 2016, 157, 77–90. [Google Scholar] [CrossRef]
- Liu, C.-J.; Hu, F.-F.; Xia, M.-X.; Han, L.; Zhang, Q.; Guo, A.-Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.; Lightfoot, H.; Forbes, S.; Sridhar, R.; Futreal, P.A.; Haber, D.; Stratton, M.; et al. Abstract 2206: Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. In Proceedings of the AACR 104th Annual Meeting, Washington, DC, USA, 6–10 April 2013. [Google Scholar] [CrossRef]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An Interactive Resource to Identify Cancer Genetic and Lineage Dependencies Targeted by Small Molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Stopeck, A.; Linden, H.; Solit, D.; Chandarlapaty, S.; Rosen, N.; D’Andrea, G.; Dickler, M.; Moynahan, M.E.; Sugarman, S.; et al. HSP90 Inhibition Is Effective in Breast Cancer: A Phase II Trial of Tanespimycin (17-AAG) Plus Trastuzumab in Patients with HER2-Positive Metastatic Breast Cancer Progressing on Trastuzumab. Clin. Cancer Res. 2011, 17, 5132–5139. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.D.; Hewlett, E.L. Niclosamide Therapy for Tapeworm Infections. Ann. Intern. Med. 1985, 102, 550. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Brana, I.; Haddad, R.; Bauman, J.; Bible, K.; Oosting, S.; Wong, D.J.; Ahn, M.-J.; Boni, V.; Even, C.; et al. Tipifarnib in Head and Neck Squamous Cell Carcinoma With HRAS Mutations. J. Clin. Oncol. 2021, 39, 1856–1864. [Google Scholar] [CrossRef]
- Dhuguru, J.; Ghoneim, O.A. Quinazoline Based HDAC Dual Inhibitors as Potential Anti-Cancer Agents. Molecules 2022, 27, 2294. [Google Scholar] [CrossRef]
- Kurokawa, Y.; Honma, Y.; Sawaki, A.; Naito, Y.; Iwagami, S.; Komatsu, Y.; Takahashi, T.; Nishida, T.; Doi, T. Pimitespib in patients with advanced gastrointestinal stromal tumor (CHAPTER-GIST-301): A randomized, double-blind, placebo-controlled phase III trial. Ann. Oncol. 2022, 33, 959–967. [Google Scholar] [CrossRef]
- Burock, S.; Daum, S.; Keilholz, U.; Neumann, K.; Walther, W.; Stein, U. Phase II trial to investigate the safety and efficacy of orally applied niclosamide in patients with metachronous or sychronous metastases of a colorectal cancer progressing after therapy: The NIKOLO trial. BMC Cancer 2018, 18, 297. [Google Scholar] [CrossRef]
- Parikh, M.; Liu, C.; Wu, C.-Y.; Evans, C.P.; Dall’Era, M.; Robles, D.; Lara, P.N.; Agarwal, N.; Gao, A.C.; Pan, C.-X. Phase Ib trial of reformulated niclosamide with abiraterone/prednisone in men with castration-resistant prostate cancer. Sci. Rep. 2021, 11, 6377. [Google Scholar] [CrossRef]
- Fakih, M.G.; Pendyala, L.; Fetterly, G.; Toth, K.; Zwiebel, J.A.; Espinoza-Delgado, I.; Litwin, A.; Rustum, Y.M.; Ross, M.E.; Holleran, J.L.; et al. A Phase I, Pharmacokinetic and Pharmacodynamic Study on Vorinostat in Combination with 5-Fluorouracil, Leucovorin, and Oxaliplatin in Patients with Refractory Colorectal Cancer. Clin. Cancer Res. 2009, 15, 3189–3195. [Google Scholar] [CrossRef]
- Kummar, S.; Gutierrez, M.; Gardner, E.R.; Donovan, E.; Hwang, K.; Chung, E.J.; Lee, M.J.; Maynard, K.; Kalnitskiy, M.; Chen, A.; et al. Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin. Cancer Res. 2007, 13, 5411–5417. [Google Scholar] [CrossRef] [PubMed]
- Lassen, U.; Molife, L.R.; Sorensen, M.; Engelholm, S.-A.; Vidal, L.; Sinha, R.; Penson, R.T.; Buhl-Jensen, P.; Crowley, E.; Tjornelund, J.; et al. A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br. J. Cancer 2010, 103, 12–17. [Google Scholar] [CrossRef]
- Steele, N.L.; Plumb, J.A.; Vidal, L.; Tjørnelund, J.; Knoblauch, P.; Rasmussen, A.; Ooi, C.E.; Buhl-Jensen, P.; Brown, R.; Evans, T.J.; et al. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 804–810. [Google Scholar] [CrossRef]
- Gojo, I.; Jiemjit, A.; Trepel, J.B.; Sparreboom, A.; Figg, W.D.; Rollins, S.; Tidwell, M.L.; Greer, J.; Chung, E.J.; Lee, M.-J.; et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 2006, 109, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Robertson, F.M.; Woodward, W.; Pickei, R.; Ye, Z.; Bornmann, W.; Pal, A.; Peng, Z.; Hall, C.S.; Cristofanilli, M.; Woodward, W.A.; et al. Suberoylanilide hydroxamic acid blocks self-renewal and homotypic aggregation of inflammatory breast cancer spheroids. Cancer 2010, 116, 2760–2767. [Google Scholar] [CrossRef]
- Robertson, F.M.; Chu, K.; Boley, K.M.; Ye, Z.; Liu, H.; Wright, M.C.; Moraes, R.; Zhang, X.; Green, T.L.; Barsky, S.H.; et al. The class I HDAC inhibitor Romidepsin targets inflammatory breast cancer tumor emboli and synergizes with paclitaxel to inhibit metastasis. J. Exp. Ther. Oncol. 2013, 10, 219–233. [Google Scholar] [PubMed]
- Correia, A.S.; Gärtner, F.; Vale, N. Drug combination and repurposing for cancer therapy: The example of breast cancer. Heliyon 2021, 7, e05948. [Google Scholar] [CrossRef]
- Modi, S.; Stopeck, A.T.; Gordon, M.S.; Mendelson, D.; Solit, D.B.; Bagatell, R.; Ma, W.; Wheler, J.; Rosen, N.; Norton, L.; et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: A phase I dose-escalation study. J. Clin. Oncol. 2007, 25, 5410–5417. [Google Scholar] [CrossRef]
- Ahmad, S.; He, Q.; Williams, K.P.; Scott, J.E. Identification of a Triple Drug Combination That Is Synergistically Cytotoxic for Triple-Negative Breast Cancer Cells Using a Novel Combination Discovery Approach. SLAS Discov. 2020, 25, 923–938. [Google Scholar] [CrossRef]
- Lohiya, G.; Katti, D. A Synergistic Combination of Niclosamide and Doxorubicin as an Efficacious Therapy for All Clinical Subtypes of Breast Cancer. Cancers 2021, 13, 3299. [Google Scholar] [CrossRef]
- Johnston, P.B.; Cashen, A.F.; Nikolinakos, P.G.; Beaven, A.W.; Barta, S.K.; Bhat, G.; Hasal, S.J.; De Vos, S.; Oki, Y.; Deng, C.; et al. Belinostat in combination with standard cyclophosphamide, doxorubicin, vincristine and prednisone as first-line treatment for patients with newly diagnosed peripheral T-cell lymphoma. Exp. Hematol. Oncol. 2021, 10, 15. [Google Scholar] [CrossRef]
- Zhao, D.; Hu, C.; Fu, Q.; Lv, H. Combined chemotherapy for triple negative breast cancer treatment by paclitaxel and niclosamide nanocrystals loaded ther-mosensitive hydrogel. Eur. J. Pharm. Sci. 2021, 167, 105992. [Google Scholar] [CrossRef] [PubMed]
- Ávalos-Moreno, M.; López-Tejada, A.; Blaya-Cánovas, J.; Cara-Lupiañez, F.; González-González, A.; Lorente, J.; Sánchez-Rovira, P.; Granados-Principal, S. Drug Repurposing for Triple-Negative Breast Cancer. J. Pers. Med. 2020, 10, 200. [Google Scholar] [CrossRef] [PubMed]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; de Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef]
- Cokol-Cakmak, M.; Cetiner, S.; Erdem, N.; Bakan, F.; Cokol, M. Guided screen for synergistic three-drug combinations. PLoS ONE 2020, 15, e0235929. [Google Scholar] [CrossRef] [PubMed]
- Katzir, I.; Cokol, M.; Aldridge, B.B.; Alon, U. Prediction of ultra-high-order antibiotic combinations based on pairwise interactions. PLoS Comput. Biol. 2019, 15, e1006774. [Google Scholar] [CrossRef]
- Meyer, C.T.; Wooten, D.J.; Lopez, C.F.; Quaranta, V. Charting the Fragmented Landscape of Drug Synergy. Trends Pharmacol. Sci. 2020, 41, 266–280. [Google Scholar] [CrossRef]
- Cairns, D.M.; Dulko, D.; Griffiths, J.K.; Golan, Y.; Cohen, T.; Trinquart, L.; Price, L.L.; Beaulac, K.R.; Selker, H.P. Efficacy of Niclosamide vs. Placebo in SARS-CoV-2 Respiratory Viral Clearance, Viral Shedding, and Duration of Symptoms Among Patients With Mild to Moderate COVID-19: A Phase 2 Randomized Clinical Trial. JAMA Netw. 2022, 5, e2144942. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Doi, T.; Kurokawa, Y.; Sawaki, A.; Komatsu, Y.; Ozaka, M.; Takahashi, T.; Naito, Y.; Ohkubo, S.; Nishida, T. Efficacy and safety of TAS-116, an oral inhibitor of heat shock protein 90, in patients with metastatic or unresectable gastro-intestinal stromal tumour refractory to imatinib, sunitinib and regorafenib: A phase II, single-arm trial. Eur. J. Cancer 2019, 121, 29–39. [Google Scholar] [CrossRef]
- Hsu, T.-J.; Nepali, K.; Tsai, C.-H.; Imtiyaz, Z.; Lin, F.-L.; Hsiao, G.; Lai, M.-J.; Cheng, Y.-W. The HDAC/HSP90 Inhibitor G570 Attenuated Blue Light-Induced Cell Migration in RPE Cells and Neovascularization in Mice through Decreased VEGF Production. Molecules 2021, 26, 4359. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Chao, M.-W.; Tu, H.-J.; Chen, L.-C.; Hsu, K.-C.; Liou, J.-P.; Yang, C.-R.; Yen, S.-C.; HuangFu, W.-C.; Pan, S.-L. A novel dual HDAC and HSP90 inhibitor, MPT0G449, downregulates oncogenic pathways in human acute leukemia in vitro and in vivo. Oncogenesis 2021, 10, 39. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef]
- Houseman, E.A.; Ince, T.A. Normal Cell-Type Epigenetics and Breast Cancer Classification: A Case Study of Cell Mixture–Adjusted Analysis of DNA Methylation Data from Tumors: Supplementary Issue: Array Platform Modeling and Analysis (A.). Cancer Inform. 2014, 13, S13980. [Google Scholar] [CrossRef]
- Houseman, E.A.; Kile, M.L.; Christiani, D.C.; Ince, T.A.; Kelsey, K.T.; Marsit, C.J. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinform. 2016, 17, 259. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Schmidt-Supprian, M.; Rad, G.S.R.; Saur, D. Tissue-specific tumorigenesis: Context matters. Nat. Rev. Cancer 2017, 17, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M.; Cichowski, K.; Elledge, S.J. Tissue-specificity in cancer: The rule, not the exception. Science 2019, 363, 1150–1151. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, J.J.; Zhao, X.; Mays, J.C.; Davoli, T. Not all cancers are created equal: Tissue specificity in cancer genes and pathways. Curr. Opin. Cell Biol. 2020, 63, 135–143. [Google Scholar] [CrossRef]
- Unberath, P.; Knell, C.; Prokosch, H.U.; Christoph, J. Developing New Analysis Functions for a Translational Research Platform: Extending the cBioPortal for Cancer Genomics. Stud. Health Technol. Inform. 2019, 258, 46–50. [Google Scholar]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal 2013, 6, 11. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drug | Phase I–II Cmax (nM) | Experimental Three-Drug (nM) | Ratio |
|---|---|---|---|
| Entinostat | 1000 | 100 | 10 |
| Vorinostat | 40,000 | 1000 | 40 |
| Belinostat | 200,000 | 1000 | 200 |
| Niclosamide | 2000 | 100 | 20 |
| Tanespimycin | 15,000 | 50 | 300 |
| Pimitespib | 7000 | 200–600 | 11–35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, U.; Kamran, M.; Manai, M.; Cristofanilli, M.; Ince, T.A. Cell-of-Origin Targeted Drug Repurposing for Triple-Negative and Inflammatory Breast Carcinoma with HDAC and HSP90 Inhibitors Combined with Niclosamide. Cancers 2023, 15, 332. https://doi.org/10.3390/cancers15020332
Bhattacharya U, Kamran M, Manai M, Cristofanilli M, Ince TA. Cell-of-Origin Targeted Drug Repurposing for Triple-Negative and Inflammatory Breast Carcinoma with HDAC and HSP90 Inhibitors Combined with Niclosamide. Cancers. 2023; 15(2):332. https://doi.org/10.3390/cancers15020332
Chicago/Turabian StyleBhattacharya, Udayan, Mohammad Kamran, Maroua Manai, Massimo Cristofanilli, and Tan A. Ince. 2023. "Cell-of-Origin Targeted Drug Repurposing for Triple-Negative and Inflammatory Breast Carcinoma with HDAC and HSP90 Inhibitors Combined with Niclosamide" Cancers 15, no. 2: 332. https://doi.org/10.3390/cancers15020332
APA StyleBhattacharya, U., Kamran, M., Manai, M., Cristofanilli, M., & Ince, T. A. (2023). Cell-of-Origin Targeted Drug Repurposing for Triple-Negative and Inflammatory Breast Carcinoma with HDAC and HSP90 Inhibitors Combined with Niclosamide. Cancers, 15(2), 332. https://doi.org/10.3390/cancers15020332