


Decoding Diffuse Midline Gliomas: A Comprehensive Review of Pathogenesis, Diagnosis and Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. The WHO Classification
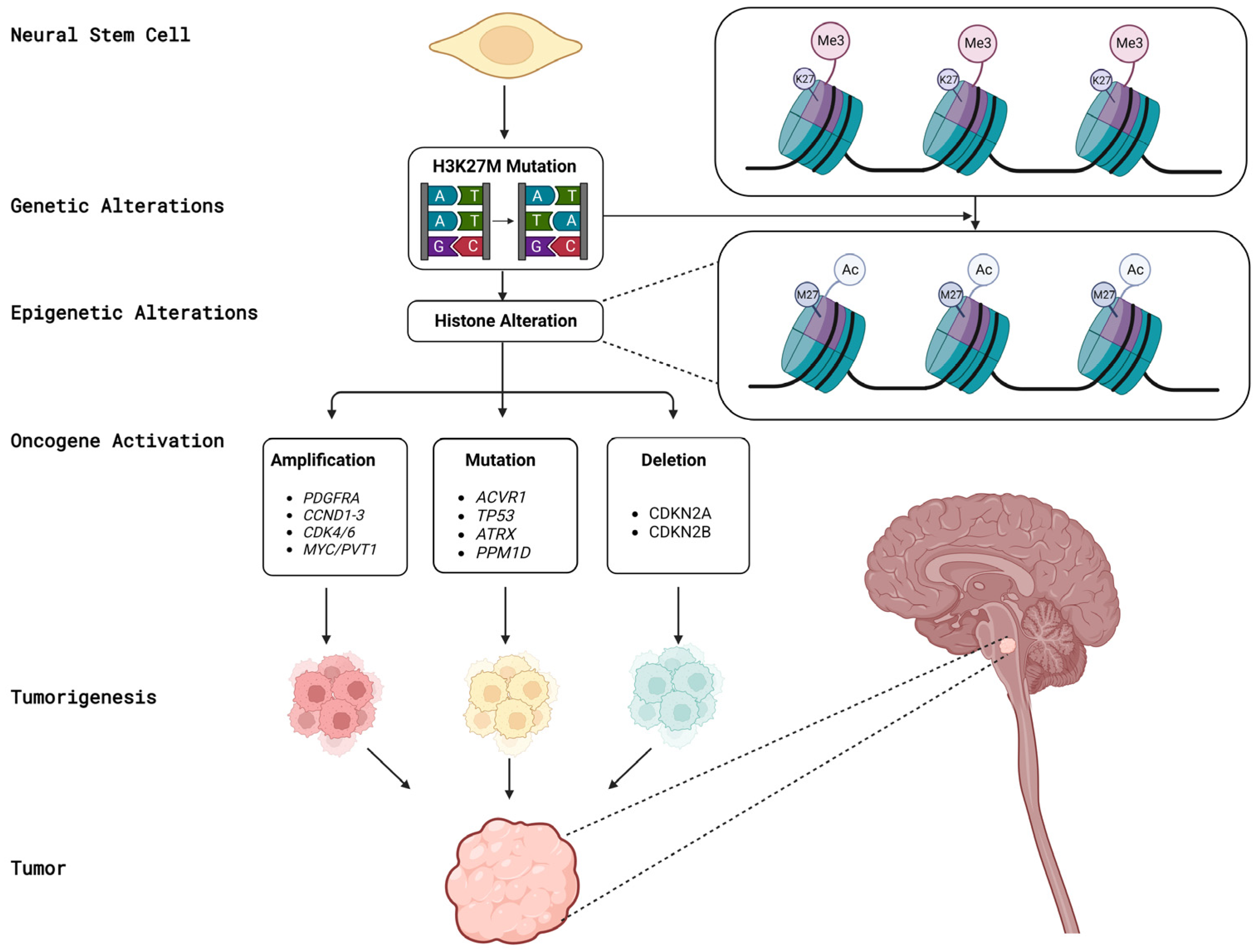
3. Biological Aspects of DMGs
4. Clinical Presentation and Prevalence
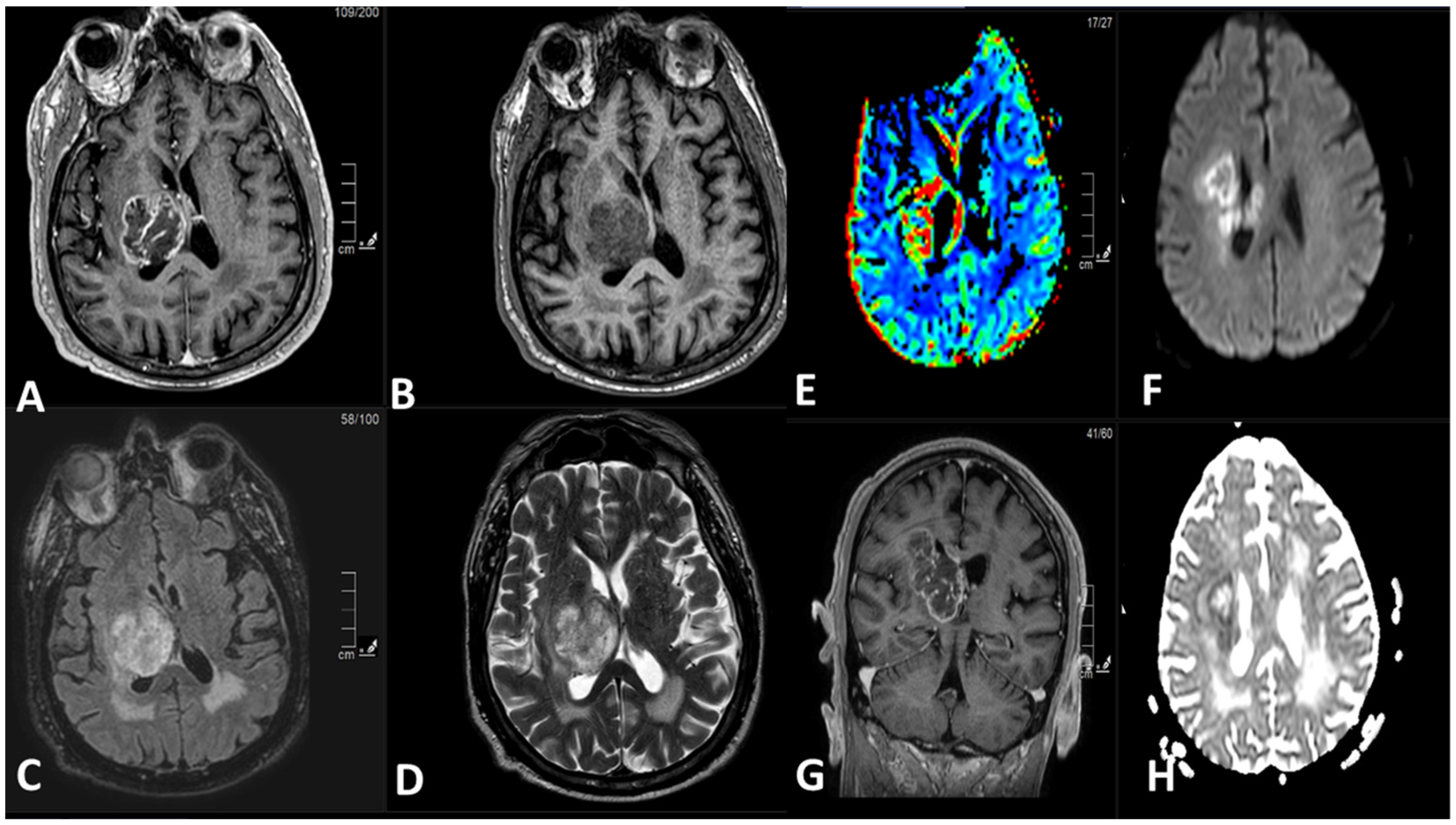
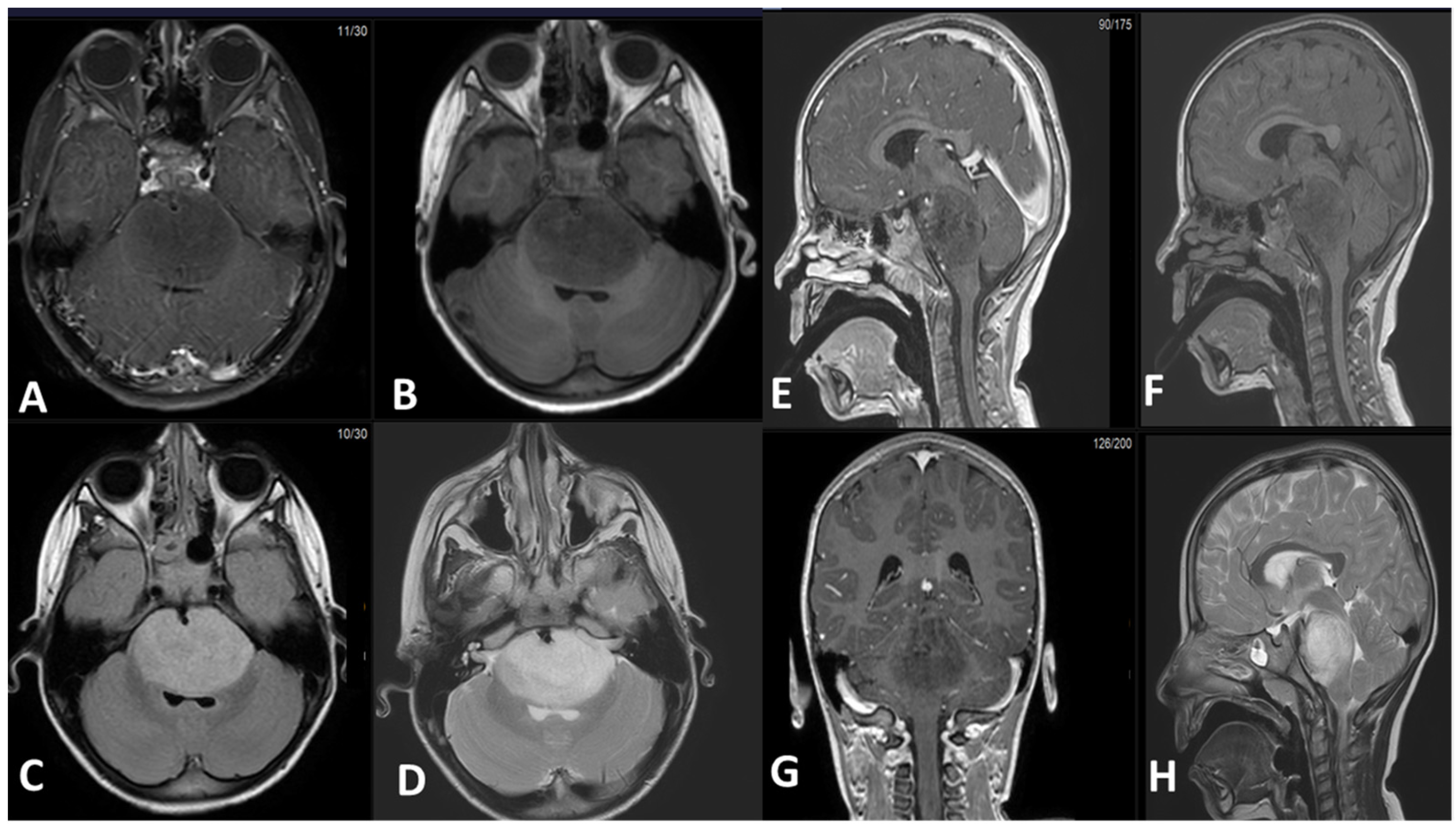
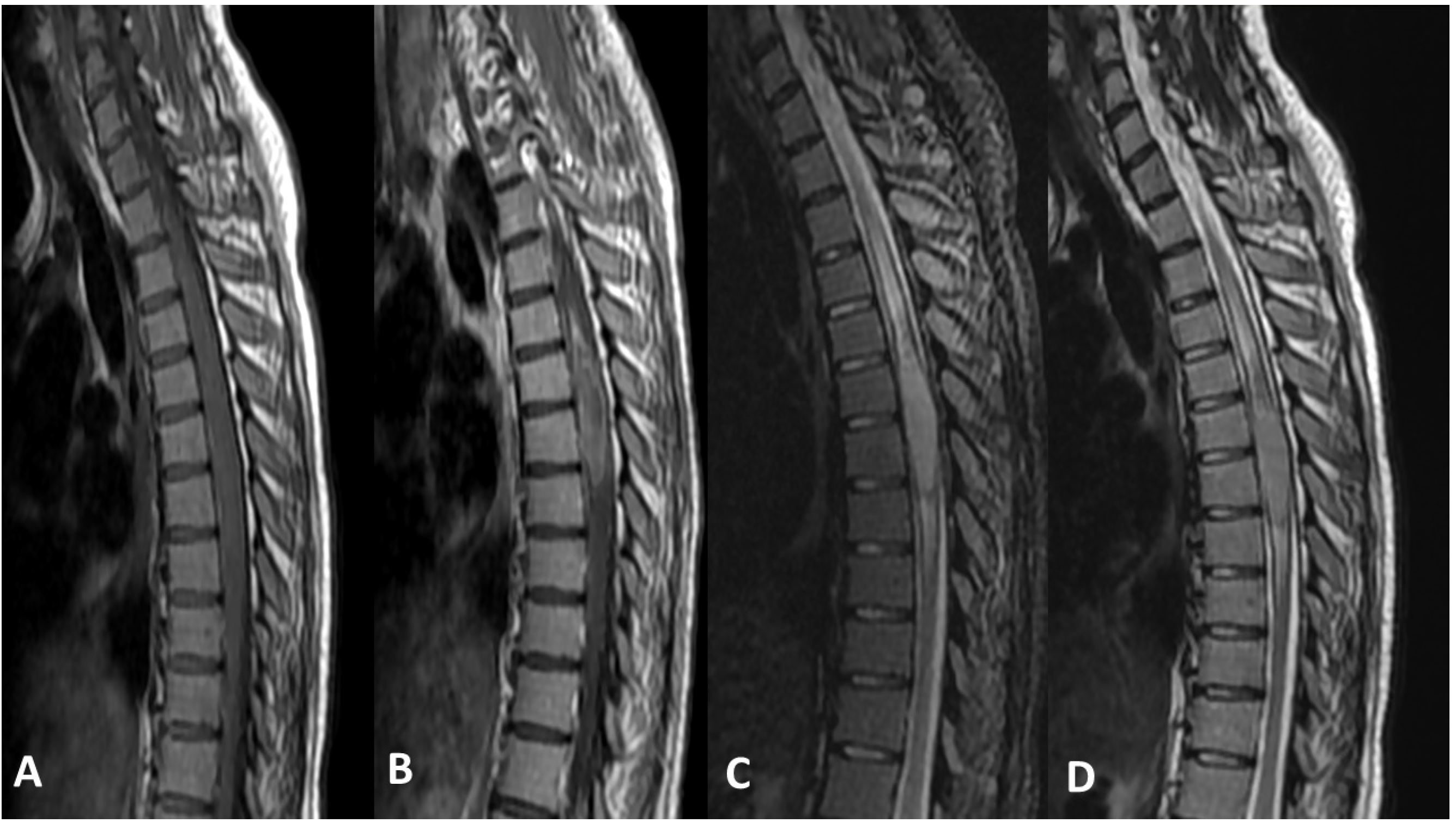
5. Radiological Features
5.1. Magnetic Resonance Imaging (MRI):
5.2. Positron Emission Tomography (PET)
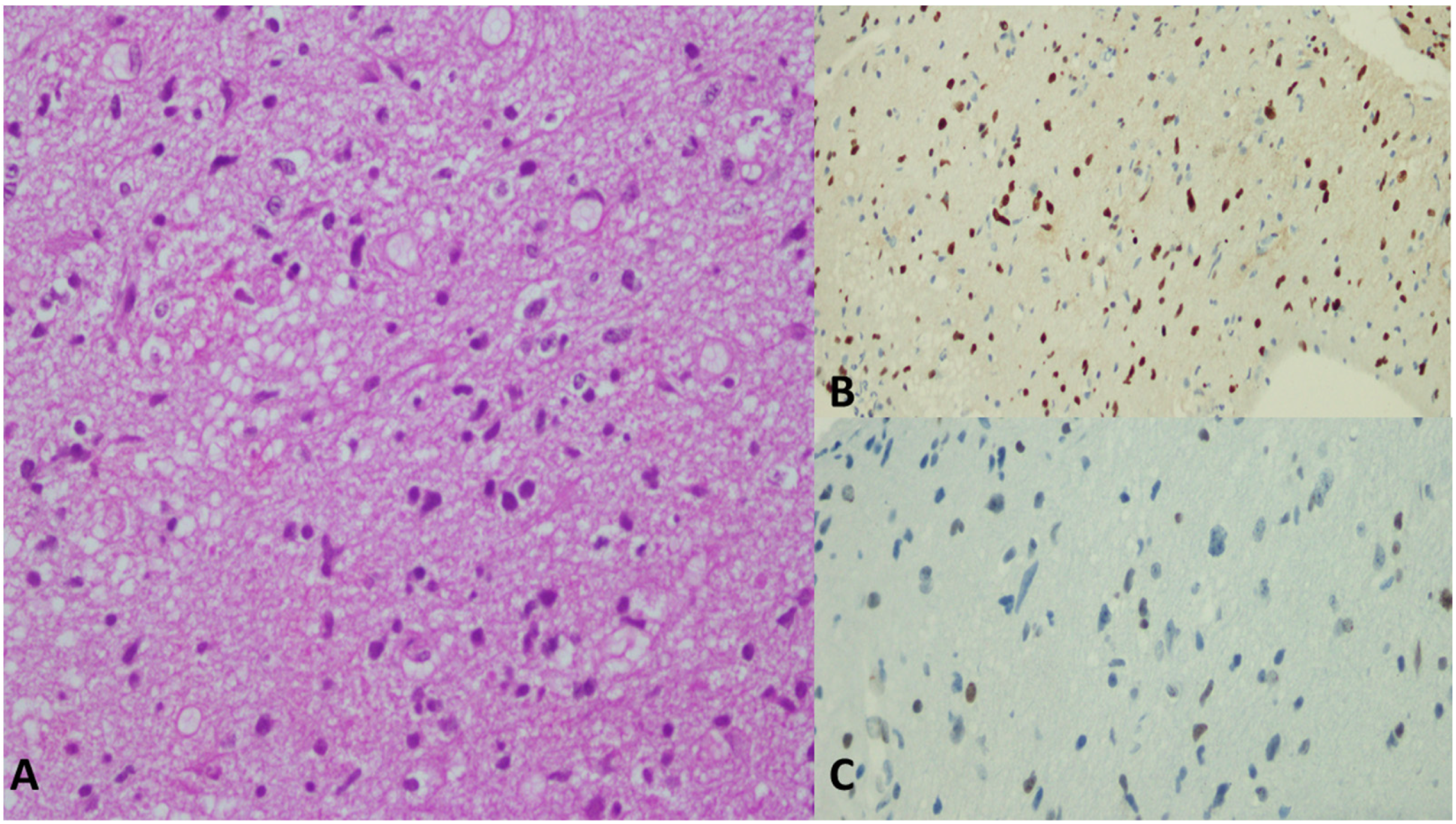
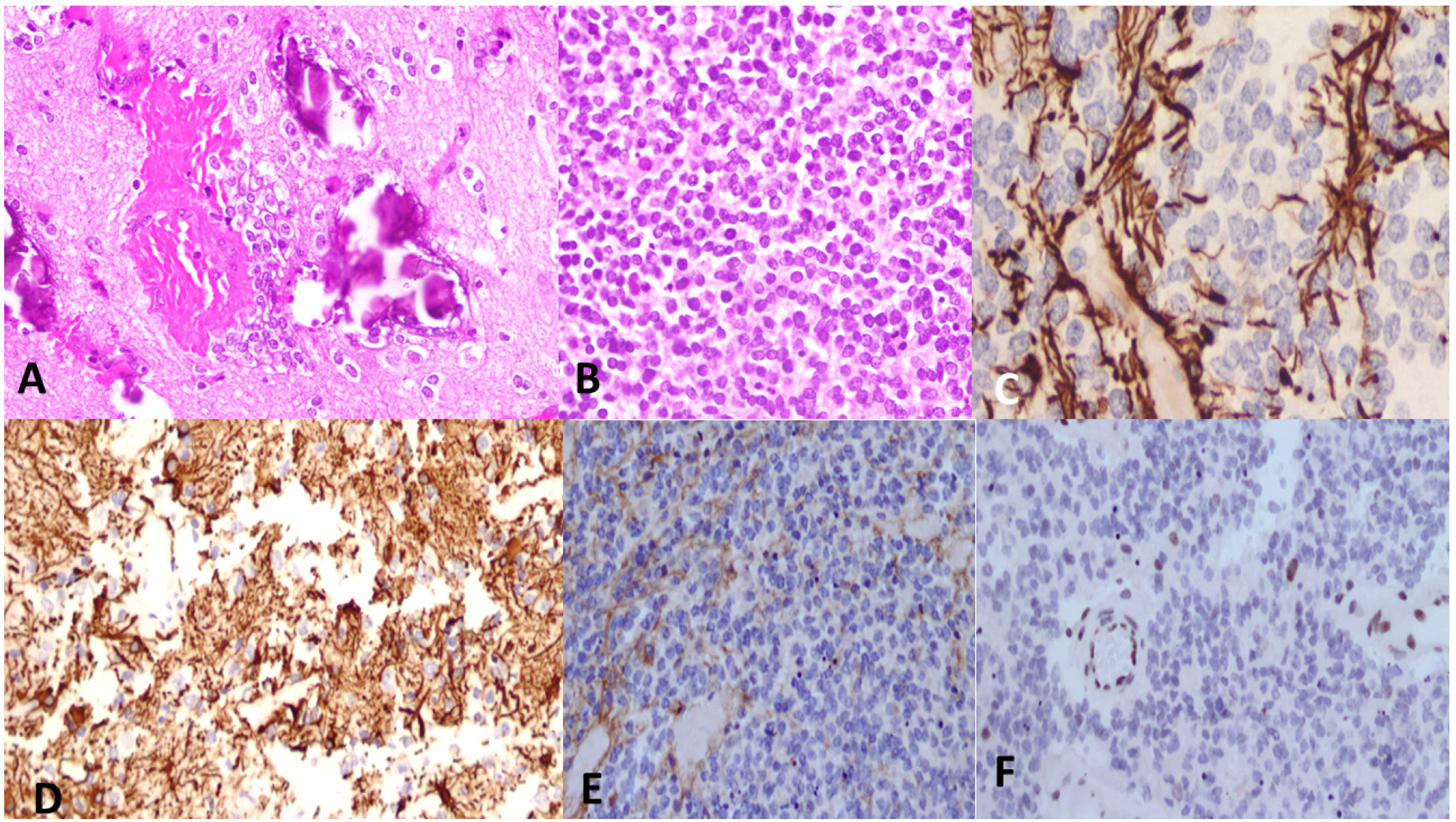
6. Histological Features
7. The Role of Biopsy in DMGs
8. Treatment Options
8.1. Chemotherapy
8.2. Radiotherapy
8.3. Targeted Therapy and Immunotherapy
9. Key Determinants of Drug Resistance
10. Prognostic Factors
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.M.; Veldhuijzen van Zanten, S.E.M.; Colditz, N.; Baugh, J.; Chaney, B.; Hoffmann, M.; Lane, A.; Fuller, C.; Miles, L.; Hawkins, C.; et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 2018, 36, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Johung, T.B.; Monje, M. Diffuse Intrinsic Pontine Glioma: New Pathophysiological Insights and Emerging Therapeutic Targets. Curr. Neuropharmacol. 2017, 15, 88–97. [Google Scholar] [CrossRef]
- Argersinger, D.P.; Rivas, S.R.; Shah, A.H.; Jackson, S.; Heiss, J.D. New Developments in the Pathogenesis, Therapeutic Targeting, and Treatment of H3K27M-Mutant Diffuse Midline Glioma. Cancers 2021, 13, 5280. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, S.; Monje, M. An active role for neurons in glioma progression: Making sense of Scherer’s structures. Neuro-Oncology 2018, 20, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Brownson, R.H. Perineuronal satellite cells in the motor cortex of aging brains. J. Neuropathol. Exp. Neurol. 1956, 15, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.K.; Zhou, S.S.; Russell, M.J.; Geddes, J.; Ellis, W.; Cotman, C.W. Perineuronal satellitosis in the human hippocampal formation. Hippocampus 1993, 3, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.J.; Jabado, N.; Grill, J. Diffuse intrinsic pontine gliomas-current management and new biologic insights. Is there a glimmer of hope? Neuro-Oncology 2017, 19, 1025–1034. [Google Scholar] [CrossRef]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers 2019, 11, 660. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Krug, B.; De Jay, N.; Harutyunyan, A.S.; Deshmukh, S.; Marchione, D.M.; Guilhamon, P.; Bertrand, K.C.; Mikael, L.G.; McConechy, M.K.; Chen, C.C.L.; et al. Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell 2019, 35, 782–797.e788. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.; Monje, M. Developmental origins and emerging therapeutic opportunities for childhood cancer. Nat. Med. 2019, 25, 367–376. [Google Scholar] [CrossRef]
- Rashed, W.M.; Maher, E.; Adel, M.; Saber, O.; Zaghloul, M.S. Pediatric diffuse intrinsic pontine glioma: Where do we stand? Cancer Metastasis Rev. 2019, 38, 759–770. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Hoffman, S.E.; Kappel, A.D.; Valdes, P.A.; Essayed, W.; Klinger, N.V.; Kang, K.D.; Totsch, S.K.; Olsen, H.E.; Schlappi, C.W.; et al. Immunotherapy approaches for the treatment of diffuse midline gliomas. Oncoimmunology 2022, 11, 2124058. [Google Scholar] [CrossRef]
- Louis, D.N.; Giannini, C.; Capper, D.; Paulus, W.; Figarella-Branger, D.; Lopes, M.B.; Batchelor, T.T.; Cairncross, J.G.; van den Bent, M.; Wick, W.; et al. cIMPACT-NOW update 2: Diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol. 2018, 135, 639–642. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Di Ruscio, V.; Del Baldo, G.; Fabozzi, F.; Vinci, M.; Cacchione, A.; de Billy, E.; Megaro, G.; Carai, A.; Mastronuzzi, A. Pediatric Diffuse Midline Gliomas: An Unfinished Puzzle. Diagnostics 2022, 12, 2064. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hawkins, C. Pathology, Molecular Genetics, and Epigenetics of Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2015, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E. Beyond the Blood:Brain Barrier: The Importance of Central Nervous System (CNS) Pharmacokinetics for the Treatment of CNS Tumors, Including Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2018, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Noon, A.; Galban, S. Therapeutic avenues for targeting treatment challenges of diffuse midline gliomas. Neoplasia 2023, 40, 100899. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef]
- Moudgil-Joshi, J.; Kaliaperumal, C. Letter regarding Louis et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 2120–2121. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef]
- Gojo, J.; Pavelka, Z.; Zapletalova, D.; Schmook, M.T.; Mayr, L.; Madlener, S.; Kyr, M.; Vejmelkova, K.; Smrcka, M.; Czech, T.; et al. Personalized Treatment of H3K27M-Mutant Pediatric Diffuse Gliomas Provides Improved Therapeutic Opportunities. Front. Oncol. 2019, 9, 1436. [Google Scholar] [CrossRef]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef]
- Paugh, B.S.; Broniscer, A.; Qu, C.; Miller, C.P.; Zhang, J.; Tatevossian, R.G.; Olson, J.M.; Geyer, J.R.; Chi, S.N.; da Silva, N.S.; et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J. Clin. Oncol. 2011, 29, 3999–4006. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Philippe, C.; Kergrohen, T.; Sill, M.; Merlevede, J.; Barret, E.; Puget, S.; Sainte-Rose, C.; Kramm, C.M.; Jones, C.; et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun. 2018, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro-Oncology 2018, 21, 83–94. [Google Scholar] [CrossRef]
- Minasi, S.; Baldi, C.; Gianno, F.; Antonelli, M.; Buccoliero, A.M.; Pietsch, T.; Massimino, M.; Buttarelli, F.R. Alternative lengthening of telomeres in molecular subgroups of paediatric high-grade glioma. Childs Nerv. Syst. 2021, 37, 809–818. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Bartels, U.; Bouffet, E.; Becher, O.; Hawkins, C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: Diagnostic and therapeutic implications. Acta Neuropathol. 2014, 128, 573–581. [Google Scholar] [CrossRef]
- Zarghooni, M.; Bartels, U.; Lee, E.; Buczkowicz, P.; Morrison, A.; Huang, A.; Bouffet, E.; Hawkins, C. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J. Clin. Oncol. 2010, 28, 1337–1344. [Google Scholar] [CrossRef]
- Truong, A.Y.; Nicolaides, T.P. Targeted Therapy for MAPK Alterations in Pediatric Gliomas. Brain Disord. Ther. 2015, (Suppl. S2), 005. [Google Scholar] [CrossRef]
- Xu, K.; Sun, Z.; Wang, L.; Guan, W. Bilateral Thalamic Gliomas Harboring Alterations of EGFR and H3K27M: An Integrated Clinicopathological Characteristics Case Series. World Neurosurg. 2022, 168, e442–e450. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Skardelly, M.; Bonzheim, I.; Ott, I.; Mühleisen, H.; Eckert, F.; Tabatabai, G.; Schittenhelm, J. ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol. Commun. 2016, 4, 60. [Google Scholar] [CrossRef]
- Li, H.; Shan, C.; Wu, S.; Cheng, B.; Fan, C.; Cai, L.; Chen, Y.; Shi, Y.; Liu, K.; Shao, Y.; et al. Genomic Profiling Identified Novel Prognostic Biomarkers in Chinese Midline Glioma Patients. Front. Oncol. 2020, 10, 607429. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.L.; Wilson, K.M.; Ceribelli, M.; Stanton, B.Z.; Woo, P.J.; Kreimer, S.; Qin, E.Y.; Zhang, X.; Lennon, J.; Nagaraja, S.; et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci. Transl. Med. 2019, 11, eaaw0064. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.K.; Smith-Cohn, M.A.; Tamrazi, B.; Ji, J.; Krieger, M.; Holdhoff, M.; Eberhart, C.G.; Margol, A.S.; Cotter, J.A. IDH-mutant brainstem gliomas in adolescent and young adult patients: Report of three cases and review of the literature. Brain Pathol. 2021, 31, e12959. [Google Scholar] [CrossRef]
- Coleman, C.; Chen, K.; Lu, A.; Seashore, E.; Stoller, S.; Davis, T.; Braunstein, S.; Gupta, N.; Mueller, S. Interdisciplinary care of children with diffuse midline glioma. Neoplasia 2023, 35, 100851. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E. Diffuse intrinsic pontine glioma: Poised for progress. Front. Oncol. 2012, 2, 205. [Google Scholar] [CrossRef]
- Boele, F.W.; Rooney, A.G.; Grant, R.; Klein, M. Psychiatric symptoms in glioma patients: From diagnosis to management. Neuropsychiatr. Dis. Treat. 2015, 11, 1413–1420. [Google Scholar] [CrossRef]
- Sharma, A.; Graber, J.J. Overview of prognostic factors in adult gliomas. Ann. Palliat. Med. 2021, 10, 863–874. [Google Scholar] [CrossRef]
- American Association of Neuropathologists, Inc. Abstracts of the 97th Annual Meeting 10–13 June 2021, St. Louis, MO, USA. J. Neuropathol. Exp. Neurol. 2021, 80, 558–607. [Google Scholar] [CrossRef]
- ’t Hart, E.; Bianco, J.; Besse, H.C.; Chin Joe Kie, L.A.; Cornet, L.; Eikelenboom, K.L.; van den Broek, T.J.M.; Derieppe, M.; Su, Y.; Hoving, E.W.; et al. Towards Standardisation of a Diffuse Midline Glioma Patient-Derived Xenograft Mouse Model Based on Suspension Matrices for Preclinical Research. Biomedicines 2023, 11, 527. [Google Scholar] [CrossRef]
- Tauziède-Espariat, A.; Siegfried, A.; Uro-Coste, E.; Nicaise, Y.; Castel, D.; Sevely, A.; Gambart, M.; Boetto, S.; Hasty, L.; Métais, A.; et al. Disseminated diffuse midline gliomas, H3K27-altered mimicking diffuse leptomeningeal glioneuronal tumors: A diagnostical challenge! Acta Neuropathol. Commun. 2022, 10, 119. [Google Scholar] [CrossRef]
- Al Sharie, S.; Talafha, M.; Abu Laban, D.; Al Awabdeh, T.; Al-Mousa, A.; Al-Masri, N.; Al-Hussaini, M. H3 K27M-mutant diffuse midline glioma with osseous metastases: A case report and a literature review. Clin. Neuropathol. 2022, 41, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, R.; Baba, A.; Kurokawa, M.; Capizzano, A.; Ota, Y.; Kim, J.; Srinivasan, A.; Moritani, T. Perfusion and diffusion-weighted imaging parameters: Comparison between pre- and postbiopsy MRI for high-grade glioma. Medicine 2022, 101, e30183. [Google Scholar] [CrossRef] [PubMed]
- Resende, L.L.; Alves, C. Imaging of brain tumors in children: The basics-a narrative review. Transl. Pediatr. 2021, 10, 1138–1168. [Google Scholar] [CrossRef] [PubMed]
- Lasocki, A.; Abdalla, G.; Chow, G.; Thust, S.C. Imaging features associated with H3 K27-altered and H3 G34-mutant gliomas: A narrative systematic review. Cancer Imaging 2022, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Raab, P.; Banan, R.; Akbarian, A.; Esmaeilzadeh, M.; Samii, M.; Samii, A.; Bertalanffy, H.; Lehmann, U.; Krauss, J.K.; Lanfermann, H.; et al. Differences in the MRI Signature and ADC Values of Diffuse Midline Gliomas with H3 K27M Mutation Compared to Midline Glioblastomas. Cancers 2022, 14, 1397. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef]
- Aboian, M.S.; Solomon, D.A.; Felton, E.; Mabray, M.C.; Villanueva-Meyer, J.E.; Mueller, S.; Cha, S. Imaging Characteristics of Pediatric Diffuse Midline Gliomas with Histone H3 K27M Mutation. AJNR Am. J. Neuroradiol. 2017, 38, 795–800. [Google Scholar] [CrossRef]
- Chiang, J.; Diaz, A.K.; Makepeace, L.; Li, X.; Han, Y.; Li, Y.; Klimo, P.; Boop, F.A.; Baker, S.J.; Gajjar, A.; et al. Clinical, imaging, and molecular analysis of pediatric pontine tumors lacking characteristic imaging features of DIPG. Acta Neuropathol. Commun. 2020, 8, 57. [Google Scholar] [CrossRef]
- Puget, S.; Blauwblomme, T.; Grill, J. Is Biopsy Safe in Children with Newly Diagnosed Diffuse Intrinsic Pontine Glioma? Am. Soc. Clin. Oncol. Educ. Book 2012, 32, 629–633. [Google Scholar] [CrossRef]
- Tam, L.T.; Yeom, K.W.; Wright, J.N.; Jaju, A.; Radmanesh, A.; Han, M.; Toescu, S.; Maleki, M.; Chen, E.; Campion, A.; et al. MRI-based radiomics for prognosis of pediatric diffuse intrinsic pontine glioma: An international study. Neurooncol. Adv. 2021, 3, vdab042. [Google Scholar] [CrossRef]
- Lovibond, S.; Gewirtz, A.N.; Pasquini, L.; Krebs, S.; Graham, M.S. The promise of metabolic imaging in diffuse midline glioma. Neoplasia 2023, 39, 100896. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.M.; Parolia, A.; Dunphy, M.P.; Venneti, S. Non-invasive metabolic imaging of brain tumours in the era of precision medicine. Nat. Rev. Clin. Oncol. 2016, 13, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Tinkle, C.L.; Duncan, E.C.; Doubrovin, M.; Han, Y.; Li, Y.; Kim, H.; Broniscer, A.; Snyder, S.E.; Merchant, T.E.; Shulkin, B.L. Evaluation of (11)C-Methionine PET and Anatomic MRI Associations in Diffuse Intrinsic Pontine Glioma. J. Nucl. Med. 2019, 60, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Masselli, G.; Casciani, E.; De Angelis, C.; Sollaku, S.; Gualdi, G. Clinical application of (18)F-DOPA PET/TC in pediatric patients. Am. J. Nucl. Med. Mol. Imaging 2021, 11, 64–76. [Google Scholar] [PubMed]
- Piccardo, A.; Tortora, D.; Mascelli, S.; Severino, M.; Piatelli, G.; Consales, A.; Pescetto, M.; Biassoni, V.; Schiavello, E.; Massollo, M.; et al. Advanced MR imaging and (18)F-DOPA PET characteristics of H3K27M-mutant and wild-type pediatric diffuse midline gliomas. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Miele, E.; Spinelli, G.P.; Tomao, F.; Zullo, A.; De Marinis, F.; Pasciuti, G.; Rossi, L.; Zoratto, F.; Tomao, S. Positron Emission Tomography (PET) radiotracers in oncology--utility of 18F-Fluoro-deoxy-glucose (FDG)-PET in the management of patients with non-small-cell lung cancer (NSCLC). J. Exp. Clin. Cancer Res. 2008, 27, 52. [Google Scholar] [CrossRef]
- Alauddin, M.M. Positron emission tomography (PET) imaging with (18)F-based radiotracers. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 55–76. [Google Scholar]
- Pun, M.; Pratt, D.; Nano, P.R.; Joshi, P.K.; Jiang, L.; Englinger, B.; Rao, A.; Cieslik, M.; Chinnaiyan, A.M.; Aldape, K.; et al. Common molecular features of H3K27M DMGs and PFA ependymomas map to hindbrain developmental pathways. Acta Neuropathol. Commun. 2023, 11, 25. [Google Scholar] [CrossRef]
- Antonelli, M.; Poliani, P.L. Adult type diffuse gliomas in the new 2021 WHO Classification. Pathologica 2022, 114, 397–409. [Google Scholar] [CrossRef]
- Komori, T. The 2016 WHO Classification of Tumours of the Central Nervous System: The Major Points of Revision. Neurol. Med. -Chir. 2017, 57, 301–311. [Google Scholar] [CrossRef]
- Jang, S.W.; Song, S.W.; Kim, Y.H.; Cho, Y.H.; Hong, S.H.; Kim, J.H.; Ra, Y.S.; Chong, S. Clinical Features and Prognosis of Diffuse Midline Glioma: A Series of 24 Cases. Brain Tumor Res. Treat. 2022, 10, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Mondal, G.; Lee, J.C.; Ravindranathan, A.; Villanueva-Meyer, J.E.; Tran, Q.T.; Allen, S.J.; Barreto, J.; Gupta, R.; Doo, P.; Van Ziffle, J.; et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020, 139, 1071–1088. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Perbet, R.; Leblond, P.; Vasseur, R.; Stechly, L.; Pierache, A.; Reyns, N.; Touzet, G.; Le Rhun, E.; Vinchon, M.; et al. Identification of prognostic markers in diffuse midline gliomas H3K27M-mutant. Brain Pathol. 2020, 30, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Kergrohen, T.; Tauziède-Espariat, A.; Mackay, A.; Ghermaoui, S.; Lechapt, E.; Pfister, S.M.; Kramm, C.M.; Boddaert, N.; Blauwblomme, T.; et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. 2020, 139, 1109–1113. [Google Scholar] [CrossRef]
- Fortin, J.; Tian, R.; Zarrabi, I.; Hill, G.; Williams, E.; Sanchez-Duffhues, G.; Thorikay, M.; Ramachandran, P.; Siddaway, R.; Wong, J.F.; et al. Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell 2020, 37, 308–323.e312. [Google Scholar] [CrossRef]
- Khadka, P.; Reitman, Z.J.; Lu, S.; Buchan, G.; Gionet, G.; Dubois, F.; Carvalho, D.M.; Shih, J.; Zhang, S.; Greenwald, N.F.; et al. PPM1D mutations are oncogenic drivers of de novo diffuse midline glioma formation. Nat. Commun. 2022, 13, 604. [Google Scholar] [CrossRef]
- Chang, R.; Tosi, U.; Voronina, J.; Adeuyan, O.; Wu, L.Y.; Schweitzer, M.E.; Pisapia, D.J.; Becher, O.J.; Souweidane, M.M.; Maachani, U.B. Combined targeting of PI3K and MEK effector pathways via CED for DIPG therapy. Neurooncol. Adv. 2019, 1, vdz004. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Islam, M.Z.; Li, Y.; Circu, M.L.; Traylor, J.; Notarianni, C.M.; Kline, C.N.; Burns, D.K. Global activation of oncogenic pathways underlies therapy resistance in diffuse midline glioma. Acta Neuropathol. Commun. 2020, 8, 111. [Google Scholar] [CrossRef]
- Yoon, H.I.; Wee, C.W.; Kim, Y.Z.; Seo, Y.; Im, J.H.; Dho, Y.-S.; Kim, K.H.; Hong, J.B.; Park, J.-S.; Choi, S.H.; et al. The Korean Society for Neuro-Oncology (KSNO) Guideline for Adult Diffuse Midline Glioma: Version 2021.1. Brain Tumor Res. Treat. 2021, 9, 1–8. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e525. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Schwartz, J.; Guerin, J.; Johnson, D.; Khatua, S.; Laack, N.; Ahmed, S.; Silvera, V.M.; Brinkmann, D.; Pafundi, D.; et al. EPCT-04. Stereotactic biopsy split-course radiation therapy for diffuse midline glioma of the pons (SPORT-DMG): Early phase II enrolling clinical trial. Neuro-Oncology 2022, 24, i36. [Google Scholar] [CrossRef]
- Krieger, M.D.; Chandrasoma, P.T.; Zee, C.S.; Apuzzo, M.L. Role of stereotactic biopsy in the diagnosis and management of brain tumors. Semin. Surg. Oncol. 1998, 14, 13–25. [Google Scholar] [CrossRef]
- Meshkini, A.; Shahzadi, S.; Alikhah, H.; Naghavi-Behzad, M. Role of stereotactic biopsy in histological diagnosis of multiple brain lesions. Asian J. Neurosurg. 2013, 8, 69–73. [Google Scholar] [CrossRef]
- Katzendobler, S.; Do, A.; Weller, J.; Dorostkar, M.M.; Albert, N.L.; Forbrig, R.; Niyazi, M.; Egensperger, R.; Thon, N.; Tonn, J.C.; et al. Diagnostic Yield and Complication Rate of Stereotactic Biopsies in Precision Medicine of Gliomas. Front. Neurol. 2022, 13, 822362. [Google Scholar] [CrossRef]
- Ekşi, M. A New Era in Stereotactic Brain Biopsy: Frameless Navigation-Based System. J. Neurosci. Rural. Pr. 2019, 10, 3. [Google Scholar] [CrossRef]
- Michela, B. Liquid Biopsy: A Family of Possible Diagnostic Tools. Diagnostics 2021, 11, 1391. [Google Scholar] [CrossRef]
- Azad, T.D.; Jin, M.C.; Bernhardt, L.J.; Bettegowda, C. Liquid biopsy for pediatric diffuse midline glioma: A review of circulating tumor DNA and cerebrospinal fluid tumor DNA. Neurosurg. Focus. 2020, 48, E9. [Google Scholar] [CrossRef]
- Li, Y.Z.; Kong, S.N.; Liu, Y.P.; Yang, Y.; Zhang, H.M. Can Liquid Biopsy Based on ctDNA/cfDNA Replace Tissue Biopsy for the Precision Treatment of EGFR-Mutated NSCLC? J. Clin. Med. 2023, 12, 1438. [Google Scholar] [CrossRef]
- Liu, A.P.; Northcott, P.A.; Robinson, G.W.; Gajjar, A. Circulating tumor DNA profiling for childhood brain tumors: Technical challenges and evidence for utility. Lab. Invest. 2022, 102, 134–142. [Google Scholar] [CrossRef]
- Wadden, J.; Ravi, K.; John, V.; Babila, C.M.; Koschmann, C. Cell-Free Tumor DNA (cf-tDNA) Liquid Biopsy: Current Methods and Use in Brain Tumor Immunotherapy. Front. Immunol. 2022, 13, 882452. [Google Scholar] [CrossRef] [PubMed]
- Hirahata, T.; Ul Quraish, R.; Quraish, A.U.; Ul Quraish, S.; Naz, M.; Razzaq, M.A. Liquid Biopsy: A Distinctive Approach to the Diagnosis and Prognosis of Cancer. Cancer Inf. 2022, 21, 11769351221076062. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.N.; Nisar, S.; Masoodi, T.; Singh, M.; Rizwan, A.; Hashem, S.; El-Rifai, W.; Bedognetti, D.; Batra, S.K.; Haris, M.; et al. Liquid biopsy: A step closer to transform diagnosis, prognosis and future of cancer treatments. Mol. Cancer 2022, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, G.; Cascone, R.; Carlucci, A.; Fiorelli, A.; Di Domenico, M.; Santini, M. Diagnostic and prognostic role of liquid biopsy in non-small cell lung cancer: Evaluation of circulating biomarkers. Explor. Target. Antitumor Ther. 2020, 1, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Greuter, L.; Frank, N.; Guzman, R.; Soleman, J. The Clinical Applications of Liquid Biopsies in Pediatric Brain Tumors: A Systematic Literature Review. Cancers 2022, 14, 2683. [Google Scholar] [CrossRef]
- Azad, T.D.; Bettegowda, C. Longitudinal monitoring of diffuse midline glioma using liquid biopsy. Neuro-Oncology 2022, 24, 1375–1376. [Google Scholar] [CrossRef]
- Armakolas, A.; Kotsari, M.; Koskinas, J. Liquid Biopsies, Novel Approaches and Future Directions. Cancers 2023, 15, 1579. [Google Scholar] [CrossRef]
- Leung, F.; Kulasingam, V.; Diamandis, E.P.; Hoon, D.S.; Kinzler, K.; Pantel, K.; Alix-Panabières, C. Circulating Tumor DNA as a Cancer Biomarker: Fact or Fiction? Clin. Chem. 2016, 62, 1054–1060. [Google Scholar] [CrossRef]
- Feng, Y.; Xu, Q.; Fang, M.; Hu, C. Anlotinib combined with temozolomide for the treatment of patients with diffuse midline glioma: A case report and literature review. Transl. Cancer Res. 2022, 11, 3876–3882. [Google Scholar] [CrossRef]
- Himes, B.T.; Zhang, L.; Daniels, D.J. Treatment Strategies in Diffuse Midline Gliomas With the H3K27M Mutation: The Role of Convection-Enhanced Delivery in Overcoming Anatomic Challenges. Front. Oncol. 2019, 9, 31. [Google Scholar] [CrossRef]
- Ruggiero, A.; Ariano, A.; Triarico, S.; Capozza, M.A.; Romano, A.; Maurizi, P.; Mastrangelo, S.; Attinà, G. Temozolomide and oral etoposide in children with recurrent malignant brain tumors. Drugs Context 2020, 9, 2020-3-1. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.; Nafziger, E.; Leung, D. Evidence-Based Practice: Temozolomide Beyond Glioblastoma. Curr. Oncol. Rep. 2019, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, Y.; Hou, X.; Gao, X.; Shi, Q.; Li, S.; Huang, H. Combined carboplatin and etoposide chemotherapy for patients with recurrent glioma. Ann. Palliat. Med. 2021, 10, 12650–12656. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Kuwashige, H.; Adachi, J.I.; Takahashi, M.; Oda, M.; Kumabe, T.; Shimizu, H. Long-term survival of a patient with diffuse midline glioma in the pineal region: A case report and literature review. Surg. Neurol. Int. 2021, 12, 612. [Google Scholar] [CrossRef]
- Hollingworth, M.; Zacharoulis, S. Infusion-related side-effects during convection enhanced delivery for brainstem-diffuse midline glioma/diffuse intrinsic pontine glioma. J. Neuro-Oncology 2022, 159, 417–424. [Google Scholar] [CrossRef]
- Ali, A.S.; Grönberg, M.; Langer, S.W.; Ladekarl, M.; Hjortland, G.O.; Vestermark, L.W.; Österlund, P.; Welin, S.; Grønbæk, H.; Knigge, U.; et al. Intravenous versus oral etoposide: Efficacy and correlation to clinical outcome in patients with high-grade metastatic gastroenteropancreatic neuroendocrine neoplasms (WHO G3). Med. Oncol. 2018, 35, 47. [Google Scholar] [CrossRef]
- Rezonja, R.; Knez, L.; Cufer, T.; Mrhar, A. Oral treatment with etoposide in small cell lung cancer—Dilemmas and solutions. Radiol. Oncol. 2013, 47, 1–13. [Google Scholar] [CrossRef]
- Heravi Shargh, V.; Luckett, J.; Bouzinab, K.; Paisey, S.; Turyanska, L.; Singleton, W.G.B.; Lowis, S.; Gershkovich, P.; Bradshaw, T.D.; Stevens, M.F.G.; et al. Chemosensitization of Temozolomide-Resistant Pediatric Diffuse Midline Glioma Using Potent Nanoencapsulated Forms of a N(3)-Propargyl Analogue. ACS Appl. Mater. Interfaces 2021, 13, 35266–35280. [Google Scholar] [CrossRef]
- Abe, H.; Natsumeda, M.; Kanemaru, Y.; Watanabe, J.; Tsukamoto, Y.; Okada, M.; Yoshimura, J.; Oishi, M.; Fujii, Y. MGMT Expression Contributes to Temozolomide Resistance in H3K27M-Mutant Diffuse Midline Gliomas and MGMT Silencing to Temozolomide Sensitivity in IDH-Mutant Gliomas. Neurol Med Chir. 2018, 58, 290–295. [Google Scholar] [CrossRef]
- MacDonald, V. Chemotherapy: Managing side effects and safe handling. Can. Vet. J. 2009, 50, 665–668. [Google Scholar]
- Pachocki, C.J.; Hol, E.M. Current perspectives on diffuse midline glioma and a different role for the immune microenvironment compared to glioblastoma. J. Neuroinflammation 2022, 19, 276. [Google Scholar] [CrossRef] [PubMed]
- UK, C.R. Having radiotherapy for brain and spinal cord tumours. Available online: https://www.cancerresearchuk.org/about-cancer/brain-tumours/treatment/radiotherapy/radiotherapy-treatment (accessed on 25 June 2023).
- Thakkar, J.; Kumthekar, P. Chapter 15—Approach to the patient with leptomeningeal metastases. In Neuro-Oncology for the Clinical Neurologist; Strowd, R.E., Ed.; Elsevier: Philadelphia, PA, USA, 2021; pp. 197–209. [Google Scholar]
- Paulino, A.C. Treatment strategies to reduce radiotherapy late effects in children. J. Radiat. Oncol. 2013, 2, 121–128. [Google Scholar] [CrossRef]
- Rutkowski, S.; Bode, U.; Deinlein, F.; Ottensmeier, H.; Warmuth-Metz, M.; Soerensen, N.; Graf, N.; Emser, A.; Pietsch, T.; Wolff, J.E.; et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N. Engl. J. Med. 2005, 352, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Lassaletta, A. Medulloblastoma in infants: The never-ending challenge. Lancet Oncol. 2018, 19, 720–721. [Google Scholar] [CrossRef]
- Remke, M.; Ramaswamy, V. Infant medulloblastoma—Learning new lessons from old strata. Nat. Rev. Clin. Oncol. 2018, 15, 659–660. [Google Scholar] [CrossRef]
- Robison, N.J.; Kieran, M.W. Diffuse intrinsic pontine glioma: A reassessment. J. Neurooncol 2014, 119, 7–15. [Google Scholar] [CrossRef]
- Murthy, V.; Gurram, L.; Kannan, S.; Gandhi, M.; Gupta, T.; Laskar, S.G.; Budrukkar, A.; Agarwal, J.P. Elective nodal dose of 60 Gy or 50 Gy in head and neck cancers: A matched pair analysis of outcomes and toxicity. Adv. Radiat. Oncol. 2017, 2, 339–345. [Google Scholar] [CrossRef]
- Zschaeck, S.; Wust, P.; Graf, R.; Misch, M.; Onken, J.; Ghadjar, P.; Badakhshi, H.; Florange, J.; Budach, V.; Kaul, D. Locally dose-escalated radiotherapy may improve intracranial local control and overall survival among patients with glioblastoma. Radiat. Oncol. 2018, 13, 251. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Wang, P.; Chen, L.; Feng, J.; Qiu, X. High-dose radiotherapy in newly diagnosed low-grade gliomas with nonmethylated O(6)-methylguanine-DNA methyltransferase. Radiat. Oncol. 2021, 16, 157. [Google Scholar] [CrossRef]
- Brook, I. Early side effects of radiation treatment for head and neck cancer. Cancer Radiother. 2021, 25, 507–513. [Google Scholar] [CrossRef]
- Al-Mefty, O.; Kersh, J.E.; Routh, A.; Smith, R.R. The long-term side effects of radiation therapy for benign brain tumors in adults. J. Neurosurg. 1990, 73, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Seidensaal, K.; Harrabi, S.B.; Uhl, M.; Debus, J. Re-irradiation with protons or heavy ions with focus on head and neck, skull base and brain malignancies. Br. J. Radiol. 2020, 93, 20190516. [Google Scholar] [CrossRef] [PubMed]
- Janssens, G.O.; Gidding, C.E.; Van Lindert, E.J.; Oldenburger, F.R.; Erasmus, C.E.; Schouten-Meeteren, A.Y.; Kaanders, J.H. The role of hypofractionation radiotherapy for diffuse intrinsic brainstem glioma in children: A pilot study. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Amsbaugh, M.J.; Mahajan, A.; Thall, P.F.; McAleer, M.F.; Paulino, A.C.; Grosshans, D.; Khatua, S.; Ketonen, L.; Fontanilla, H.; McGovern, S.L. A Phase 1/2 Trial of Reirradiation for Diffuse Intrinsic Pontine Glioma. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 144–148. [Google Scholar] [CrossRef]
- Lassaletta, A.; Strother, D.; Laperriere, N.; Hukin, J.; Vanan, M.I.; Goddard, K.; Lafay-Cousin, L.; Johnston, D.L.; Zelcer, S.; Zapotocky, M.; et al. Reirradiation in patients with diffuse intrinsic pontine gliomas: The Canadian experience. Pediatr. Blood Cancer 2018, 65, e26988. [Google Scholar] [CrossRef]
- Lu, V.M.; Welby, J.P.; Mahajan, A.; Laack, N.N.; Daniels, D.J. Reirradiation for diffuse intrinsic pontine glioma: A systematic review and meta-analysis. Childs Nerv. Syst. 2019, 35, 739–746. [Google Scholar] [CrossRef]
- Hayden, E.; Holliday, H.; Lehmann, R.; Khan, A.; Tsoli, M.; Rayner, B.S.; Ziegler, D.S. Therapeutic Targets in Diffuse Midline Gliomas-An Emerging Landscape. Cancers 2021, 13, 6251. [Google Scholar] [CrossRef]
- Dhar, S.; Gadd, S.; Patel, P.; Vaynshteyn, J.; Raju, G.P.; Hashizume, R.; Brat, D.J.; Becher, O.J. A tumor suppressor role for EZH2 in diffuse midline glioma pathogenesis. Acta Neuropathol. Commun. 2022, 10, 47. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Baer-Dubowska, W. Chapter 2—Pharmacoepigenetics: Basic Principles for Personalized Medicine. In Pharmacoepigenetics; Cacabelos, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 10, pp. 101–112. [Google Scholar]
- Tinkle, C.L.; Broniscer, A.; Chiang, J.; Campagne, O.; Huang, J.; Orr, B.A.; Li, X.; Patay, Z.; Zhang, J.; Baker, S.J.; et al. Phase I study using crenolanib to target PDGFR kinase in children and young adults with newly diagnosed DIPG or recurrent high-grade glioma, including DIPG. Neurooncol. Adv. 2021, 3, vdab179. [Google Scholar] [CrossRef]
- Broniscer, A.; Baker, S.D.; Wetmore, C.; Pai Panandiker, A.S.; Huang, J.; Davidoff, A.M.; Onar-Thomas, A.; Panetta, J.C.; Chin, T.K.; Merchant, T.E.; et al. Phase I trial, pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clin. Cancer Res. 2013, 19, 3050–3058. [Google Scholar] [CrossRef]
- Pollack, I.F.; Jakacki, R.I.; Blaney, S.M.; Hancock, M.L.; Kieran, M.W.; Phillips, P.; Kun, L.E.; Friedman, H.; Packer, R.; Banerjee, A.; et al. Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: A Pediatric Brain Tumor Consortium report. Neuro-Oncology 2007, 9, 145–160. [Google Scholar] [CrossRef]
- Geyer, J.R.; Stewart, C.F.; Kocak, M.; Broniscer, A.; Phillips, P.; Douglas, J.G.; Blaney, S.M.; Packer, R.J.; Gururangan, S.; Banerjee, A.; et al. A phase I and biology study of gefitinib and radiation in children with newly diagnosed brain stem gliomas or supratentorial malignant gliomas. Eur. J. Cancer 2010, 46, 3287–3293. [Google Scholar] [CrossRef]
- Flannery, P.C.; DeSisto, J.A.; Amani, V.; Venkataraman, S.; Lemma, R.T.; Prince, E.W.; Donson, A.; Moroze, E.E.; Hoffman, L.; Levy, J.M.M.; et al. Preclinical analysis of MTOR complex 1/2 inhibition in diffuse intrinsic pontine glioma. Oncol. Rep. 2018, 39, 455–464. [Google Scholar] [CrossRef]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The dual mTOR kinase inhibitor TAK228 inhibits tumorigenicity and enhances radiosensitization in diffuse intrinsic pontine glioma. Cancer Lett. 2017, 400, 110–116. [Google Scholar] [CrossRef]
- Li, G.; Mitra, S.S.; Monje, M.; Henrich, K.N.; Bangs, C.D.; Nitta, R.T.; Wong, A.J. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J. Neurooncol. 2012, 108, 395–402. [Google Scholar] [CrossRef]
- Massimino, M.; Biassoni, V.; Miceli, R.; Schiavello, E.; Warmuth-Metz, M.; Modena, P.; Casanova, M.; Pecori, E.; Giangaspero, F.; Antonelli, M.; et al. Results of nimotuzumab and vinorelbine, radiation and re-irradiation for diffuse pontine glioma in childhood. J. Neurooncol. 2014, 118, 305–312. [Google Scholar] [CrossRef]
- Kline, C.; Liu, S.J.; Duriseti, S.; Banerjee, A.; Nicolaides, T.; Raber, S.; Gupta, N.; Haas-Kogan, D.; Braunstein, S.; Mueller, S. Reirradiation and PD-1 inhibition with nivolumab for the treatment of recurrent diffuse intrinsic pontine glioma: A single-institution experience. J. Neurooncol. 2018, 140, 629–638. [Google Scholar] [CrossRef]
- Price, G.; Bouras, A.; Hambardzumyan, D.; Hadjipanayis, C.G. Current knowledge on the immune microenvironment and emerging immunotherapies in diffuse midline glioma. eBioMedicine 2021, 69, 103453. [Google Scholar] [CrossRef]
- Franson, A.; McClellan, B.L.; Varela, M.L.; Comba, A.; Syed, M.F.; Banerjee, K.; Zhu, Z.; Gonzalez, N.; Candolfi, M.; Lowenstein, P.; et al. Development of immunotherapy for high-grade gliomas: Overcoming the immunosuppressive tumor microenvironment. Front. Med. 2022, 9, 966458. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharmacol. 2018, 9, 185. [Google Scholar] [CrossRef]
- Werbrouck, C.; Evangelista, C.C.S.; Lobón-Iglesias, M.J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Garcia-Fabiani, M.B.; Carney, S.; Altshuler, D.; Núñez, F.J.; Méndez, F.M.; Núñez, F.; Lowenstein, P.R.; Castro, M.G. Mutant ATRX: Uncovering a new therapeutic target for glioma. Expert. Opin. Ther. Targets 2018, 22, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia 2020, 22, 725–744. [Google Scholar] [CrossRef] [PubMed]
- El-Soussi, S.; Hanna, R.; Semaan, H.; Khater, A.R.; Abdallah, J.; Abou-Kheir, W.; Abou-Antoun, T. A Novel Therapeutic Mechanism of Imipridones ONC201/ONC206 in MYCN-Amplified Neuroblastoma Cells via Differential Expression of Tumorigenic Proteins. Front. Pediatr. 2021, 9, 693145. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Kawakibi, A.R.; Ji, S.; Waszak, S.M.; Sweha, S.R.; Mota, M.; Pun, M.; Deogharkar, A.; Chung, C.; Tarapore, R.S.; et al. Clinical efficacy of ONC201 in H3K27M-mutant diffuse midline gliomas is driven by disruption of integrated metabolic and epigenetic pathways. Cancer Discov. 2023, OF1–OF24. [Google Scholar] [CrossRef] [PubMed]
- Purow, B. ONC201 and ONC206: Metabolically ClipPing the wings of diffuse midline glioma. Neuro-Oncology 2022, 24, 1452–1453. [Google Scholar] [CrossRef]
- Bonner, E.R.; Waszak, S.M.; Grotzer, M.A.; Mueller, S.; Nazarian, J. Mechanisms of imipridones in targeting mitochondrial metabolism in cancer cells. Neuro-Oncology 2021, 23, 542–556. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Y.; Yin, Y.; Fan, Y.; Sun, W.; Zhao, X.; Tucker, K.; Staley, A.; Paraghamian, S.; Hawkins, G.; et al. ONC206, an Imipridone Derivative, Induces Cell Death Through Activation of the Integrated Stress Response in Serous Endometrial Cancer In Vitro. Front. Oncol. 2020, 10, 577141. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational implications of tumor heterogeneity. Clin. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef]
- Lim, Z.F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Line, S.; Sajesh, B.V.; Schwinghamer, K.; Yathindranath, V.; Tsoli, M.; Ziegler, D.; Siahaan, T.; Miller, D.; Vanan, M.I. DIPG-59. Blood Brain Barrier (BBB) modulation using Cadherin peptides in the treatment of Diffuse Midline Glioma of the Pons (DMG-P). Neuro-Oncology 2022, 24, i32. [Google Scholar] [CrossRef]
- Jovanovich, N.; Habib, A.; Head, J.; Hameed, F.; Agnihotri, S.; Zinn, P.O. Pediatric diffuse midline glioma: Understanding the mechanisms and assessing the next generation of personalized therapeutics. Neurooncol. Adv. 2023, 5, vdad040. [Google Scholar] [CrossRef]
- Ozair, A.; Bhat, V.; Alisch, R.S.; Khosla, A.A.; Kotecha, R.R.; Odia, Y.; McDermott, M.W.; Ahluwalia, M.S. DNA Methylation and Histone Modification in Low-Grade Gliomas: Current Understanding and Potential Clinical Targets. Cancers 2023, 15, 1342. [Google Scholar] [CrossRef]
- Li, Q.; Li, Z.; Luo, T.; Shi, H. Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol. Biomed. 2022, 3, 47. [Google Scholar] [CrossRef]
- Da-Veiga, M.A.; Rogister, B.; Lombard, A.; Neirinckx, V.; Piette, C. Glioma Stem Cells in Pediatric High-Grade Gliomas: From Current Knowledge to Future Perspectives. Cancers 2022, 14, 2296. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, C.; Li, S.; Wang, J.; Zhang, H. Immune Microenvironment and Immunotherapies for Diffuse Intrinsic Pontine Glioma. Cancers 2023, 15, 602. [Google Scholar] [CrossRef]
- Rechberger, J.S.; Power, B.T.; Power, E.A.; Nesvick, C.L.; Daniels, D.J. H3K27-altered diffuse midline glioma: A paradigm shifting opportunity in direct delivery of targeted therapeutics. Expert. Opin. Ther. Targets 2023, 27, 9–17. [Google Scholar] [CrossRef]
- Pedersen, H.; Schmiegelow, K.; Hamerlik, P. Radio-Resistance and DNA Repair in Pediatric Diffuse Midline Gliomas. Cancers 2020, 12, 2813. [Google Scholar] [CrossRef]
- Solomon, D.A.; Wood, M.D.; Tihan, T.; Bollen, A.W.; Gupta, N.; Phillips, J.J.; Perry, A. Diffuse Midline Gliomas with Histone H3-K27M Mutation: A Series of 47 Cases Assessing the Spectrum of Morphologic Variation and Associated Genetic Alterations. Brain Pathol. 2016, 26, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Duffner, P.K.; Cohen, M.E.; Freeman, A.I. Pediatric brain tumors: An overview. CA Cancer J. Clin. 1985, 35, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Broniscer, A.; Laningham, F.H.; Sanders, R.P.; Kun, L.E.; Ellison, D.W.; Gajjar, A. Young age may predict a better outcome for children with diffuse pontine glioma. Cancer 2008, 113, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.E.; Duffner, P.K.; Heffner, R.R.; Lacey, D.J.; Brecher, M. Prognostic factors in brainstem gliomas. Neurology 1986, 36, 602–605. [Google Scholar] [CrossRef]
- Poussaint, T.Y.; Kocak, M.; Vajapeyam, S.; Packer, R.I.; Robertson, R.L.; Geyer, R.; Haas-Kogan, D.; Pollack, I.F.; Vezina, G.; Zimmerman, R.; et al. MRI as a central component of clinical trials analysis in brainstem glioma: A report from the Pediatric Brain Tumor Consortium (PBTC). Neuro-Oncology 2011, 13, 417–427. [Google Scholar] [CrossRef]
- Leach, J.L.; Roebker, J.; Schafer, A.; Baugh, J.; Chaney, B.; Fuller, C.; Fouladi, M.; Lane, A.; Doughman, R.; Drissi, R.; et al. MR imaging features of diffuse intrinsic pontine glioma and relationship to overall survival: Report from the International DIPG Registry. Neuro-Oncology 2020, 22, 1647–1657. [Google Scholar] [CrossRef]
- Erker, C.; Tamrazi, B.; Poussaint, T.Y.; Mueller, S.; Mata-Mbemba, D.; Franceschi, E.; Brandes, A.A.; Rao, A.; Haworth, K.B.; Wen, P.Y.; et al. Response assessment in paediatric high-grade glioma: Recommendations from the Response Assessment in Pediatric Neuro-Oncology (RAPNO) working group. Lancet Oncol. 2020, 21, e317–e329. [Google Scholar] [CrossRef]
- Ueoka, D.I.; Nogueira, J.; Campos, J.C.; Maranhão Filho, P.; Ferman, S.; Lima, M.A. Brainstem gliomas--retrospective analysis of 86 patients. J. Neurol. Sci. 2009, 281, 20–23. [Google Scholar] [CrossRef]
- Cooney, T.M.; Cohen, K.J.; Guimaraes, C.V.; Dhall, G.; Leach, J.; Massimino, M.; Erbetta, A.; Chiapparini, L.; Malbari, F.; Kramer, K.; et al. Response assessment in diffuse intrinsic pontine glioma: Recommendations from the Response Assessment in Pediatric Neuro-Oncology (RAPNO) working group. Lancet Oncol. 2020, 21, e330–e336. [Google Scholar] [CrossRef]
- Löbel, U.; Sedlacik, J.; Reddick, W.E.; Kocak, M.; Ji, Q.; Broniscer, A.; Hillenbrand, C.M.; Patay, Z. Quantitative diffusion-weighted and dynamic susceptibility-weighted contrast-enhanced perfusion MR imaging analysis of T2 hypointense lesion components in pediatric diffuse intrinsic pontine glioma. AJNR Am. J. Neuroradiol. 2011, 32, 315–322. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Sharie, S.; Abu Laban, D.; Al-Hussaini, M. Decoding Diffuse Midline Gliomas: A Comprehensive Review of Pathogenesis, Diagnosis and Treatment. Cancers 2023, 15, 4869. https://doi.org/10.3390/cancers15194869
Al Sharie S, Abu Laban D, Al-Hussaini M. Decoding Diffuse Midline Gliomas: A Comprehensive Review of Pathogenesis, Diagnosis and Treatment. Cancers. 2023; 15(19):4869. https://doi.org/10.3390/cancers15194869
Chicago/Turabian StyleAl Sharie, Sarah, Dima Abu Laban, and Maysa Al-Hussaini. 2023. "Decoding Diffuse Midline Gliomas: A Comprehensive Review of Pathogenesis, Diagnosis and Treatment" Cancers 15, no. 19: 4869. https://doi.org/10.3390/cancers15194869
APA StyleAl Sharie, S., Abu Laban, D., & Al-Hussaini, M. (2023). Decoding Diffuse Midline Gliomas: A Comprehensive Review of Pathogenesis, Diagnosis and Treatment. Cancers, 15(19), 4869. https://doi.org/10.3390/cancers15194869