
RNA-Seq-Based Molecular Classification Analyses in Colorectal Cancer and Synchronous Adenoma

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Samples
2.2. Sample Preparation, Library Construction, and RNA-Seq
2.3. Mapping Preparation and Clean-Up Reads
2.4. CMS Prediction and CiberSortx
2.5. Variant Calling and Annotation
3. Results
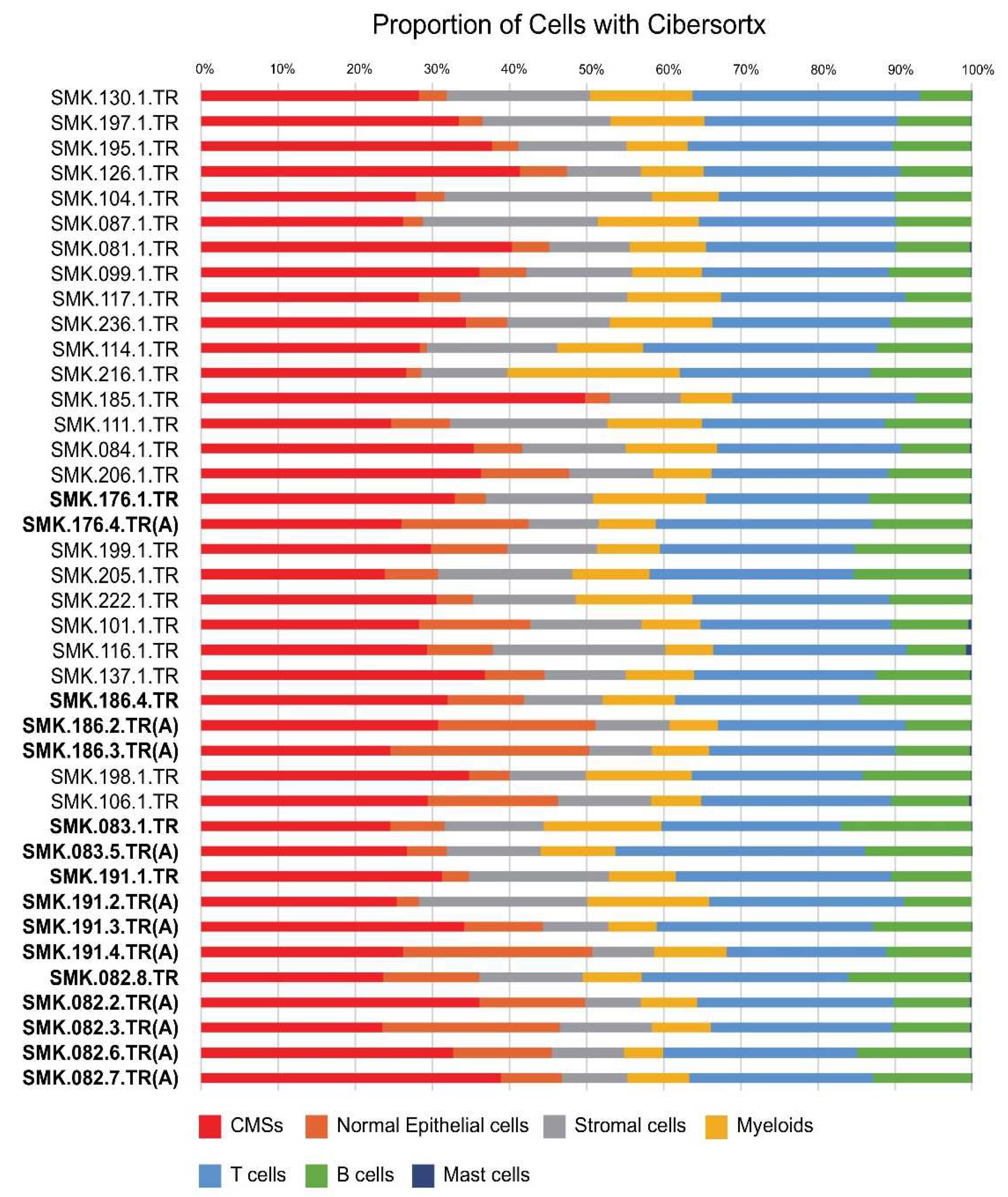
3.1. In Silico Cell Fraction Estimation Using CiberSortx
3.2. Classification by iCMS System
3.3. CMS and iCMS Classification of Adenomas
3.4. CRC Characteristics and Association with CMS Classification
3.5. Variant Calling and Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Joanito, I.; Wirapati, P.; Zhao, N.; Nawaz, Z.; Yeo, G.; Lee, F.; Eng, C.L.P.; Macalinao, D.C.; Kahraman, M.; Srinivasan, H.; et al. Single-Cell and Bulk Transcriptome Sequencing Identifies Two Epithelial Tumor Cell States and Refines the Consensus Molecular Classification of Colorectal Cancer. Nat. Genet. 2022, 54, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Morson, B. The Polyp-Cancer Sequence in the Large Bowel. Proc. R. Soc. Med. 1974, 67, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Mojarad, E.N.; Kuppen, P.J.K.; Aghdaei, H.A.; Zali, M.R. The CpG Island Methylator Phenotype (CIMP) in Colorectal Cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar]
- Nojadeh, J.N.; Sharif, S.B.; Sakhinia, E. Microsatellite Instability in Colorectal Cancer. EXCLI J. 2018, 17, 159. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus Molecular Subtypes and the Evolution of Precision Medicine in Colorectal Cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Salas, N.; Dominguez, G.; Barderas, R.; Mendiola, M.; García-Albéniz, X.; Maurel, J.; Batlle, J.F. Clinical Relevance of Colorectal Cancer Molecular Subtypes. Crit. Rev. Oncol. Hematol. 2017, 109, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.O.; Hong, Y.; Etlioglu, H.E.; Cho, Y.B.; Pomella, V.; Van den Bosch, B.; Vanhecke, J.; Verbandt, S.; Hong, H.; Min, J.W.; et al. Lineage-Dependent Gene Expression Programs Influence the Immune Landscape of Colorectal Cancer. Nat. Genet. 2020, 52, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.H.; Watanabe, T. Colorectal Cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Li, B.; Cai, X.; Lv, Z.; Cai, S.; Zhong, Y.; Wen, B. Transcriptomic Analyses of the Adenoma-Carcinoma Sequence Identify Hallmarks Associated with the Onset of Colorectal Cancer. Front. Oncol. 2021, 11, 704531. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [PubMed]
- Sparks, A.B.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Mutational Analysis of the APC/β-Catenin/Tcf Pathway in Colorectal Cancer. Cancer Res. 1998, 58, 1130–1134. [Google Scholar]
- Al-Kuraya, K.S. KRAS and TP53 Mutations in Colorectal Carcinoma. Saudi J. Gastroenterol. Off. J. Saudi Gastroenterol. Assoc. 2009, 15, 217–219. [Google Scholar] [CrossRef]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 Mutations in Colorectal Cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef]
- Rad, R.; Cadiñanos, J.; Rad, L.; Varela, I.; Strong, A.; Kriegl, L.; Constantino-Casas, F.; Eser, S.; Hieber, M.; Seidler, B.; et al. A Genetic Progression Model of BrafV600E-Induced Intestinal Tumorigenesis Reveals Targets for Therapeutic Intervention. Cancer Cell 2013, 24, 15–29. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Gender | Age | CMS1 | CMS2 | CMS3 | CMS4 | RF Nearest | iCMS | MSI Status | Differentiation | Initial TNM Stage | Final Metastasis | Location | Size | Adenoma | Location | Size | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SMK.130.1.TR | F | 53 | 0.05% | 93.51% | 5.86% | 0.58% | 2 | 2 | MSS | MD | pT3N0M0 | IIA | M0 | SC | 4.2 | |||
| SMK.197.1.TR | M | 59 | 0.00% | 71.25% | 28.53% | 0.23% | 4 | 2 | MSS | MD | pT3N0M0 | IIA | M0 | SC | 4.9 | |||
| SMK.195.1.TR | M | 50 | 0.00% | 69.85% | 30.06% | 0.09% | 4 | 2 | MSS | MD | pT3N0M0 | IIA | M0 | REC | 7 | |||
| SMK.126.1.TR | M | 54 | 0.00% | 68.70% | 31.18% | 0.12% | 2 | 2 | MSS | WD | T4bN3M1 | IVA | M1 | RSJ | 8.3 | |||
| SMK.104.1.TR | M | 57 | 0.00% | 67.65% | 32.35% | 0.00% | 4 | 2 | MSS | MD | pT3N1cM0 | IIIB | M0 | AC | 5.0 | |||
| SMK.087.1.TR | M | 60 | 0.00% | 67.33% | 32.67% | 0.00% | 4 | 2 | MSS | MD | pT3N0M0 | IIA | M0 | RSJ | 11.7 | |||
| SMK.081.1.TR | F | 76 | 0.00% | 65.96% | 33.67% | 0.37% | 2 | 2 | MSS | MD | pT3N2aM0 | IIIB | M0 | AC | 5.3 | |||
| SMK.099.1.TR | F | 55 | 0.00% | 65.83% | 33.93% | 0.24% | 2 | 2 | MSS | MD | pT4aN2bM1 | IVB | M1 | SC | ||||
| SMK.117.1.TR | M | 42 | 0.00% | 64.92% | 34.86% | 0.22% | 4 | 2 | MSS | MD | pT3N1bM1 | IVB | M1 | RSJ | 5.3 | |||
| SMK.236.1.TR | M | 64 | 0.00% | 60.90% | 38.59% | 0.51% | 4 | 2 | MSS | MD | pT3N0M0 | IIA | M0 | SC | ||||
| SMK.114.1.TR | F | 75 | 0.00% | 60.19% | 39.74% | 0.06% | 4 | 2 | MSS | MD | pT3N0M0 | IIA | M0 | AC | 4.3 | |||
| SMK.216.1.TR | F | 63 | 16.23% | 56.75% | 27.02% | 0.00% | 1 | 2 | MSI | PD | pT3N0M0 | IIA | M0 | AC | ||||
| SMK.185.1.TR | F | 51 | 0.00% | 51.18% | 48.70% | 0.12% | 2 | 2 | MSS | WD | T3N2aM0 | IIIB | M1 | REC | 4.2 | |||
| SMK.111.1.TR | M | 68 | 0.00% | 45.54% | 54.46% | 0.00% | 4 | 2 | MSS | MD | pT3N1aM0 | IIIB | M0 | SC | 3.8 | |||
| SMK.084.1.TR | M | 54 | 0.00% | 36.88% | 62.75% | 0.36% | 4 | 3 | MSS | MD | pT3N1aM0 | IIIB | M1 | TC | 3.7 | |||
| SMK.206.1.TR | M | 67 | 0.00% | 29.07% | 70.69% | 0.23% | 2 | 3 | MSS | MD | pT3N1bM0 | IIIB | M0 | SC | ||||
| SMK.176.1.TR | M | 66 | 0.00% | 27.38% | 72.62% | 0.00% | 4 | 3 | MSS | WD | pT3N0M0 | IIA | M0 | SC | ||||
| SMK.176.4.TR(A) | M | 66 | 0.00% | 7.09% | 92.74% | 0.17% | 3 | 3 | REC | 1 | TA, LGD | REC | 1.0 | |||||
| SMK.199.1.TR | M | 58 | 0.00% | 26.03% | 73.94% | 0.04% | 2 | 3 | MSS | MD | T3N1M0 | IIIB | M0 | REC | 5.5 | |||
| SMK.205.1.TR | M | 60 | 0.00% | 20.06% | 79.94% | 0.00% | 4 | 3 | MSS | MD | pT3N2aM0 | IIIB | M0 | REC | ||||
| SMK.222.1.TR | M | 61 | 8.92% | 19.49% | 71.60% | 0.00% | 1 | 3 | MSI | Mucinous | pT3N0M0 | IIA | M0 | AC | 7.0 | |||
| SMK.101.1.TR | F | 64 | 0.00% | 18.99% | 80.89% | 0.12% | 4 | 3 | MSS | MD | pT4aN1bM1 | IVA | M1 | RSJ | 4.8 | |||
| SMK.116.1.TR | M | 58 | 0.00% | 17.78% | 82.22% | 0.00% | 4 | 3 | MSS | MD | pT4aN1aM0 | IIIC | M1 | SC | ||||
| SMK.137.1.TR | M | 60 | 0.00% | 17.13% | 82.66% | 0.21% | 3 | 3 | MSS | MD | pT3N1cM0 | IIIB | NA | REC | 8.5 | |||
| SMK.186.4.TR | M | 54 | 0.00% | 15.87% | 84.05% | 0.08% | 2 | 3 | MSS | MD | pT3N0M0 | IIA | M0 | SC | ||||
| SMK.186.2.TR(A) | M | 54 | 0.00% | 6.29% | 93.67% | 0.04% | 3 | 3 | HF | 1 | TA, LGD | TC | 1.0 | |||||
| SMK.186.3.TR(A) | M | 54 | 0.00% | 5.02% | 94.52% | 0.45% | 3 | 3 | DC | 0.7 | TA, LGD | DC | 0.8 | |||||
| SMK.198.1.TR | M | 83 | 2.57% | 13.38% | 83.97% | 0.08% | 1 | 3 | MSI | MD, mucinous | pT3N1bM0 | IIIB | M0 | SC | 4.6 | |||
| SMK.106.1.TR | F | 53 | 0.00% | 11.81% | 88.15% | 0.04% | 2 | 3 | MSS | MD | pT3N0M0 | IIA | M0 | RSJ | 2.5 | |||
| SMK.083.1.TR | M | 79 | 0.00% | 9.81% | 90.12% | 0.07% | 3 | 3 | MSS | MD | pT2N0M0 | I | M0 | TC | 2.0 | |||
| SMK.083.5.TR(A) | M | 79 | 0.00% | 46.77% | 52.91% | 0.32% | 3 | 3 | REC | 2 | TA, HGD | REC | 2.0 | |||||
| SMK.191.1.TR | M | 77 | 2.50% | 0.83% | 96.67% | 0.00% | 1 | 3 | MSI | MD | pT3N0M0 | IIA | M0 | TC | ||||
| SMK.191.2.TR(A) | M | 77 | 7.13% | 5.00% | 87.57% | 0.30% | 4 | 3 | TC | 0.7 | TA, LGD | TC | 0.7 | |||||
| SMK.191.3.TR(A) | M | 77 | 0.40% | 0.00% | 99.45% | 0.16% | 3 | 3 | TC | 0.7 | TSA, LGD | TC | 0.7 | |||||
| SMK.191.4.TR(A) | M | 77 | 0.00% | 0.00% | 100% | 0.00% | 3 | 3 | SC | 1 | TA, LGD | SC | 1.0 | |||||
| SMK.082.8.TR | M | 87 | 0.00% | 0.00% | 100% | 0.00% | 4 | 3 | MSS | MD | pT4aN0 | IIB | NA | SC | 4.0 | |||
| SMK.082.2.TR(A) | M | 87 | 0.00% | 16.83% | 83.00% | 0.18% | 3 | 3 | TC | 1.5 | TA, LGD | TC | 1.5 | |||||
| SMK.082.3.TR(A) | M | 87 | 0.00% | 0.00% | 99.92% | 0.08% | 4 | 3 | TC | 0.8 | TA, LGD | TC | 0.8 | |||||
| SMK.082.6.TR(A) | M | 87 | 0.00% | 17.44% | 82.33% | 0.22% | 3 | 3 | DC | 2 | TA, HGD | DC | 2.0 | |||||
| Stage | Location | |||
|---|---|---|---|---|
| <RF> | I, II (n = 14) | III, IV (n = 15) | Left (n = 21) | Right (n = 8) |
| CMS 1 | 3 (21.4%) | 1 (6.7%) | CMS 1 | 3 (21.4%) |
| CMS 2 | 3 (21.4%) | 6 (40.0%) | CMS 2 | 3 (21.4%) |
| CMS 3 | 1 (7.1%) | 1 (6.7%) | CMS 3 | 1 (7.1%) |
| CMS 4 | 7 (50%) | 7 (46.7%) | CMS 4 | 7 (50%) |
| <iCMS> | <iCMS> | |||
| iCMS 2 | 7 (50%) | 7 (46.7%) | iCMS 2 | 7 (50%) |
| iCMS 3 | 7 (50%) | 8 (53.3%) | iCMS 3 | 7 (50%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.W.; Lee, G.-Y.; Kim, S.; Ahn, K.; Do, I.-G.; Jung, K.-U.; Kim, H.-O.; Kim, H.; Park, D.-I.; Park, S.-k. RNA-Seq-Based Molecular Classification Analyses in Colorectal Cancer and Synchronous Adenoma. Cancers 2023, 15, 4851. https://doi.org/10.3390/cancers15194851
Choi JW, Lee G-Y, Kim S, Ahn K, Do I-G, Jung K-U, Kim H-O, Kim H, Park D-I, Park S-k. RNA-Seq-Based Molecular Classification Analyses in Colorectal Cancer and Synchronous Adenoma. Cancers. 2023; 15(19):4851. https://doi.org/10.3390/cancers15194851
Chicago/Turabian StyleChoi, Ji Won, Gi-Young Lee, Sangsoo Kim, Kwangsung Ahn, In-Gu Do, Kyung-Uk Jung, Hyung-Ook Kim, Hungdai Kim, Dong-Il Park, and Soo-kyung Park. 2023. "RNA-Seq-Based Molecular Classification Analyses in Colorectal Cancer and Synchronous Adenoma" Cancers 15, no. 19: 4851. https://doi.org/10.3390/cancers15194851
APA StyleChoi, J. W., Lee, G.-Y., Kim, S., Ahn, K., Do, I.-G., Jung, K.-U., Kim, H.-O., Kim, H., Park, D.-I., & Park, S.-k. (2023). RNA-Seq-Based Molecular Classification Analyses in Colorectal Cancer and Synchronous Adenoma. Cancers, 15(19), 4851. https://doi.org/10.3390/cancers15194851