

Automated Cellular-Level Dual Global Fusion of Whole-Slide Imaging for Lung Adenocarcinoma Prognosis

 ,
,  , , , ,
, , , ,  , , , ,
, , , ,  add
Show full author list
add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
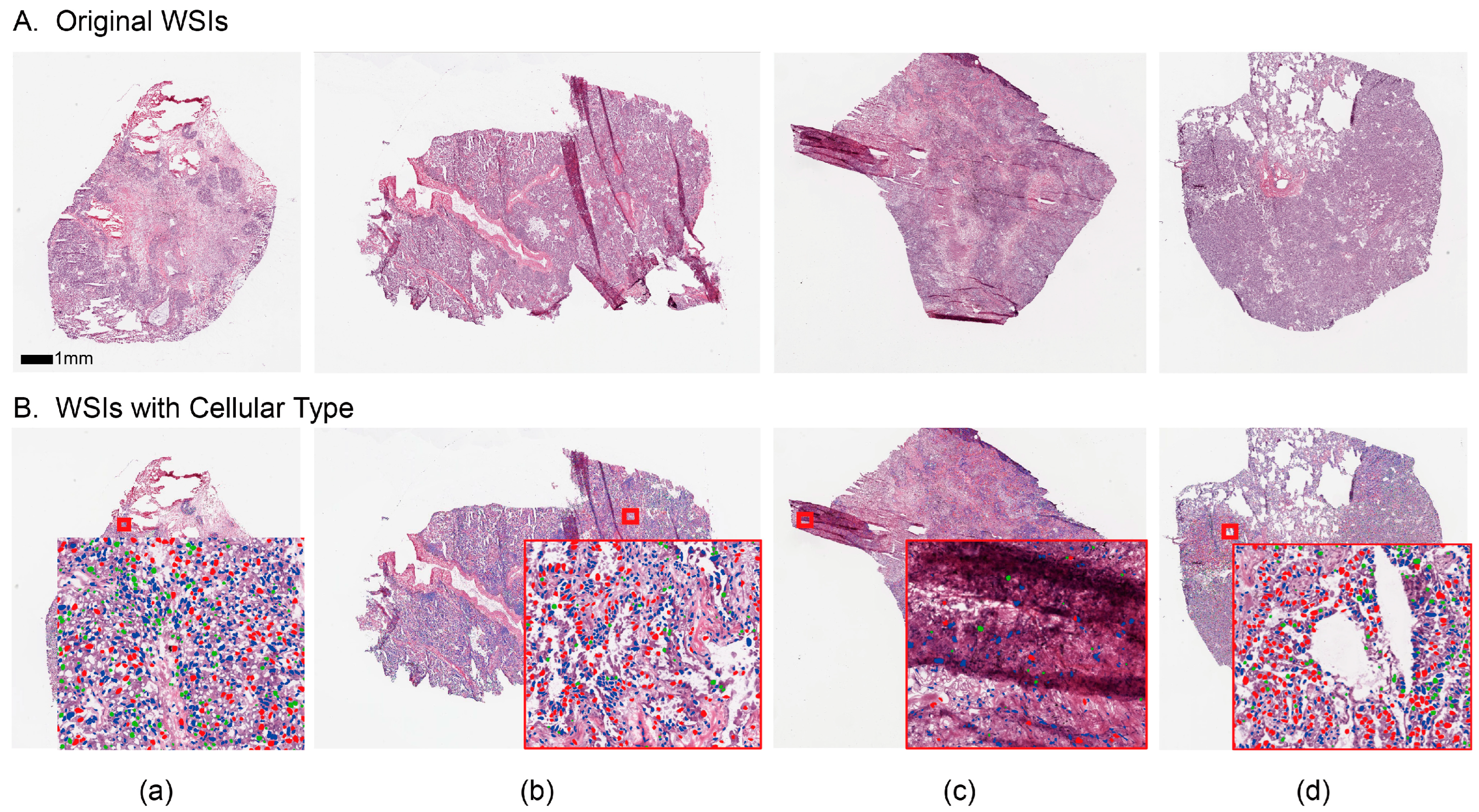
2.1. Data Curation and Pre-Processing
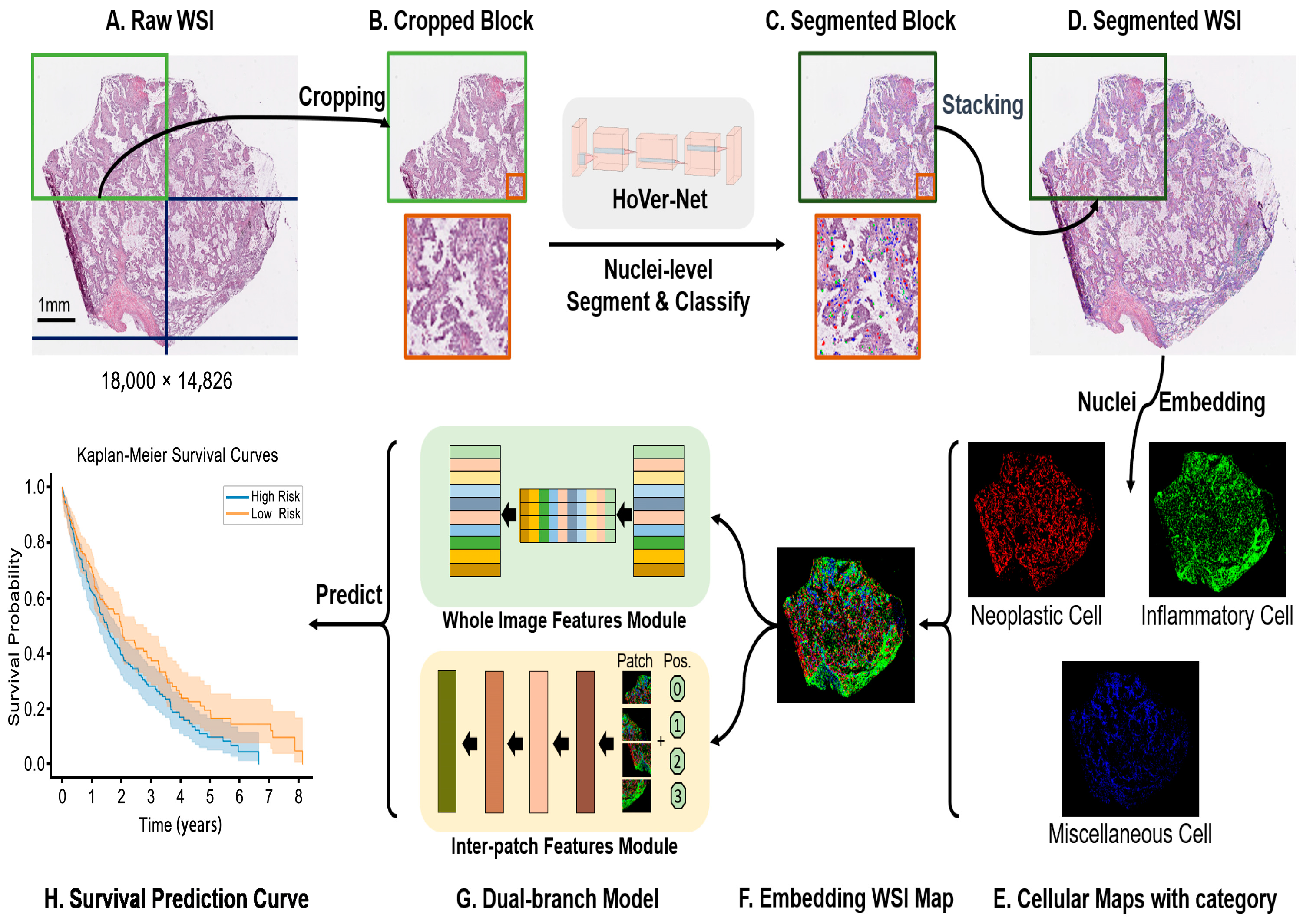
2.2. Overview of the Proposed Pipeline
2.3. Block-Based Nuclei Segmentation
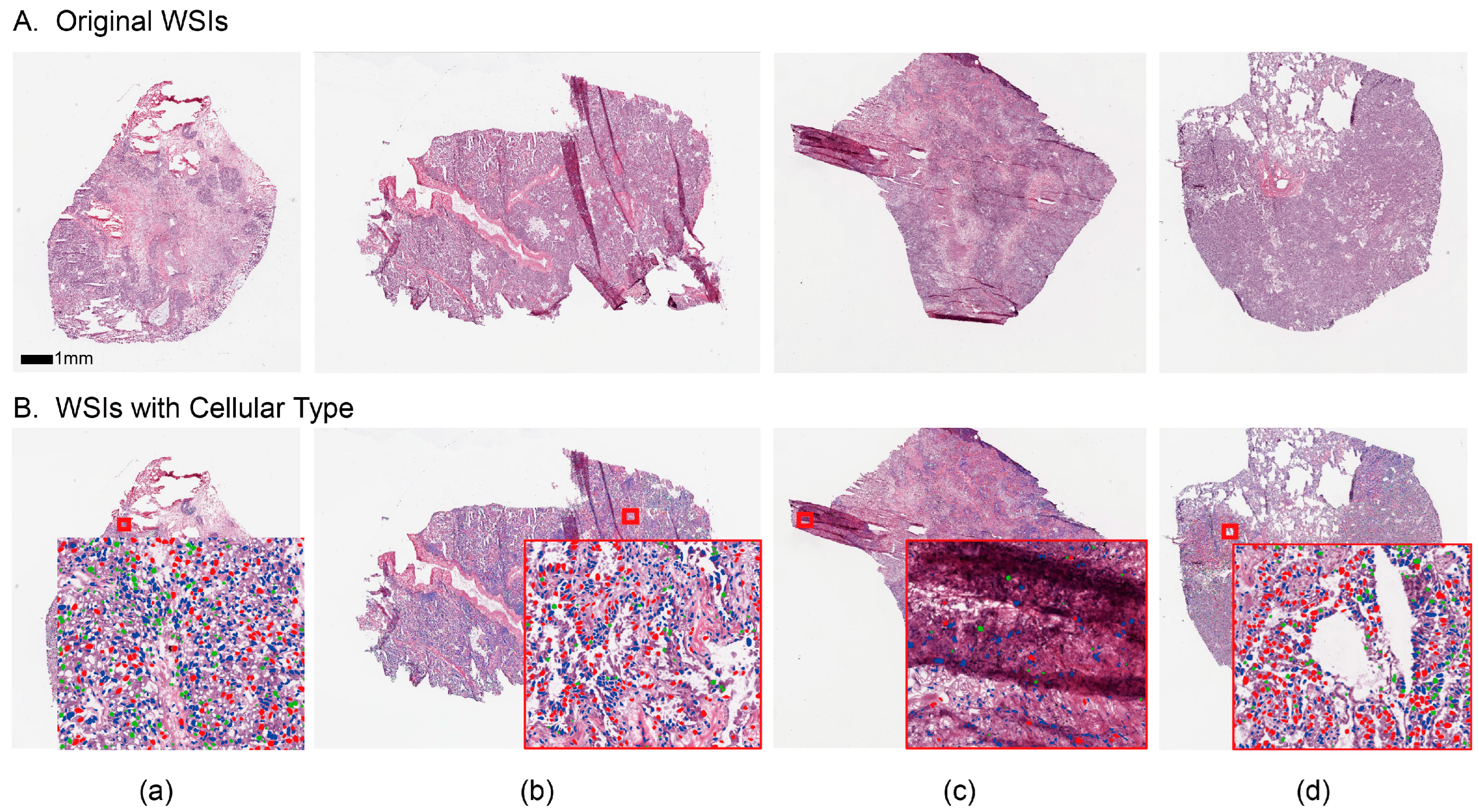
2.4. Cellular Embedding of Segmented WSI
2.5. Dual Global Fusion for Survival Prediction
3. Results
3.1. Experiment Implementation Details
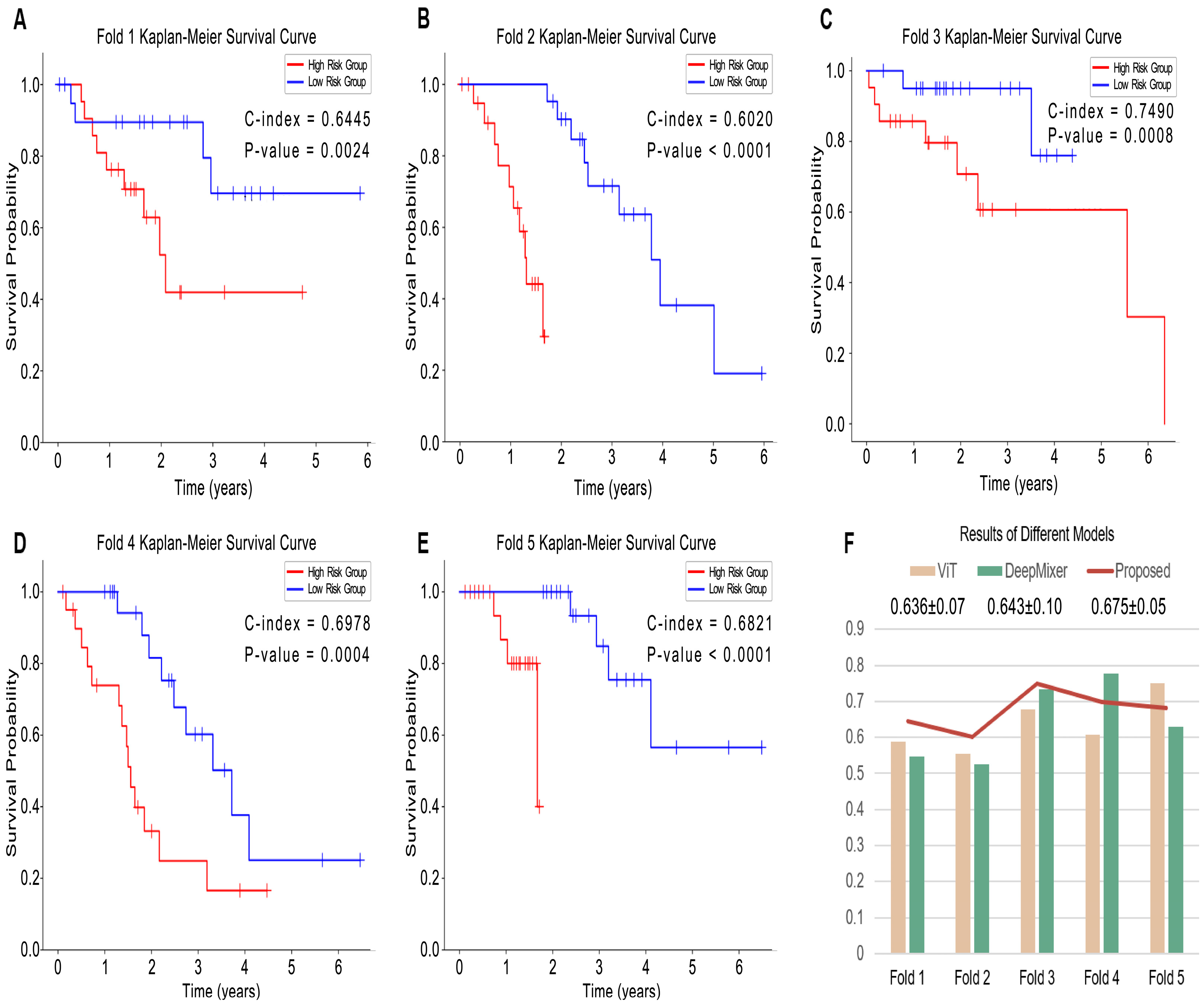
3.2. Evaluation of Prognosis Performance
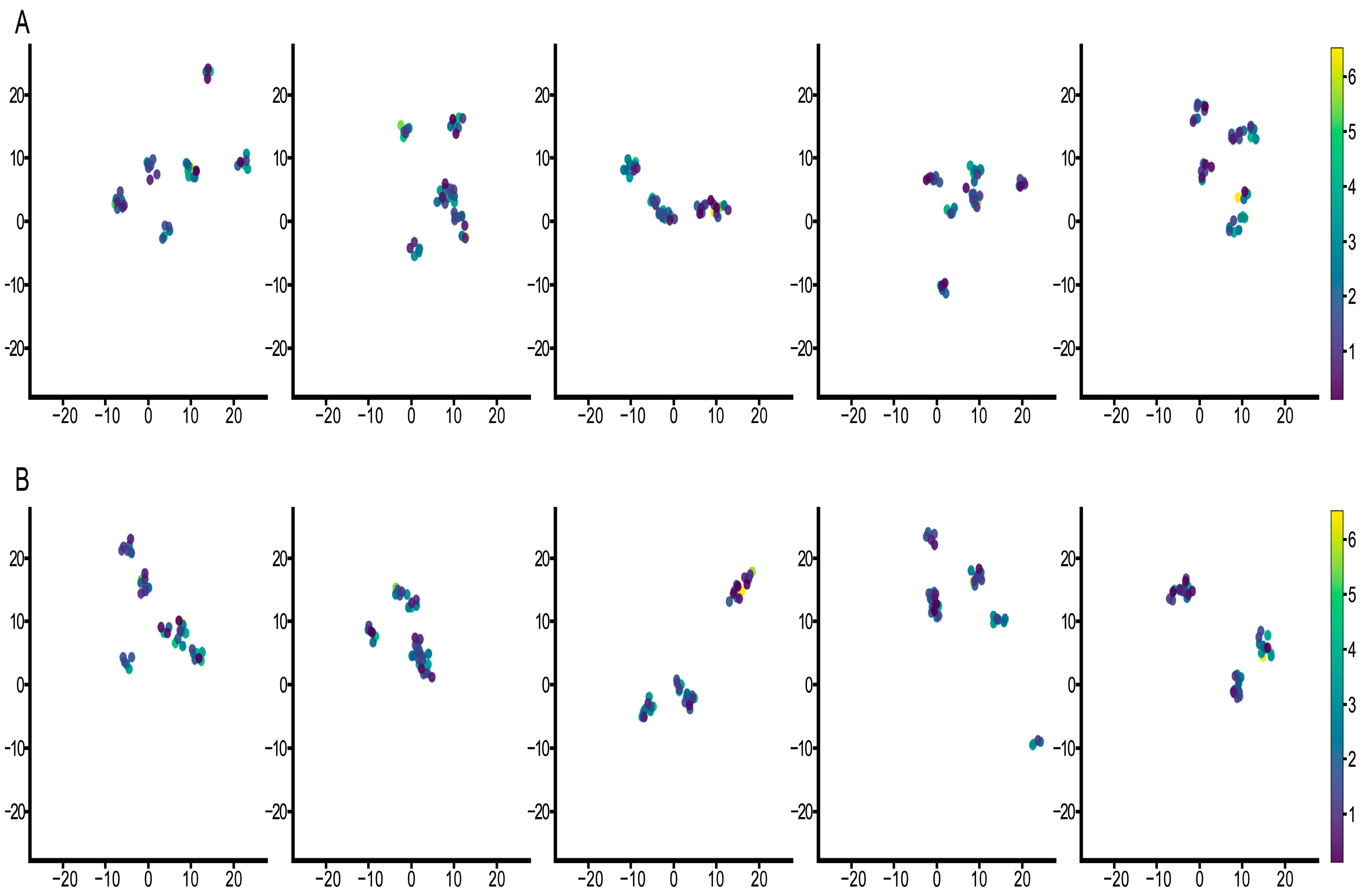
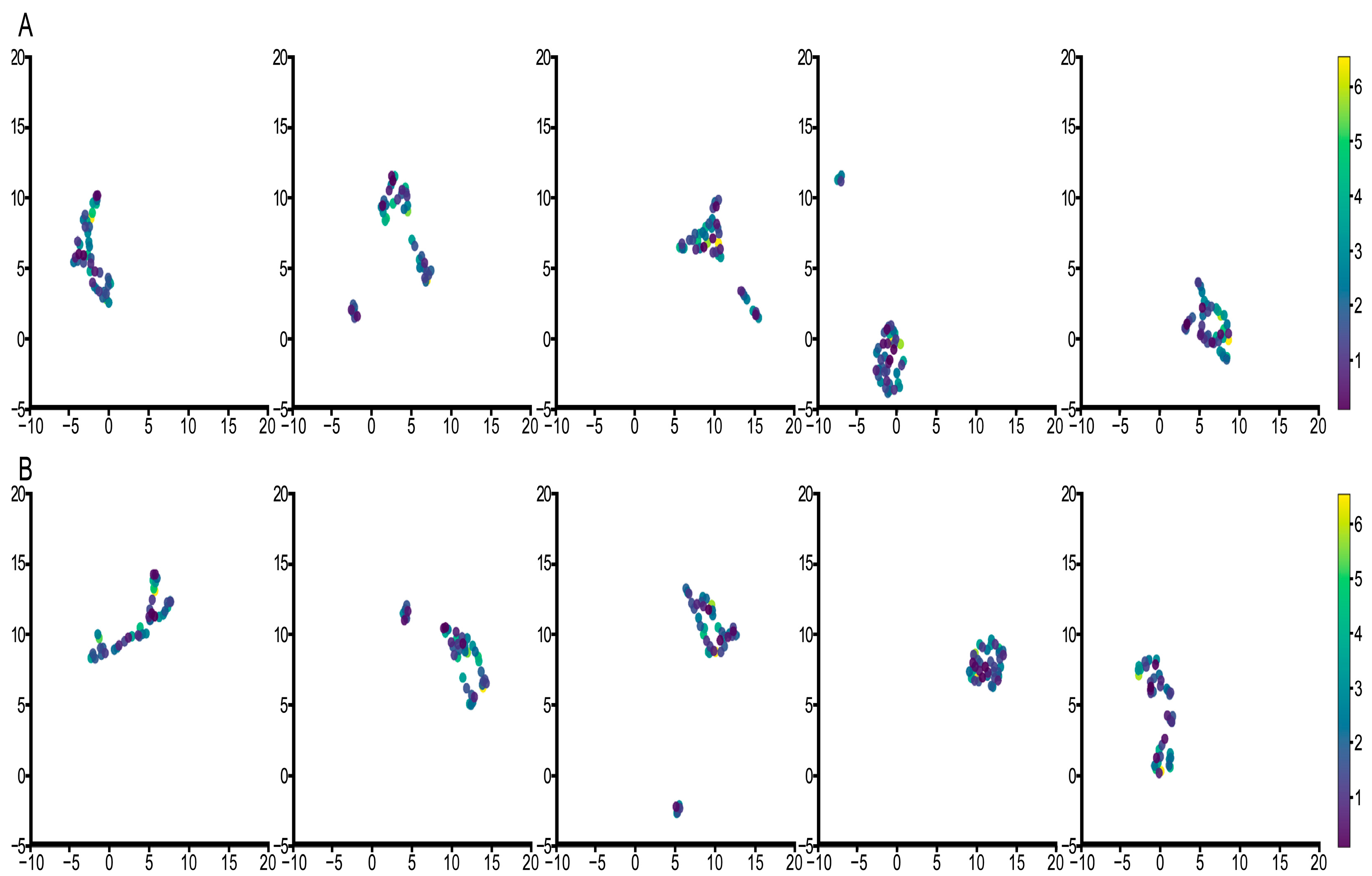
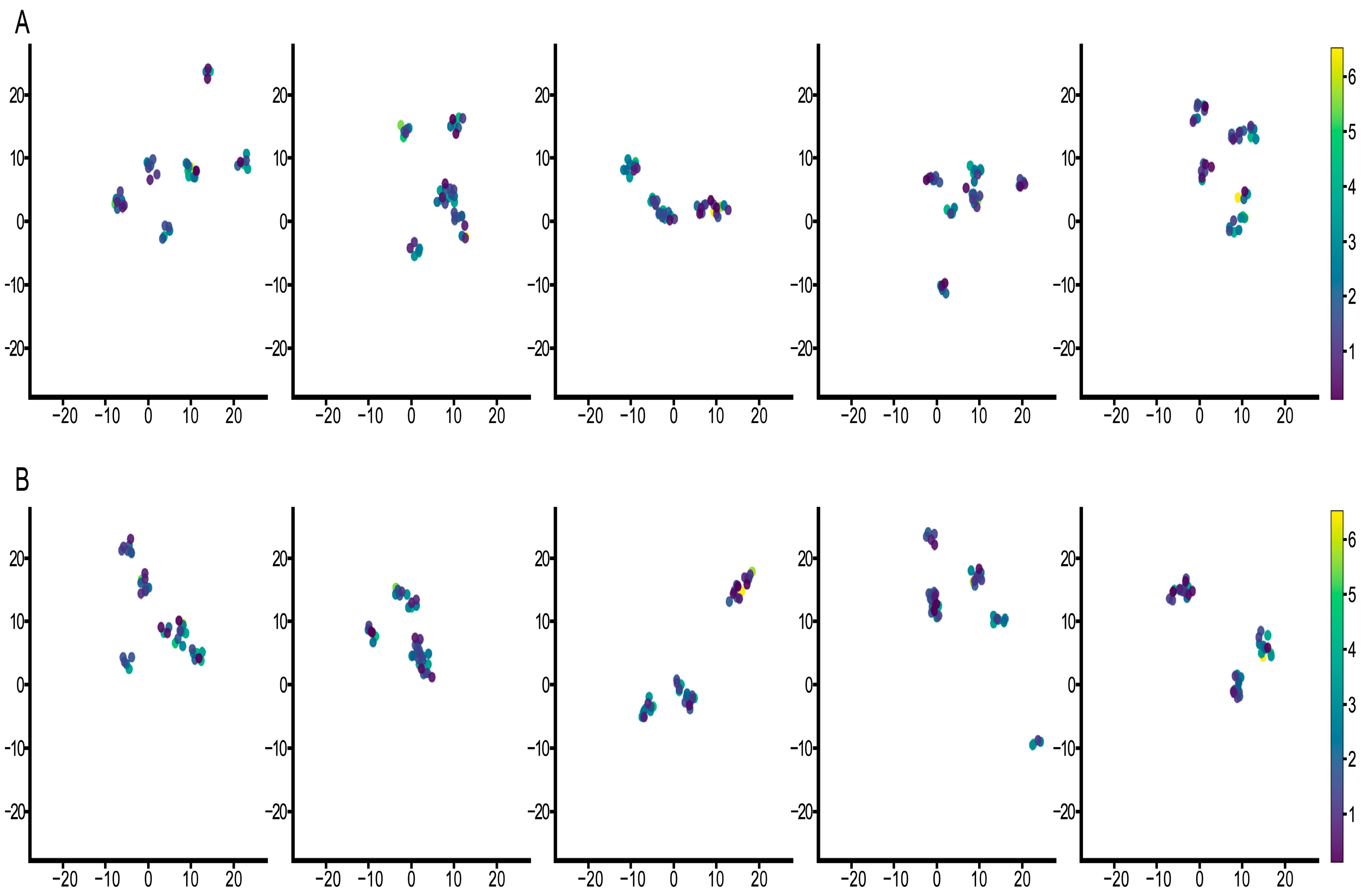
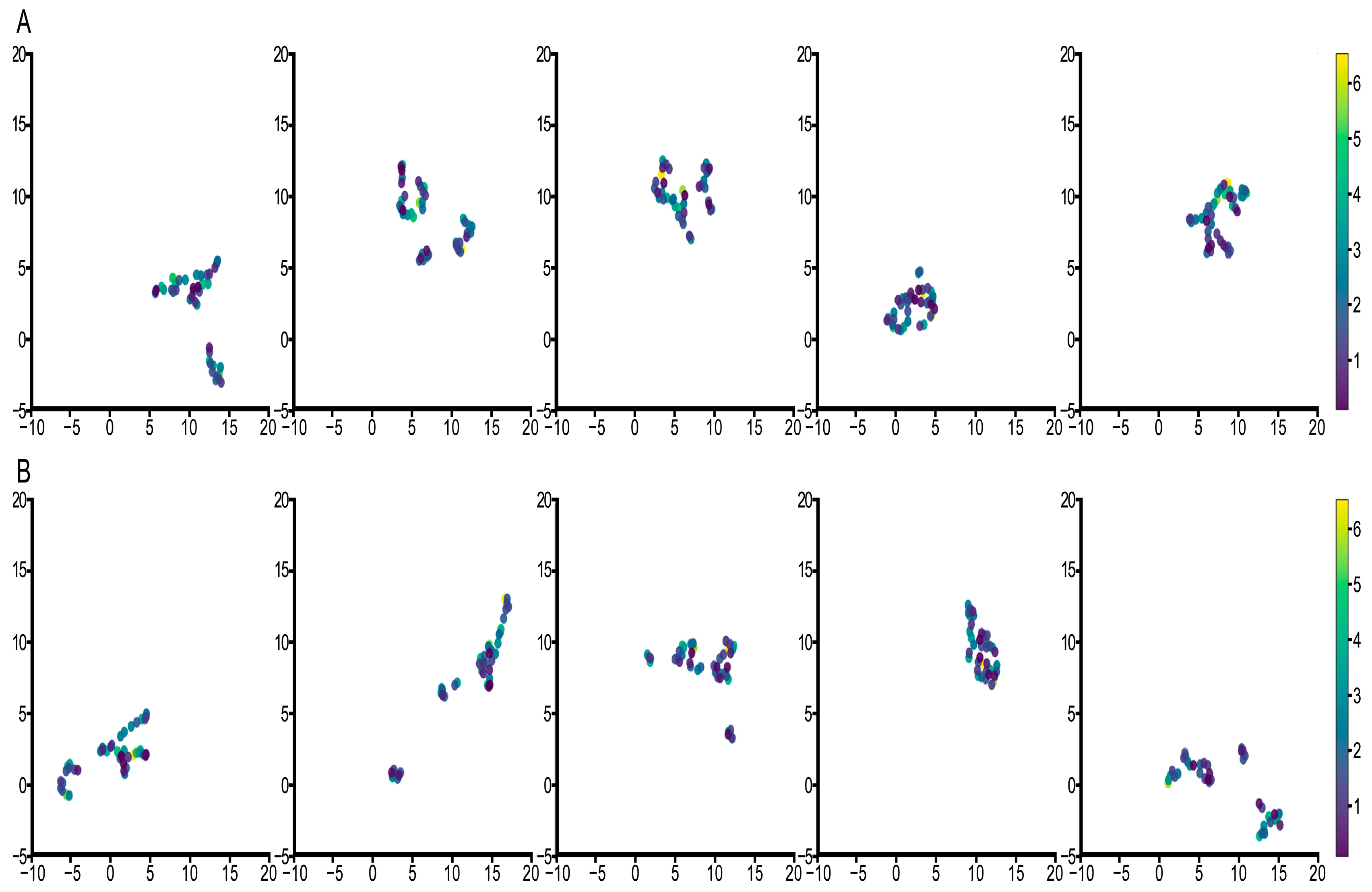
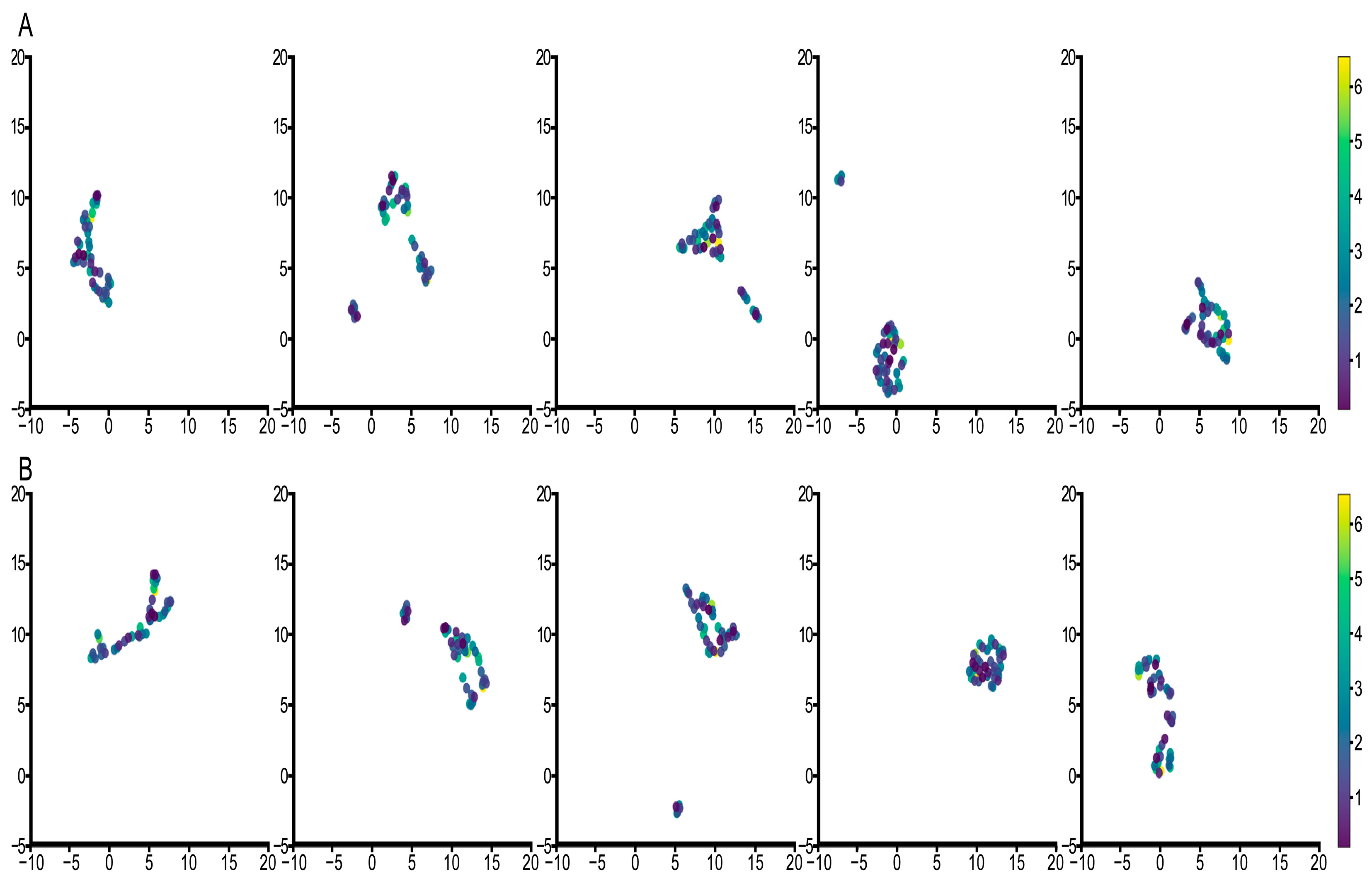
3.3. Feature Visualization Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non–small cell lung cancer: Epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Zengin, T.; Önal-Süzek, T. Analysis of genomic and transcriptomic variations as prognostic signature for lung adenocarcinoma. BMC Bioinform. 2020, 21, 368. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, M.; Yang, M.; Yang, Y.; Song, F.; Zhang, W.; Li, X.; Chen, K. Analysis of immune-related signatures of lung adenocarcinoma identified two distinct subtypes: Implications for immune checkpoint blockade therapy. Aging 2020, 12, 3312. [Google Scholar] [CrossRef]
- Altman, D.G. Prognostic models: A methodological framework and review of models for breast cancer. Cancer Investig. 2009, 27, 235–243. [Google Scholar] [CrossRef]
- Rorke, L.B. Pathologic diagnosis as the gold standard. Cancer 1997, 79, 665–667. [Google Scholar] [CrossRef]
- Chen, P.; Saad, M.B.; Rojas, F.R.; Salehjahromi, M.; Aminu, M.; Bandyopadhyay, R.; Hong, L.; Ebare, K.; Behrens, C.; Gibbons, D.L. Cellular Architecture on Whole Slide Images Allows the Prediction of Survival in Lung Adenocarcinoma. In Proceedings of the International Workshop on Computational Mathematics Modeling in Cancer Analysis, Singapore, 18 September 2022; pp. 1–10. [Google Scholar]
- Xu, Z.; Lim, S.; Shin, H.-K.; Uhm, K.-H.; Lu, Y.; Jung, S.-W.; Ko, S.-J. Risk-aware survival time prediction from whole slide pathological images. Sci. Rep. 2022, 12, 21948. [Google Scholar] [CrossRef]
- Diao, S.; Hou, J.; Yu, H.; Zhao, X.; Sun, Y.; Lambo, R.L.; Xie, Y.; Liu, L.; Qin, W.; Luo, W. Computer-aided pathologic diagnosis of nasopharyngeal carcinoma based on deep learning. Am. J. Pathol. 2020, 190, 1691–1700. [Google Scholar] [CrossRef]
- Singh, P.; Cirrone, J. A data-efficient deep learning framework for segmentation and classification of histopathology images. In Proceedings of the European Conference on Computer Vision, Tel Aviv, Israel, 23–27 October 2022; pp. 385–405. [Google Scholar]
- Lu, M.Y.; Chen, T.Y.; Williamson, D.F.; Zhao, M.; Shady, M.; Lipkova, J.; Mahmood, F. AI-based pathology predicts origins for cancers of unknown primary. Nature 2021, 594, 106–110. [Google Scholar] [CrossRef]
- Diao, S.; Tian, Y.; Hu, W.; Hou, J.; Lambo, R.; Zhang, Z.; Xie, Y.; Nie, X.; Zhang, F.; Racoceanu, D. Weakly supervised framework for cancer region detection of hepatocellular carcinoma in whole-slide pathologic images based on multiscale attention convolutional neural network. Am. J. Pathol. 2022, 192, 553–563. [Google Scholar] [CrossRef]
- Hekler, A.; Utikal, J.S.; Enk, A.H.; Berking, C.; Klode, J.; Schadendorf, D.; Jansen, P.; Franklin, C.; Holland-Letz, T.; Krahl, D. Pathologist-level classification of histopathological melanoma images with deep neural networks. Eur. J. Cancer 2019, 115, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zang, X.; Yang, L.; Huang, J.; Liang, F.; Rodriguez-Canales, J.; Wistuba, I.I.; Gazdar, A.; Xie, Y.; Xiao, G. Comprehensive computational pathological image analysis predicts lung cancer prognosis. J. Thorac. Oncol. 2017, 12, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, D.M.; Rong, R.; Zhan, X.; Fujimoto, J.; Liu, H.; Minna, J.; Wistuba, I.I.; Xie, Y.; Xiao, G. Artificial intelligence in lung cancer pathology image analysis. Cancers 2019, 11, 1673. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhu, X.; Jonnagaddala, J.; Hawkins, N.; Huang, J. Whole slide images based cancer survival prediction using attention guided deep multiple instance learning networks. Med. Image Anal. 2020, 65, 101789. [Google Scholar] [CrossRef]
- Lu, C.; Koyuncu, C.; Corredor, G.; Prasanna, P.; Leo, P.; Wang, X.; Janowczyk, A.; Bera, K.; Lewis, J., Jr.; Velcheti, V. Feature-driven local cell graph (FLocK): New computational pathology-based descriptors for prognosis of lung cancer and HPV status of oropharyngeal cancers. Med. Image Anal. 2021, 68, 101903. [Google Scholar] [CrossRef]
- Othman, N.A.; Abdel-Fattah, M.A.; Ali, A.T. A Hybrid Deep Learning Framework with Decision-Level Fusion for Breast Cancer Survival Prediction. Big Data Cogn. Comput. 2023, 7, 50. [Google Scholar] [CrossRef]
- Sandarenu, P.; Millar, E.K.; Song, Y.; Browne, L.; Beretov, J.; Lynch, J.; Graham, P.H.; Jonnagaddala, J.; Hawkins, N.; Huang, J. Survival prediction in triple negative breast cancer using multiple instance learning of histopathological images. Sci. Rep. 2022, 12, 14527. [Google Scholar] [CrossRef]
- Diao, S.; Tang, L.; He, J.; Zhao, H.; Luo, W.; Xie, Y.; Qin, W. Automatic Computer-Aided Histopathologic Segmentation for Nasopharyngeal Carcinoma Using Transformer Framework. In Proceedings of the International Workshop on Computational Mathematics Modeling in Cancer Analysis, Singapore, 18 September 2022; pp. 141–149. [Google Scholar]
- Lu, C.; Lewis, J.S., Jr.; Dupont, W.D.; Plummer, W.D., Jr.; Janowczyk, A.; Madabhushi, A. An oral cavity squamous cell carcinoma quantitative histomorphometric-based image classifier of nuclear morphology can risk stratify patients for disease-specific survival. Mod. Pathol. 2017, 30, 1655–1665. [Google Scholar] [CrossRef]
- Lu, C.; Romo-Bucheli, D.; Wang, X.; Janowczyk, A.; Ganesan, S.; Gilmore, H.; Rimm, D.; Madabhushi, A. Nuclear shape and orientation features from H&E images predict survival in early-stage estrogen receptor-positive breast cancers. Lab. Investig. 2018, 98, 1438–1448. [Google Scholar]
- Alsubaie, N.M.; Snead, D.; Rajpoot, N.M. Tumour nuclear morphometrics predict survival in lung adenocarcinoma. IEEE Access 2021, 9, 12322–12331. [Google Scholar] [CrossRef]
- Rosenbaum, P.R.; Rubin, D.B. The central role of the propensity score in observational studies for causal effects. Biometrika 1983, 70, 41–55. [Google Scholar] [CrossRef]
- Zhuang, F.; Qi, Z.; Duan, K.; Xi, D.; Zhu, Y.; Zhu, H.; Xiong, H.; He, Q. A comprehensive survey on transfer learning. Proc. IEEE 2020, 109, 43–76. [Google Scholar] [CrossRef]
- Tsuneki, M.; Abe, M.; Kanavati, F. A deep learning model for prostate adenocarcinoma classification in needle biopsy whole-slide images using transfer learning. Diagnostics 2022, 12, 768. [Google Scholar] [CrossRef]
- Graham, S.; Vu, Q.D.; Raza, S.E.A.; Azam, A.; Tsang, Y.W.; Kwak, J.T.; Rajpoot, N. Hover-net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med. Image Anal. 2019, 58, 101563. [Google Scholar] [CrossRef] [PubMed]
- Gamper, J.; Alemi Koohbanani, N.; Benet, K.; Khuram, A.; Rajpoot, N. Pannuke: An open pan-cancer histology dataset for nuclei instance segmentation and classification. In Proceedings of the Digital Pathology: 15th European Congress, ECDP 2019, Proceedings 15, Warwick, UK, 10–13 April 2019; pp. 11–19. [Google Scholar]
- Gupta, R.; Le, H.; Van Arnam, J.; Belinsky, D.; Hasan, M.; Samaras, D.; Kurc, T.; Saltz, J.H. Characterizing immune responses in whole slide images of cancer with digital pathology and pathomics. Curr. Pathobiol. Rep. 2020, 8, 133–148. [Google Scholar] [CrossRef]
- Wei, Z.; Zhao, X.; Chen, J.; Sun, Q.; Wang, Z.; Wang, Y.; Ye, Z.; Yuan, Y.; Sun, L.; Jing, J. Deep learning-based stratification of gastric cancer patients from hematoxylin and eosin-stained whole slide images by predicting molecular features for immunotherapy response. Am. J. Pathol. 2023, 193, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Rodriguez, E.; Da Silva, A.C.A.L.; Hsu, W. Using Graph Neural Networks to Capture Tumor Spatial Relationships for Lung Adenocarcinoma Recurrence Prediction. In Proceedings of the 2023 IEEE 20th International Symposium on Biomedical Imaging (ISBI), Cartagena de Indias, Colombia, 18–21 April 2023; pp. 1–5. [Google Scholar]
- Chen, P.; Rojas, F.R.; Hu, X.; Serrano, A.; Zhu, B.; Chen, H.; Hong, L.; Bandyoyadhyay, R.; Aminu, M.; Kalhor, N. Pathomic Features Reveal Immune and Molecular Evolution from Lung Preneoplasia to Invasive Adenocarcinoma. Mod. Pathol. 2023, 36, 100326. [Google Scholar] [CrossRef]
- Majurski, M.; Manescu, P.; Padi, S.; Schaub, N.; Hotaling, N.; Simon, C., Jr.; Bajcsy, P. Cell image segmentation using generative adversarial networks, transfer learning, and augmentations. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops, Long Beach, CA, USA, 16–17 June 2019. [Google Scholar]
- Riana, D.; Jamil, M.; Hadianti, S.; Na’am, J.; Sutanto, H.; Sukwadi, R. Model of Watershed Segmentation in Deep Learning Method to Improve Identification of Cervical Cancer at Overlay Cells. TEM J. 2023, 12, 813–819. [Google Scholar] [CrossRef]
- Herbsthofer, L.; Ehall, B.; Tomberger, M.; Prietl, B.; Pieber, T.R.; López-García, P. Procedural generation of synthetic multiplex immunohistochemistry images using cell-based image compression and conditional generative adversarial networks. In Proceedings of the Medical Imaging 2022: Digital and Computational Pathology, San Diego, CA, USA, 4 April 2022; pp. 140–148. [Google Scholar]
- Tolstikhin, I.O.; Houlsby, N.; Kolesnikov, A.; Beyer, L.; Zhai, X.; Unterthiner, T.; Yung, J.; Steiner, A.; Keysers, D.; Uszkoreit, J. Mlp-mixer: An all-mlp architecture for vision. Adv. Neural Inf. Process. Syst. 2021, 34, 24261–24272. [Google Scholar]
- Lai, Z.; Deng, H. Medical image classification based on deep features extracted by deep model and statistic feature fusion with multilayer perceptron. Comput. Intell. Neurosci. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Gao, J.; Lan, J.; Wang, B.; Li, F. SDANet: Spatial deep attention-based for point cloud classification and segmentation. Mach. Learn. 2022, 111, 1327–1348. [Google Scholar] [CrossRef]
- Hendrycks, D.; Gimpel, K. Gaussian error linear units (gelus). arXiv 2016, arXiv:1606.08415. [Google Scholar]
- Ba, J.L.; Kiros, J.R.; Hinton, G.E. Layer normalization. arXiv 2016, arXiv:1607.06450. [Google Scholar]
- Dosovitskiy, A.; Beyer, L.; Kolesnikov, A.; Weissenborn, D.; Zhai, X.; Unterthiner, T.; Dehghani, M.; Minderer, M.; Heigold, G.; Gelly, S. An image is worth 16x16 words: Transformers for image recognition at scale. arXiv 2020, arXiv:2010.11929. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30. [Google Scholar]
- Zhu, X.; Yao, J.; Huang, J. Deep convolutional neural network for survival analysis with pathological images. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15–18 December 2016; pp. 544–547. [Google Scholar]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Győrffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef]
- Antolini, L.; Boracchi, P.; Biganzoli, E. A time-dependent discrimination index for survival data. Stat. Med. 2005, 24, 3927–3944. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Zhang, Y.; Liu, H.; Hu, Q. Transfuse: Fusing transformers and cnns for medical image segmentation. In Proceedings of the Medical Image Computing and Computer Assisted Intervention–MICCAI 2021: 24th International Conference, Proceedings, Part I 24, Strasbourg, France, 27 September–1 October 2021; pp. 14–24. [Google Scholar]





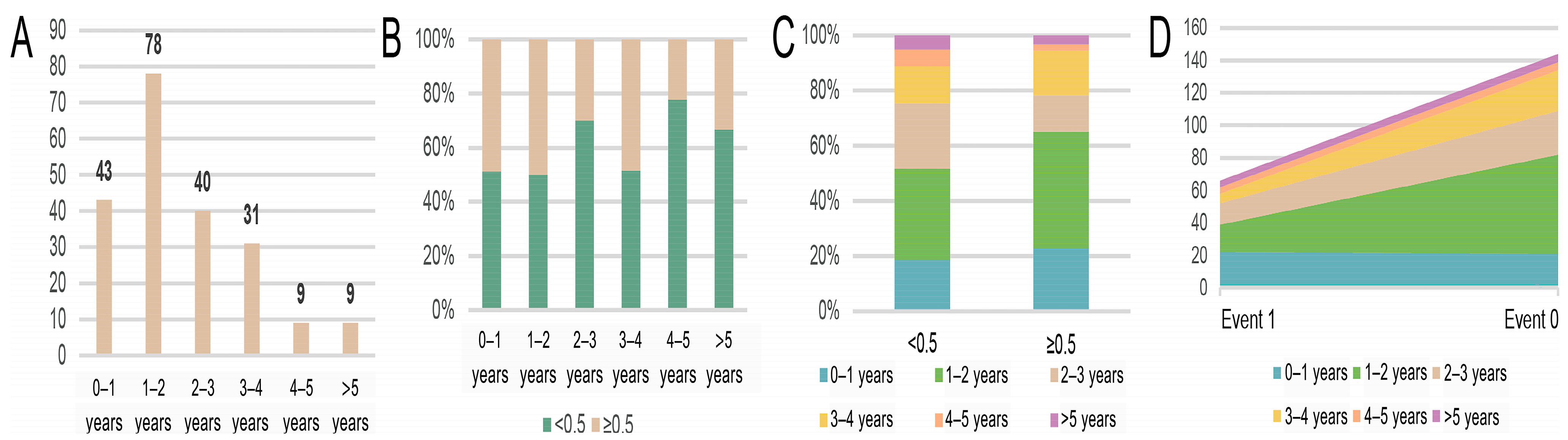{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | C-Index | p-Value |
|---|---|---|
| ViT | 0.636 ± 0.07 | 1.8 × 10−6 |
| DeepMixer | 0.643 ± 0.10 | 3.7 × 10−5 |
| Our | 0.675 ± 0.05 | 5.6 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diao, S.; Chen, P.; Showkatian, E.; Bandyopadhyay, R.; Rojas, F.R.; Zhu, B.; Hong, L.; Aminu, M.; Saad, M.B.; Salehjahromi, M.; et al. Automated Cellular-Level Dual Global Fusion of Whole-Slide Imaging for Lung Adenocarcinoma Prognosis. Cancers 2023, 15, 4824. https://doi.org/10.3390/cancers15194824
Diao S, Chen P, Showkatian E, Bandyopadhyay R, Rojas FR, Zhu B, Hong L, Aminu M, Saad MB, Salehjahromi M, et al. Automated Cellular-Level Dual Global Fusion of Whole-Slide Imaging for Lung Adenocarcinoma Prognosis. Cancers. 2023; 15(19):4824. https://doi.org/10.3390/cancers15194824
Chicago/Turabian StyleDiao, Songhui, Pingjun Chen, Eman Showkatian, Rukhmini Bandyopadhyay, Frank R. Rojas, Bo Zhu, Lingzhi Hong, Muhammad Aminu, Maliazurina B. Saad, Morteza Salehjahromi, and et al. 2023. "Automated Cellular-Level Dual Global Fusion of Whole-Slide Imaging for Lung Adenocarcinoma Prognosis" Cancers 15, no. 19: 4824. https://doi.org/10.3390/cancers15194824
APA StyleDiao, S., Chen, P., Showkatian, E., Bandyopadhyay, R., Rojas, F. R., Zhu, B., Hong, L., Aminu, M., Saad, M. B., Salehjahromi, M., Muneer, A., Sujit, S. J., Behrens, C., Gibbons, D. L., Heymach, J. V., Kalhor, N., Wistuba, I. I., Solis Soto, L. M., Zhang, J., ... Wu, J. (2023). Automated Cellular-Level Dual Global Fusion of Whole-Slide Imaging for Lung Adenocarcinoma Prognosis. Cancers, 15(19), 4824. https://doi.org/10.3390/cancers15194824