

Deletion of PGAM5 Downregulates FABP1 and Attenuates Long-Chain Fatty Acid Uptake in Hepatocellular Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Cell Growth and ATP Quantification Assays
2.3. Quantification of Fatty Acid Uptake
2.4. Quantitative PCR
2.5. Western Blotting
2.6. Statistical Analyses
3. Results
3.1. Deletion of PGAM5 Attenuates Hepatocellular Carcinoma Cell Growth
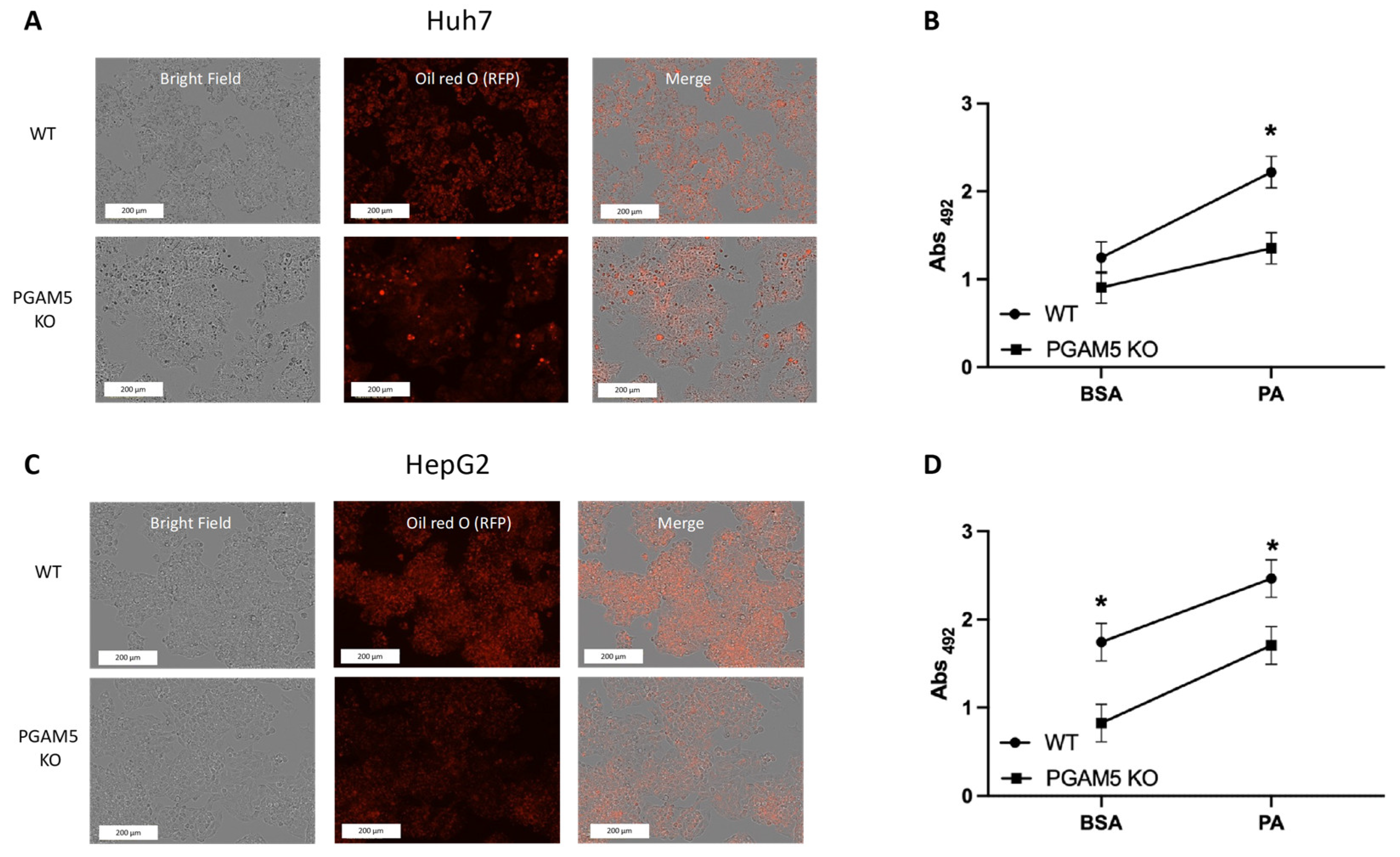
3.2. Long-Chain Fatty Acid-Induced Steatosis Is Ameliorated with PGAM5 Knockout
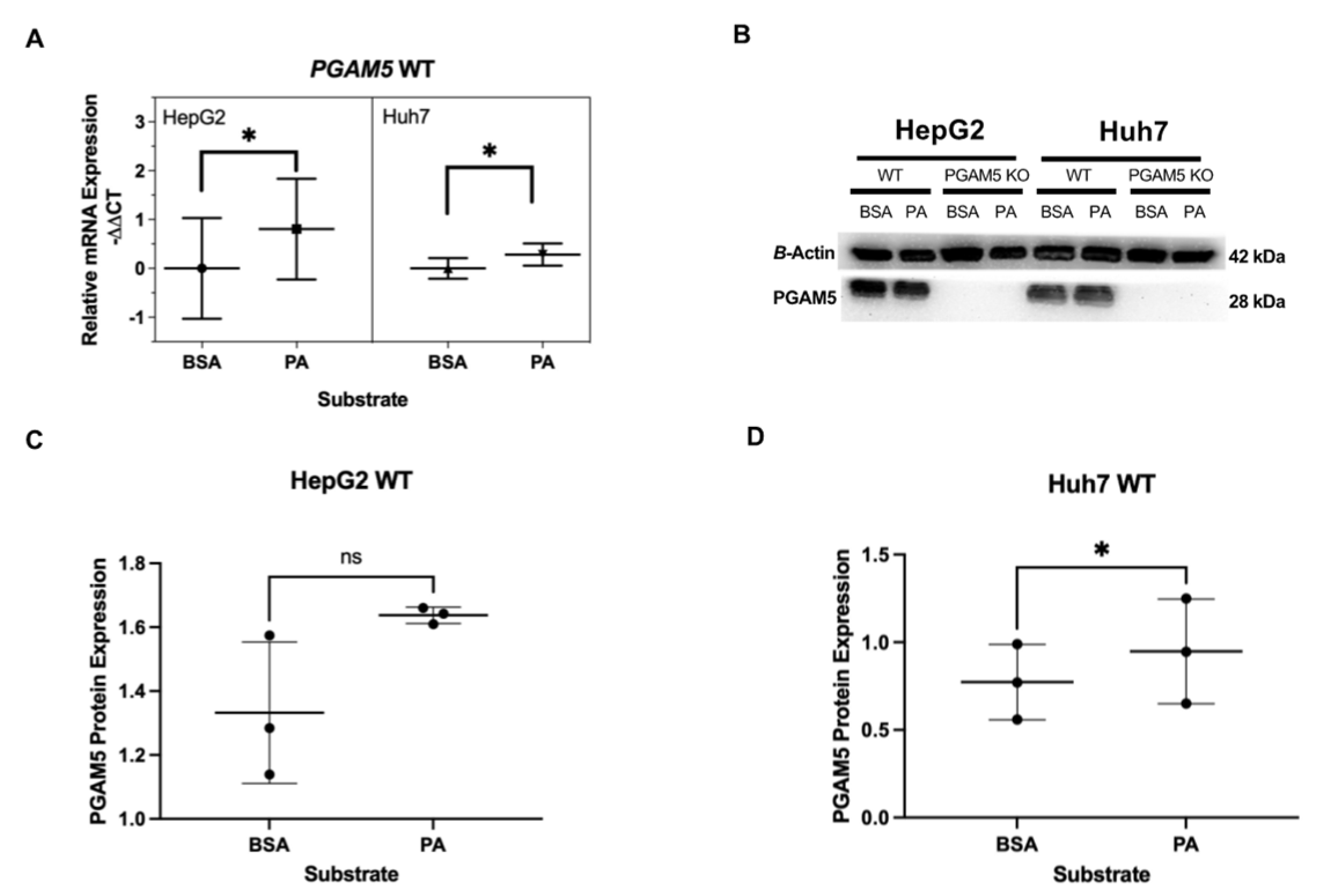
3.3. Long-Chain Fatty Acid Treatment Upregulates PGAM5 mRNA Expression
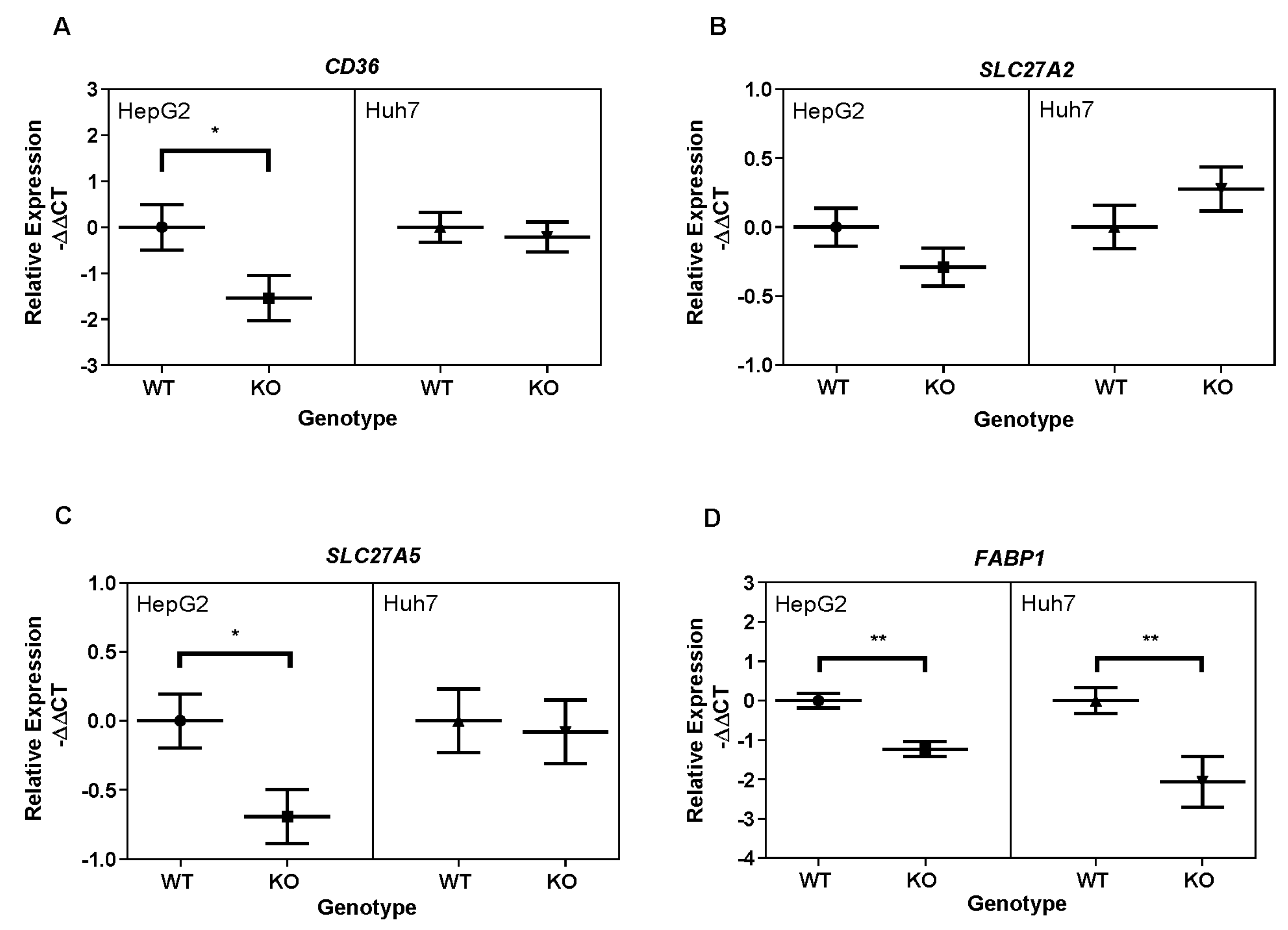
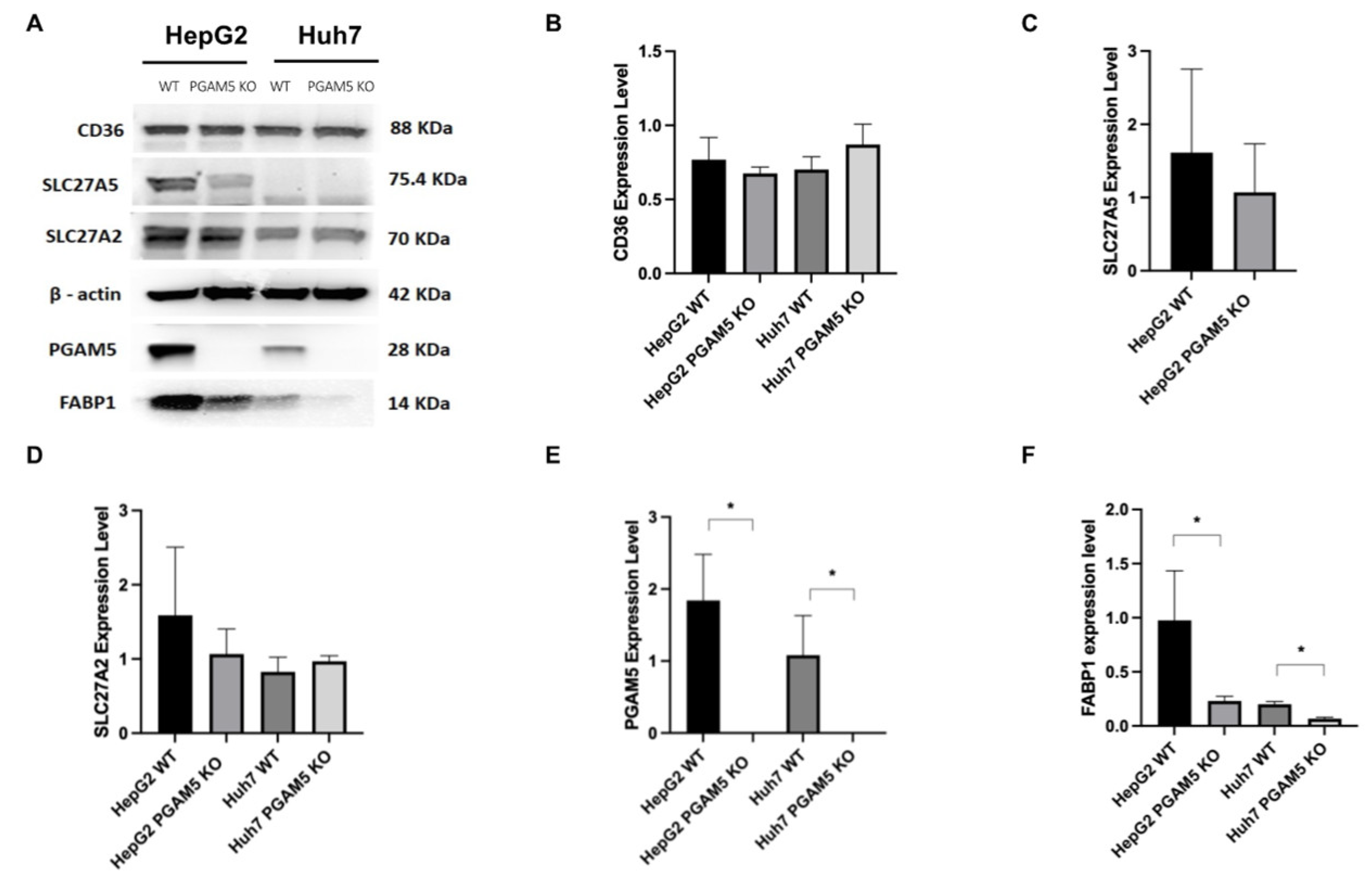
3.4. Hepatocellular Fatty Acid Transporter Expression Is Downregulated with PGAM5 Knockout
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wang, T.; Yue, C.; Luo, X.; Xiao, P. PGAM5: A necroptosis gene associated with poor tumor prognosis that promotes cutaneous melanoma progression. Front. Oncol. 2022, 12, 1004511. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, Z.; Wang, L.; Yang, Q.; Huang, J.; Huang, J. A Mitophagy-Related Gene Signature for Subtype Identification and Prognosis Prediction of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 12123. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gu, L.; Lin, X.; Liu, C.; Lu, B.; Cui, K.; Zhou, F.; Zhao, Q.; Prochownik, E.V.; Fan, C.; et al. Dynamic Regulation of ME1 Phosphorylation and Acetylation Affects Lipid Metabolism and Colorectal Tumorigenesis. Mol. Cell 2020, 77, 138–149. [Google Scholar] [CrossRef]
- Cheng, J.; Qian, D.; Ding, X.; Song, T.; Cai, M.; Xie, D.; Wang, Y.; Zhao, J.; Liu, Z.; Wu, Z.; et al. High PGAM5 expression induces chemoresistance by enhancing Bcl-xL-mediated anti-apoptotic signaling and predicts poor prognosis in hepatocellular carcinoma patients. Cell Death Dis. 2018, 9, 991. [Google Scholar] [CrossRef]
- Liang, S.; Zhu, C.; Suo, C.; Wei, H.; Yu, Y.; Gu, X.; Chen, L.; Yuan, M.; Shen, S.; Li, S.; et al. Mitochondrion-Localized SND1 Promotes Mitophagy and Liver Cancer Progression through PGAM5. Front. Oncol. 2022, 12, 857968. [Google Scholar] [CrossRef]
- Meng, L.; Hu, P.; Xu, A. PGAM5 promotes tumorigenesis of gastric cancer cells through PI3K/AKT pathway. Pathol. Res. Pract. 2023, 244, 154405. [Google Scholar] [CrossRef]
- Okuno, H.; Okuzono, H.; Hayase, A.; Kumagai, F.; Tanii, S.; Hino, N.; Okada, Y.; Tachibana, K.; Doi, T.; Ishimoto, K. Lipin-1 is a novel substrate of protein phosphatase PGAM5. Biochem. Biophys. Res. Commun. 2019, 509, 886–891. [Google Scholar] [CrossRef]
- Zhang, S.L.; Tang, H.B.; Hu, J.T.; Zang, Z.L.; Ding, X.; Li, S.; Yang, H. PGAM5-CypD pathway is involved in bromocriptine-induced RIP3/MLKL-dependent necroptosis of prolactinoma cells. Biomed. Pharmacother. 2019, 111, 638–648. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Yu, P.; Wang, Y.; Zhang, J.; Hang, W.; Yin, Z.-X.; Liu, G.; Chen, J.; Werle, K.D.; Quan, C.-S.; et al. AMP-activated protein kinase protects against necroptosis via regulation of Keap1-PGAM5 complex. Int. J. Cardiol. 2018, 259, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Lin, N.; Dong, D.; Ma, J.; Su, J.; Sun, L. PGAM5: A crucial role in mitochondrial dynamics and programmed cell death. Eur. J. Cell Biol. 2020, 100, 151144. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Gu, X.; Li, G.; Bao, Z.; Li, L. Mitochondrial Mechanisms of Necroptosis in Liver Diseases. Int. J. Mol. Sci. 2020, 22, 66. [Google Scholar] [CrossRef]
- Yao, R.-Q.; Ren, C.; Xia, Z.-F.; Yao, Y.-M. Organelle-specific autophagy in inflammatory diseases: A potential therapeutic target underlying the quality control of multiple organelles. Autophagy 2020, 17, 385–401. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, M.; Liu, X.; Ren, Y.; Liu, G.; Qin, S. Research progress of phosphoglycerate mutase 5-mediated mitophagy and necroptosis. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2022, 34, 890–896. [Google Scholar] [CrossRef]
- Scaturro, P.; Pichlmair, A. Oxeiptosis: A discreet way to respond to radicals. Curr. Opin. Immunol. 2018, 56, 37–43. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Holze, C.; Michaudel, C.; Mackowiak, C.; Haas, D.A.; Benda, C.; Hubel, P.; Pennemann, F.L.; Schnepf, D.; Wettmarshausen, J.; Braun, M.; et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 2017, 19, 130–140. [Google Scholar] [CrossRef]
- Kang, Y.J.; Bang, B.-R.; Han, K.H.; Hong, L.; Shim, E.-J.; Ma, J.; Lerner, R.A.; Otsuka, M. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat. Commun. 2015, 6, 8371. [Google Scholar] [CrossRef]
- Ma, K.; Zhang, Z.; Chang, R.; Cheng, H.; Mu, C.; Zhao, T.; Chen, L.; Zhang, C.; Luo, Q.; Lin, J.; et al. Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death Differ. 2020, 27, 1036–1051. [Google Scholar] [CrossRef] [PubMed]
- Augustine, D.; Khan, W.; Rao, R.S.; Patil, S.; Awan, K.H.; Sowmya, S.V.; Haragannavar, V.C.; Prasad, K. Lipid metabolism in cancer: A systematic review. J. Carcinog. 2021, 20, 4. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Su, X.; Abumrad, N.A. Cellular fatty acid uptake: A pathway under construction. Trends Endocrinol. Metab. 2009, 20, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2019, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Sadatomi, D.; Tanimura, S.; Ozaki, K.-I.; Takeda, K. Atypical Protein Phosphatases: Emerging Players in Cellular Signaling. Int. J. Mol. Sci. 2013, 14, 4596–4612. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Zhou, L.; Elf, S.; Fan, J.; Kang, H.-B.; Seo, J.H.; Shan, C.; Dai, Q.; Zhang, L.; Xie, J.; et al. Phosphoglycerate Mutase 1 Coordinates Glycolysis and Biosynthesis to Promote Tumor Growth. Cancer Cell 2012, 22, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Ban, Y.; Tan, Y.; Xiong, W.; Li, G.; Xiang, B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: A pair of valves for fine-tuning of glucose metabolism in human cancer. Mol. Metab. 2018, 20, 1–13. [Google Scholar] [CrossRef]
- Chaikuad, A.; Filippakopoulos, P.; Marcsisin, S.R.; Picaud, S.; Schröder, M.; Sekine, S.; Ichijo, H.; Engen, J.R.; Takeda, K.; Knapp, S. Structures of PGAM5 Provide Insight into Active Site Plasticity and Multimeric Assembly. Structure 2017, 25, 1089–1099.e3. [Google Scholar] [CrossRef]
- Storch, J.; Corsico, B. The Emerging Functions and Mechanisms of Mammalian Fatty Acid–Binding Proteins. Annu. Rev. Nutr. 2008, 28, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Coe, N.R.; Bernlohr, D.A. Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1998, 1391, 287–306. [Google Scholar] [CrossRef]
- Martin, G.G.; Danneberg, H.; Kumar, L.S.; Atshaves, B.P.; Erol, E.; Bader, M.; Schroeder, F.; Binas, B. Decreased Liver Fatty Acid Binding Capacity and Altered Liver Lipid Distribution in Mice Lacking the Liver Fatty Acid-binding Protein Gene. J. Biol. Chem. 2003, 278, 21429–21438. [Google Scholar] [CrossRef] [PubMed]
- Weisiger, R.A. Cytosolic fatty acid binding proteins catalyze two distinct steps in intracellular transport of their ligands. Mol. Cell Biochem. 2002, 239, 35–43. [Google Scholar] [CrossRef]
- Atshaves, B.P.; Storey, S.M.; Huang, H.; Schroeder, F. Liver fatty acid binding protein expression enhances branched-chain fatty acid metabolism. Mol. Cell Biochem. 2004, 259, 115–129. [Google Scholar] [CrossRef]
- Mukai, T.; Egawa, M.; Takeuchi, T.; Yamashita, H.; Kusudo, T. Silencing of FABP1 ameliorates hepatic steatosis, inflammation, and oxidative stress in mice with nonalcoholic fatty liver disease. FEBS Open Bio 2017, 7, 1009–1016. [Google Scholar] [CrossRef]
- Bordewick, U.; Heese, M.; Börchers, T.; Robenek, H.; Spener, F. Compartmentation of Hepatic Fatty-Acid-Binding Protein in Liver Cells and Its Effect on Microsomal Phosphatidic Acid Biosynthesis. Biol. Chem. Hoppe-Seyler 1989, 370, 229–238. [Google Scholar] [CrossRef]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef]
- Storch, J.; McDermott, L. Structural and functional analysis of fatty acid-binding proteins. J. Lipid Res. 2009, 50, S126–S131. [Google Scholar] [CrossRef]
- Atshaves, B.P.; Storey, S.M.; Petrescu, A.; Greenberg, C.C.; Lyuksyutova, O.I.; Smith, R., 3rd; Schroeder, F. Expression of fatty acid binding proteins inhibits lipid accumulation and alters toxicity in L cell fibroblasts. Am. J. Physiol. Cell Physiol. 2002, 283, C688–C703. [Google Scholar] [CrossRef]
- Ek, B.A.; Cistola, D.P.; Hamilton, J.A.; Kaduce, T.L.; Spector, A.A. Fatty acid binding proteins reduce 15-lipoxygenase-induced oxygenation of linoleic acid and arachidonic acid. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1997, 1346, 75–85. [Google Scholar] [CrossRef]
- Raza, H.; Pongubala, J.R.; Sorof, S. Specific high affinity binding of lipoxygenase metabolites of arachidonic acid by liver fatty acid binding protein. Biochem. Biophys. Res. Commun. 1989, 161, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Dum, D.; Ocokoljic, A.; Lennartz, M.; Hube-Magg, C.; Reiswich, V.; Höflmayer, D.; Jacobsen, F.; Bernreuther, C.; Lebok, P.; Sauter, G.; et al. FABP1 expression in human tumors: A tissue microarray study on 17,071 tumors. Virchows Arch. 2022, 481, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.-Y.; Liu, Y.-H.; Lin, H.-Y.; Lu, S.-C.; Lin, J.-Y. Liver fatty acid-binding protein (L-FABP) promotes cellular angiogenesis and migration in hepatocellular carcinoma. Oncotarget 2016, 7, 18229–18246. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, C.; Benet, M.; Pisonero-Vaquero, S.; Moya, M.; García-Mediavilla, M.V.; Martínez-Chantar, M.L.; González-Gallego, J.; Castell, J.V.; Sánchez-Campos, S.; Jover, R. The human liver fatty acid binding protein (FABP1) gene is activated by FOXA1 and PPARalpha; and repressed by C/EBPalpha: Implications in FABP1 down-regulation in nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2013, 1831, 803–818. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Torres-Ruiz, R.; Puig-Serra, P.; Moreno-Gaona, P.; Martin, M.C.; Moya, F.J.; Quintana-Bustamante, O.; Garcia-Silva, S.; Carcaboso, A.M.; Petazzi, P.; et al. In vivo CRISPR/Cas9 targeting of fusion oncogenes for selective elimination of cancer cells. Nat. Commun. 2020, 11, 5060. [Google Scholar] [CrossRef]
- Tang, W.; Liu, J.; Ding, B. Nucleic acid nanostructure for delivery of CRISPR/Cas9-based gene editing system. Interdiscip. Med. 2023, 1, e20220014. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muthusamy, G.; Liu, C.-C.; Johnston, A.N. Deletion of PGAM5 Downregulates FABP1 and Attenuates Long-Chain Fatty Acid Uptake in Hepatocellular Carcinoma. Cancers 2023, 15, 4796. https://doi.org/10.3390/cancers15194796
Muthusamy G, Liu C-C, Johnston AN. Deletion of PGAM5 Downregulates FABP1 and Attenuates Long-Chain Fatty Acid Uptake in Hepatocellular Carcinoma. Cancers. 2023; 15(19):4796. https://doi.org/10.3390/cancers15194796
Chicago/Turabian StyleMuthusamy, Ganesan, Chin-Chi Liu, and Andrea N. Johnston. 2023. "Deletion of PGAM5 Downregulates FABP1 and Attenuates Long-Chain Fatty Acid Uptake in Hepatocellular Carcinoma" Cancers 15, no. 19: 4796. https://doi.org/10.3390/cancers15194796
APA StyleMuthusamy, G., Liu, C.-C., & Johnston, A. N. (2023). Deletion of PGAM5 Downregulates FABP1 and Attenuates Long-Chain Fatty Acid Uptake in Hepatocellular Carcinoma. Cancers, 15(19), 4796. https://doi.org/10.3390/cancers15194796