Is Insulin Receptor Substrate4 (IRS4) a Platform Involved in the Activation of Several Oncogenes?


 , ,
, ,  , and
, and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Compilation of the Bibliography: Search Engines
2.2. Selection Criteria and Organization of the Results
3. Results and Discussion
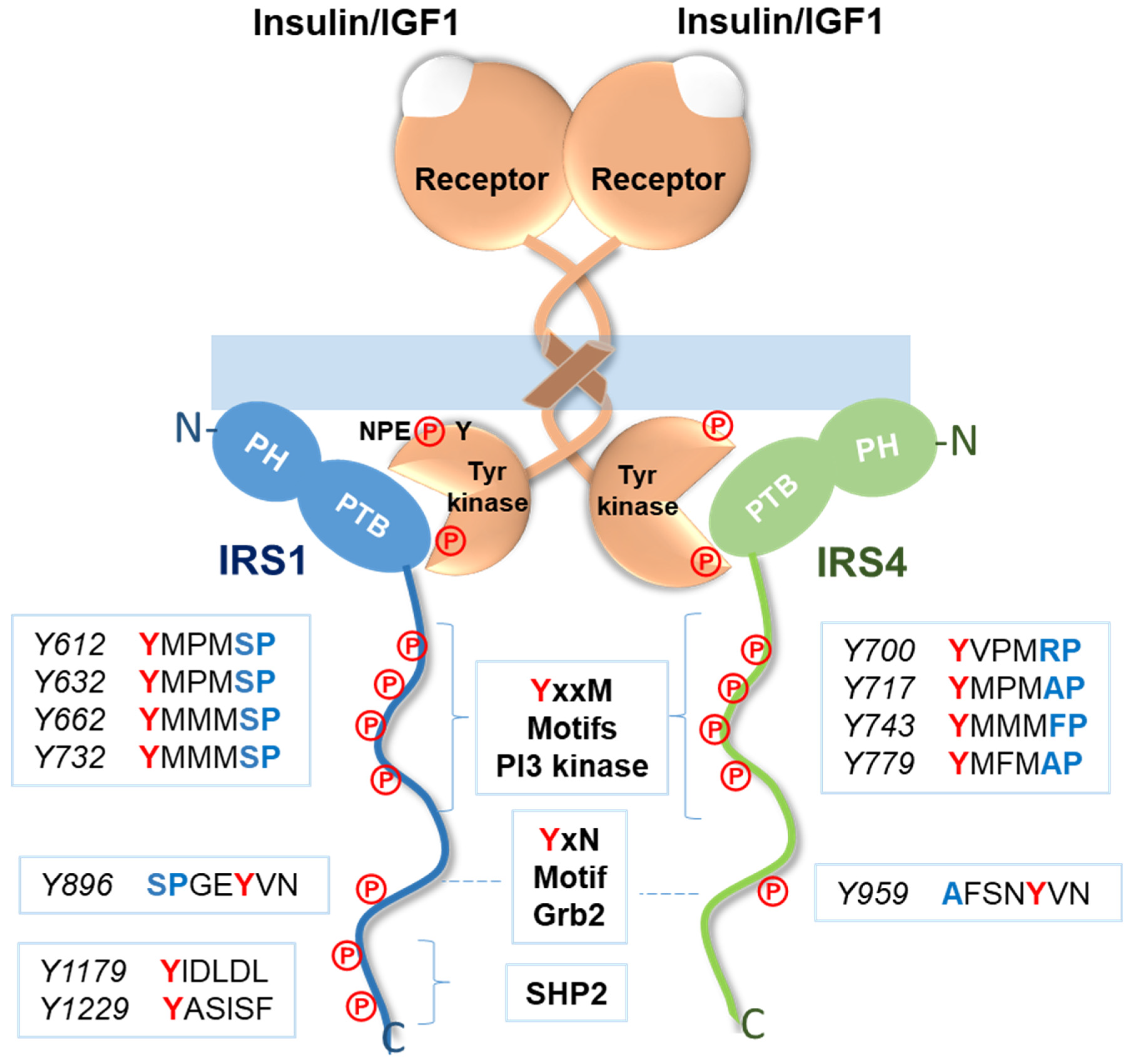
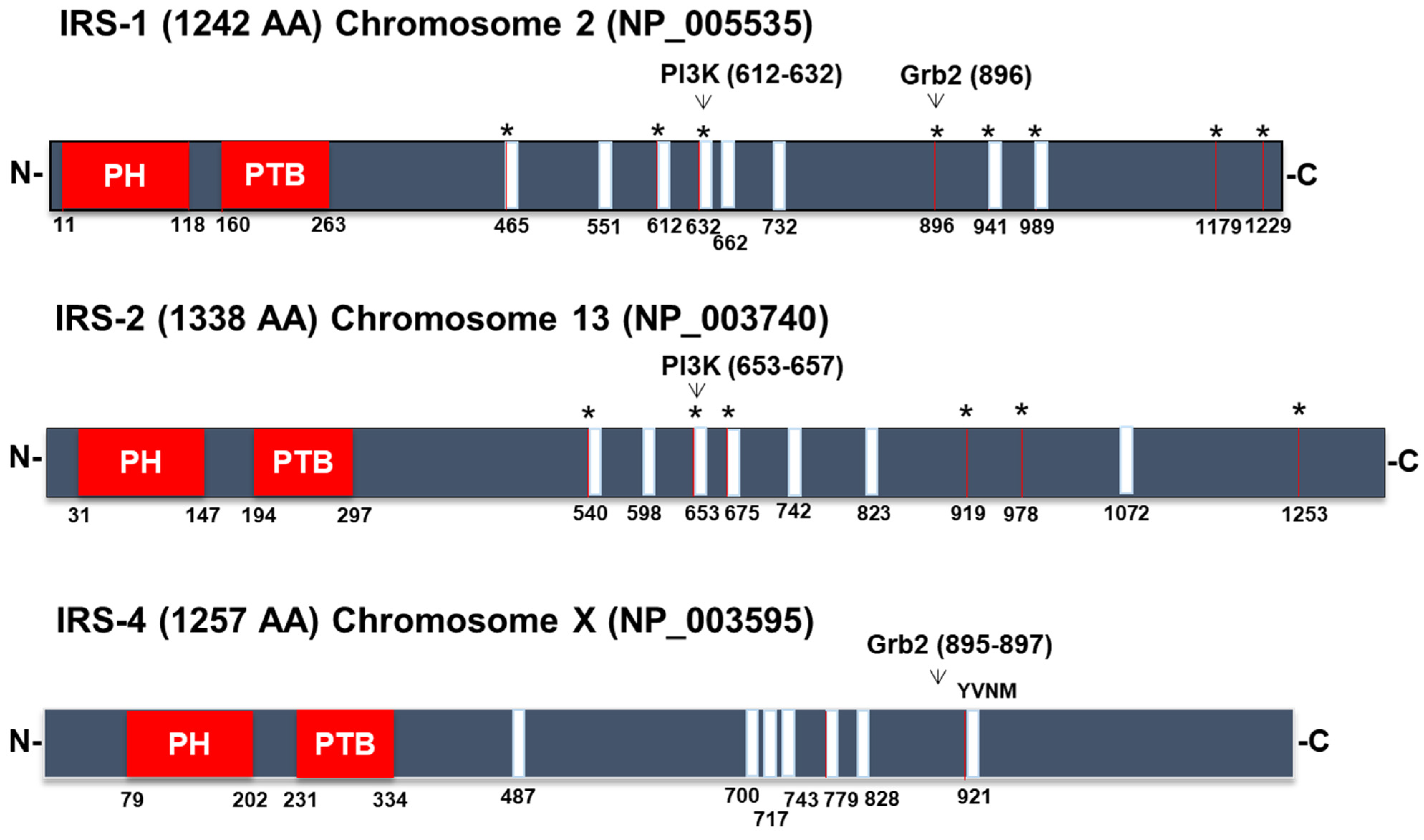
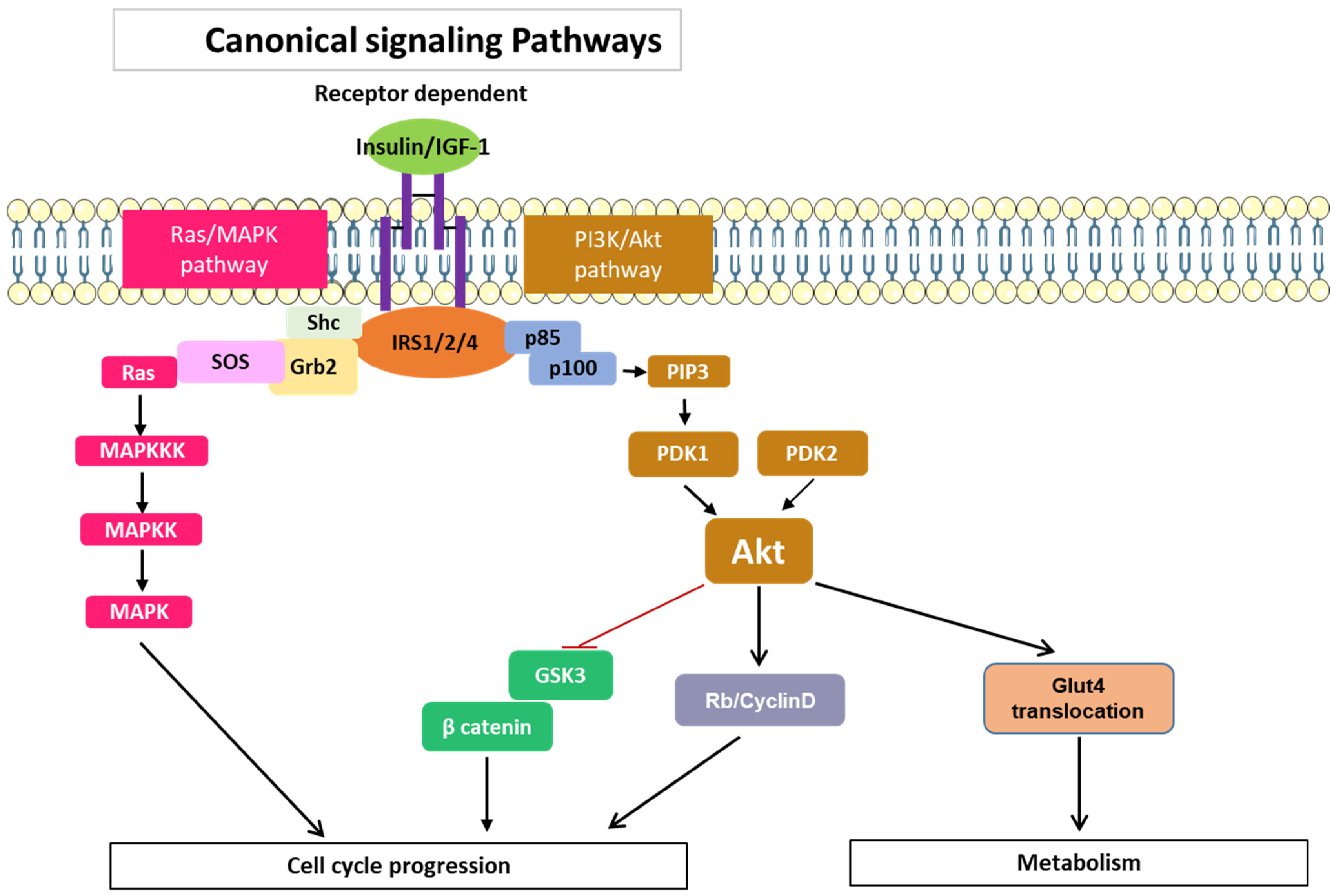
3.1. Signaling Pathways Common to All Members of the IRS Family
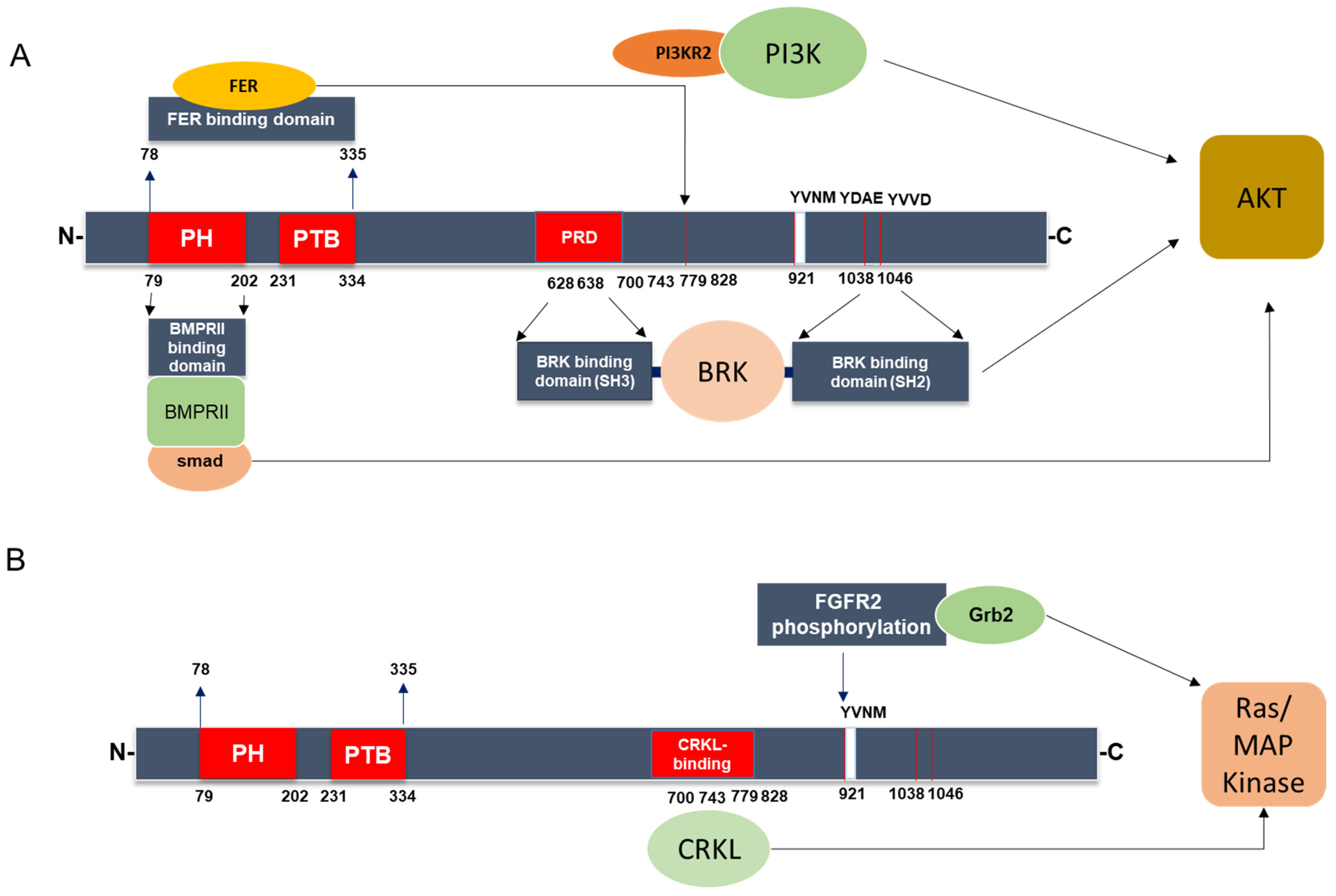
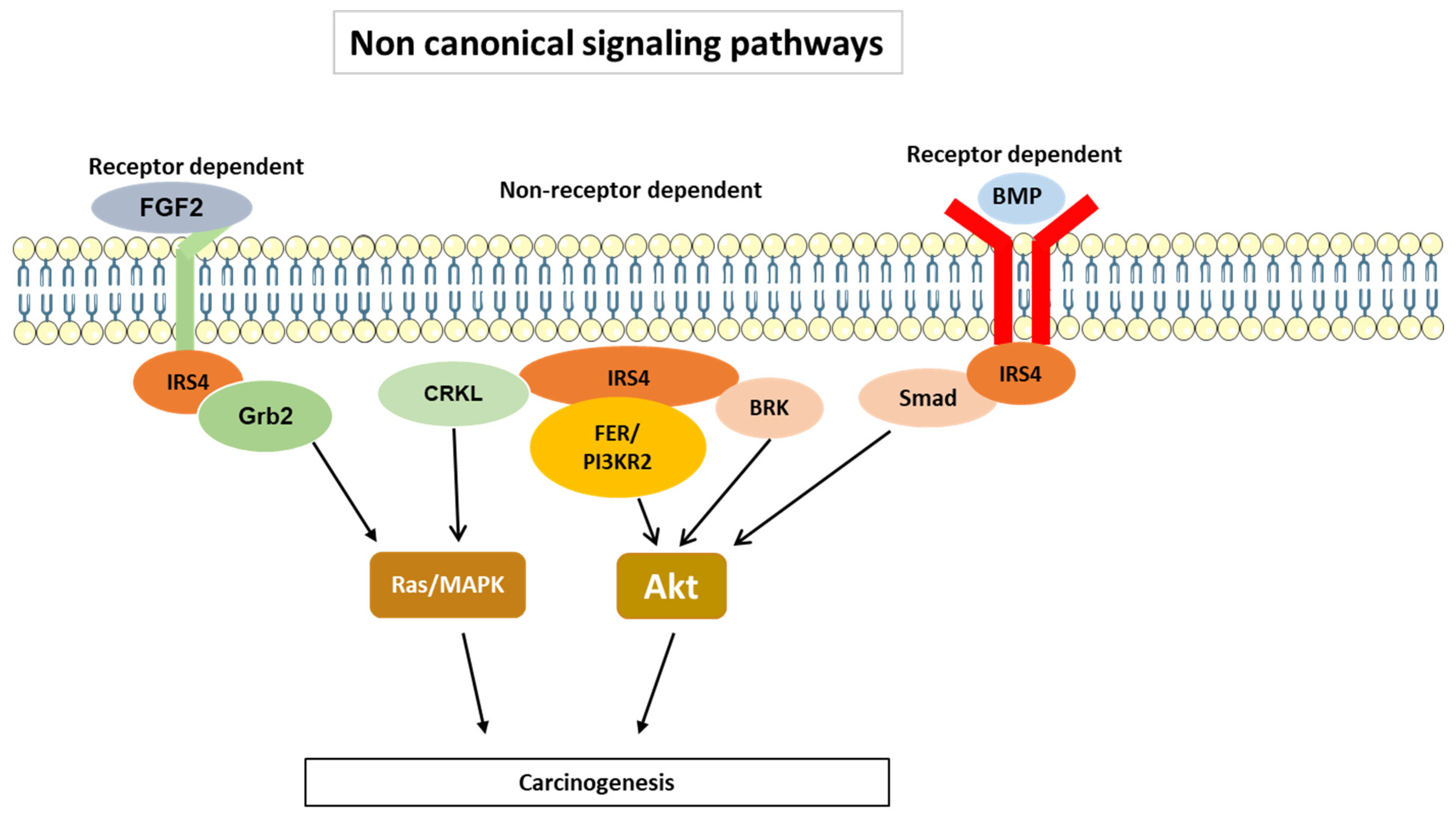
3.2. IRS-4-Specific Signaling Pathways
3.3. Regulation of IRS4 Levels
- At the transcriptional level
- Regulation of mRNA stability
- Regulation of IRS4 protein levels via proteolysis and the involvement of ser/thr phosphorylation
3.4. Regulation of Cell Growth
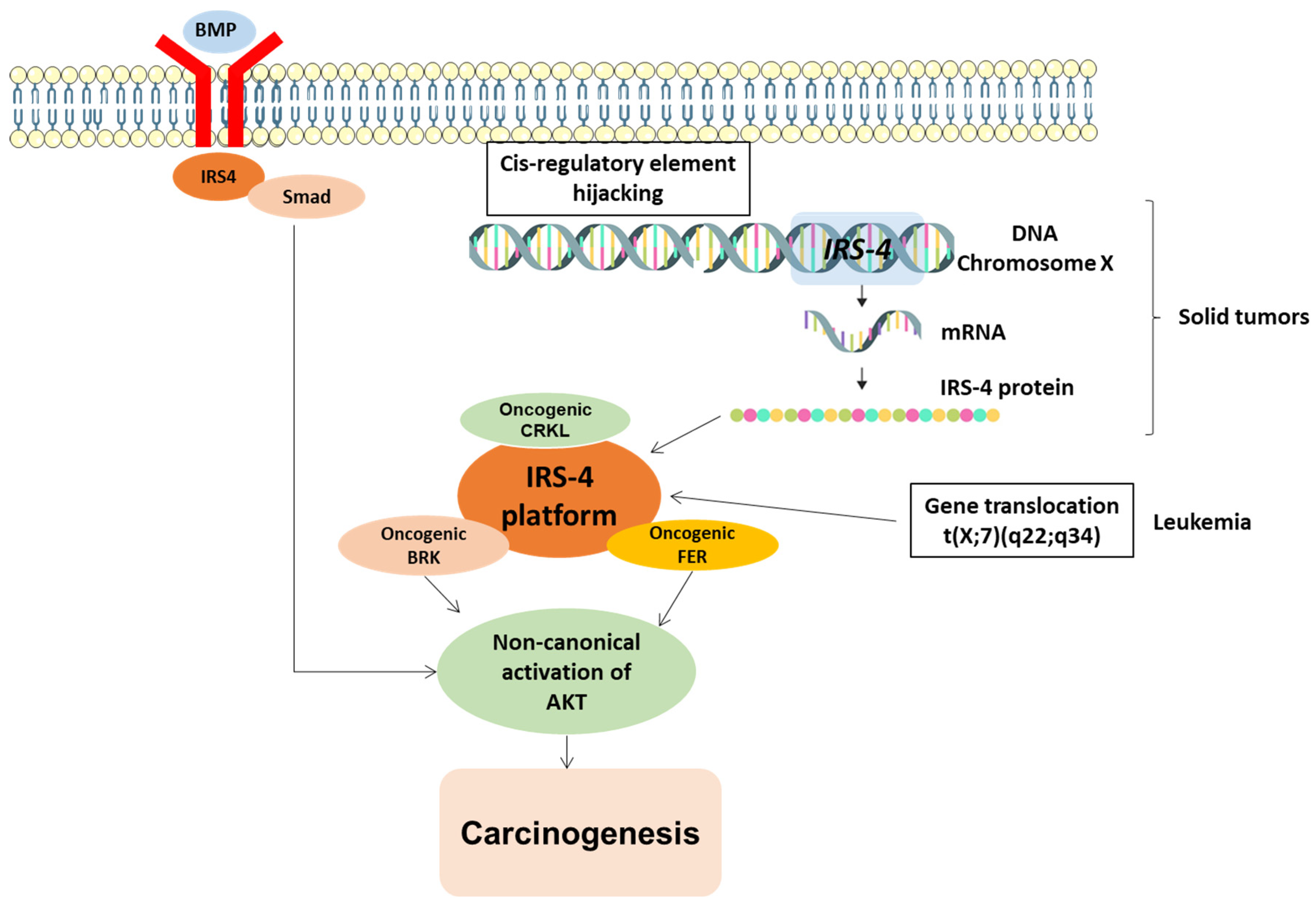
3.5. Overexpression of IRS4 in Cancer Cells
3.6. IRS-4 in Digestive Tumors
- Colorectal cancer
- Gastric cancer
- Hepatocellular carcinoma
- Pancreatic cancer
3.7. IRS-4 in Gynecological Neoplasms and Cancers
- Breast cancer
- Ovarian cancer
- Leiomyomas
3.8. IRS-4 in Lung Cancer
3.9. IRS-4 in Melanoma
3.10. IRS-4 in Hematological Tumors
3.11. IRS-4 in Other Malignancies and Proliferative Processes
- Subungual exostosis
- Meningiomas
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mardilovich, K.; Pankratz, S.L.; Shaw, L.M. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun. Signal. 2009, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Lavin, D.P.; White, M.F.; Brazil, D.P. IRS proteins and diabetic complications. Diabetologia 2016, 59, 2280–2291. [Google Scholar] [CrossRef] [PubMed]
- White, M.F.; Kahn, C.R. Insulin action at a molecular level—100 years of progress. Mol. Metab. 2021, 52, 101304. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P.; Whittaker, J. Structural Biology of Insulin and IGF1 Receptors: Implications for Drug Design. Nat. Rev. Drug Discov. 2002, 1, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. The Insulin Receptor: Both a Prototypical and Atypical Receptor Tyrosine Kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946. [Google Scholar] [CrossRef]
- Tamemoto, H.; Kadowaki, T.; Tobe, K.; Yagi, T.; Sakura, H.; Hayakawa, T.; Terauchi, Y.; Ueki, K.; Kaburagi, Y.; Satoh, S.; et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 1994, 372, 182–186. [Google Scholar] [CrossRef]
- Araki, E.; Lipes, M.A.; Patti, M.E.; Brüning, J.C.; Haag, B., 3rd; Johnson, R.S.; Kahn, C.R. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994, 372, 186–190. [Google Scholar] [CrossRef]
- Withers, D.J.; Gutierrez, J.S.; Towery, H.; Burks, D.J.; Ren, J.M.; Previs, S.; Zhang, Y.; Bernal, D.; Pons, S.; Shulman, G.I.; et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998, 391, 900–904. [Google Scholar] [CrossRef]
- Hao, Y.; Zhao, S.; Wang, Z. Targeting the protein-protein interaction between IRS1 and mutant p110α for cancer therapy. Toxicol. Pathol. 2014, 42, 140–147. [Google Scholar] [CrossRef][Green Version]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Reiss, K.; Del Valle, L.; Lassak, A.; Trojanek, J. Nuclear IRS-1 and cancer. J. Cell. Physiol. 2012, 227, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Björnholm, M.; He, A.R.; Attersand, A.; Lake, S.; Liu, S.C.H.; Lienhard, G.E.; Taylor, S.; Arner, P.; Zierath, J.R. Absence of Functional Insulin Receptor Substrate-3 (IRS-3) Gene in Humans. Diabetologia 2002, 45, 1697–1702. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Dhe-Paganon, S.; Melendez, P.A.; Lee, J.; Shoelson, S.E. Two New Substrates in Insulin Signaling, IRS5/DOK4 and IRS6/DOK5. J. Biol. Chem. 2003, 278, 25323–25330. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.; Sachs, M.; Britsch, S.; Di Cesare, S.; Schwarz-Romond, T.; Alitalo, K.; Birchmeier, W. Novel P62dok Family Members, Dok-4 and Dok-5, Are Substrates of the c-Ret Receptor Tyrosine Kinase and Mediate Neuronal Differentiation. J. Cell Biol. 2001, 154, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; Wang, Q.; Lienhard, G.E.; Keller, S.R. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E127–E133. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A.; Kwasniewski, W.; Adamek, A.; Gozdzicka-Jozefiak, A. Insulin-like growth factor (IGF) axis in cancerogenesis. Mutat. Res. Rev. Mutat. Res. 2017, 772, 78–104. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, T.A.; He, W.; Craparo, A.; Schaub, C.D.; O’Neill, T.J. Phosphotyrosine-dependent interaction of SHC and insulin receptor substrate 1 with the NPEY motif of the insulin receptor via a novel non-SH2 domain. Mol. Cell. Biol. 1995, 15, 2500–2508. [Google Scholar] [CrossRef]
- Sun, X.J.; Rothenberg, P.; Kahn, C.R.; Backer, J.M.; Araki, E.; Wilden, P.A.; Cahill, D.A.; Goldstein, B.J.; White, M.F. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991, 352, 73–77. [Google Scholar] [CrossRef]
- Fantin, V.R.; Lavan, B.E.; Wang, Q.; Jenkins, N.A.; Gilbert, D.J.; Copeland, N.G.; Keller, S.R.; Lienhard, G.E. Cloning, tissue expression, and chromosomal location of the mouse insulin receptor substrate 4 gene. Endocrinology 1999, 140, 1329–1337. [Google Scholar] [CrossRef]
- Giovannone, B.; Scaldaferri, M.L.; Federici, M.; Porzio, O.; Lauro, D.; Fusco, A.; Sbraccia, P.; Borboni, P.; Lauro, R.; Sesti, G. Insulin Receptor Substrate (IRS) Transduction System: Distinct and Overlapping Signaling Potential. Diabetes Metab. Res. Rev. 2000, 16, 434–441. [Google Scholar] [CrossRef]
- Yenush, L.; Makati, K.J.; Smith-Hall, J.; Ishibashi, O.; Myers, M.G.; White, M.F. The Pleckstrin Homology Domain Is the Principal Link between the Insulin Receptor and IRS-1. J. Biol. Chem. 1996, 271, 24300–24306. [Google Scholar] [CrossRef] [PubMed]
- Numan, S.; Russell, D.S. Discrete expression of insulin receptor substrate-4 mRNA in adult rat brain. Brain Res. Mol. Brain Res. 1999, 72, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Ikink, G.J.; Boer, M.; Bakker, E.R.; Hilkens, J. IRS4 induces mammary tumorigenesis and confers resistance to HER2-targeted therapy through constitutive PI3K/AKT-pathway hyperactivation. Nat. Commun. 2016, 7, 13567. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G.; Mendez, R.; Shi, P.; Pierce, J.H.; Rhoads, R.; White, M.F. The COOH-Terminal Tyrosine Phosphorylation Sites on IRS-1 Bind SHP-2 and Negatively Regulate Insulin Signaling. J. Biol. Chem. 1998, 273, 26908–26914. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G.; Wang, L.M.; Sun, X.J.; Zhang, Y.; Yenush, L.; Schlessinger, J.; Pierce, J.H.; White, M.F. Role of IRS-1-GRB-2 Complexes in Insulin Signaling. Mol. Cell. Biol. 1994, 14, 3577–3587. [Google Scholar] [CrossRef] [PubMed]
- Lavan, B.E.; Fantin, V.R.; Chang, E.T.; Lane, W.S.; Keller, S.R.; Lienhard, G.E. A Novel 160-KDa Phosphotyrosine Protein in Insulin-Treated Embryonic Kidney Cells Is a New Member of the Insulin Receptor Substrate Family. J. Biol. Chem. 1997, 272, 21403–21407. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Dissanayake, K.; MacKintosh, C. Effect of IRS4 levels on PI 3-kinase signalling. PLoS ONE 2013, 8, e73327. [Google Scholar] [CrossRef]
- Gorgisen, G.; Gulacar, I.M.; Ozes, O.N. The role of insulin receptor substrate (IRS) proteins in oncogenic transformation. Cell. Mol. Biol. 2017, 63, 1–5. [Google Scholar] [CrossRef]
- Fang, Z.; Meng, Q.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Zhao, Y.; Yu, X.; et al. Signaling pathways in cancer-associated fibroblasts: Recent advances and future perspectives. Cancer Commun. 2023, 43, 3–41. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic Activation of PI3K-AKT-MTOR Signaling Suppresses Ferroptosis via SREBP-Mediated Lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef] [PubMed]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/MTOR Interactive Pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/MTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Fantin, V.R.; Sparling, J.D.; Slot, J.W.; Keller, S.R.; Lienhard, G.E.; Lavan, B.E. Characterization of insulin receptor substrate 4 in human embryonic kidney 293 cells. J. Biol. Chem. 1998, 273, 10726–10732. [Google Scholar] [CrossRef]
- Hao, P.; Huang, Y.; Peng, J.; Yu, J.; Guo, X.; Bao, F.; Dian, Z.; An, S.; Xu, T.R. IRS4 Promotes the Progression of Non-Small Cell Lung Cancer and Confers Resistance to EGFR-TKI through the Activation of PI3K/Akt and Ras-MAPK Pathways. Exp. Cell Res. 2021, 403, 112615. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, X.; Zhu, Q.; Zhang, J.; Chen, S.; Wang, Y.; Cao, J.; Chen, L.; Hou, L.; Zhao, X.; et al. FER-mediated phosphorylation and PIK3R2 recruitment on IRS4 promotes AKT activation and tumorigenesis in ovarian cancer cells. eLife 2022, 11, e76183. [Google Scholar] [CrossRef]
- Qiu, H.; Zappacosta, F.; Su, W.; Annan, R.S.; Miller, W.T. Interaction between Brk kinase and insulin receptor substrate-4. Oncogene 2005, 24, 5656–5664. [Google Scholar] [CrossRef]
- Koval, A.P.; Karas, M.; Zick, Y.; LeRoith, D. Interplay of the proto-oncogene proteins CrkL and CrkII in insulin-like growth factor-I receptor-mediated signal transduction. J. Biol. Chem. 1998, 273, 14780–14787. [Google Scholar] [CrossRef]
- Park, T. Crk and CrkL as Therapeutic Targets for Cancer Treatment. Cells 2021, 10, 739. [Google Scholar] [CrossRef]
- Watanabe, T.; Tsuda, M.; Makino, Y.; Ichihara, S.; Sawa, H.; Minami, A.; Mochizuki, N.; Nagashima, K.; Tanaka, S. Adaptor molecule Crk is required for sustained phosphorylation of Grb2-associated binder 1 and hepatocyte growth factor-induced cell motility of human synovial sarcoma cell lines. Mol. Cancer Res. 2006, 4, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Díaz, J.; Chagoyen, M.; Olazabal-Morán, M.; González-García, A.; Carrera, A.C. The Opposing Roles of PIK3R1/p85α and PIK3R2/p85β in Cancer. Trends Cancer 2019, 5, 233–244. [Google Scholar] [CrossRef]
- Dörpholz, G.; Murgai, A.; Jatzlau, J.; Horbelt, D.; Belverdi, M.P.; Heroven, C.; Schreiber, I.; Wendel, G.; Ruschke, K.; Stricker, S.; et al. IRS4, a novel modulator of BMP/Smad and Akt signalling during early muscle differentiation. Sci. Rep. 2017, 7, 8778. [Google Scholar] [CrossRef] [PubMed]
- Ang, H.L.; Yuan, Y.; Lai, X.; Tan, T.Z.; Wang, L.; Huang, B.B.; Pandey, V.; Huang, R.Y.; Lobie, P.E.; Goh, B.C.; et al. Putting the BRK on breast cancer: From molecular target to therapeutics. Theranostics 2021, 11, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.W. FES/FER kinase signaling in hematopoietic cells and leukemias. Front. Biosci. Landmark Ed. 2012, 17, 861–875. [Google Scholar] [CrossRef]
- Guijarro, L.G.; Sanmartin-Salinas, P.; Pérez-Cuevas, E.; Toledo-Lobo, M.V.; Monserrat, J.; Zoullas, S.; Sáez, M.A.; Álvarez-Mon, M.A.; Bujan, J.; Noguerales-Fraguas, F.; et al. Possible Role of IRS-4 in the Origin of Multifocal Hepatocellular Carcinoma. Cancers 2021, 13, 2560. [Google Scholar] [CrossRef]
- Sanmartín-Salinas, P.; Guijarro, L.G. Overexpression of IRS-4 Correlates with Procaspase 3 Levels in Tumoural Tissue of Patients with Colorectal Cancer. J. Oncol. 2018, 2018, 3812581. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, H.; Xu, P.; Cong, L.N.; Sciacchitano, S.; Li, Y.; Graham, D.; Jacobs, A.R.; Taylor, S.I.; Quon, M.J. Action of insulin receptor substrate-3 (IRS-3) and IRS-4 to stimulate translocation of GLUT4 in rat adipose cells. Mol. Endocrinol. 1999, 13, 505–514. [Google Scholar] [CrossRef]
- Hinsby, A.M.; Olsen, J.V.; Mann, M. Tyrosine phosphoproteomics of fibroblast growth factor signaling: A role for insulin receptor substrate-4. J. Biol. Chem. 2004, 279, 46438–46447. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, C.; Jin, C.; Xie, R.; Wang, F.; McKeehan, W.L. Novel phosphotyrosine targets of FGFR2IIIb signaling. Cell Signal. 2009, 21, 1370–1378. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Barral, A.; Déjardin, J. The chromatin signatures of enhancers and their dynamic regulation. Nucleus 2023, 14, 2160551. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Koutsi, M.A.; Pouliou, M.; Champezou, L.; Vatsellas, G.; Giannopoulou, A.I.; Piperi, C.; Agelopoulos, M. Typical Enhancers, Super-Enhancers, and Cancers. Cancers 2022, 14, 4375. [Google Scholar] [CrossRef] [PubMed]
- Weischenfeldt, J.; Dubash, T.; Drainas, A.P.; Mardin, B.R.; Chen, Y.; Stütz, A.M.; Waszak, S.M.; Bosco, G.; Halvorsen, A.R.; Raeder, B.; et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 2017, 49, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Goodall, G.J.; Wickramasinghe, V.O. RNA in cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.; Jin, Z.; Gao, Z.; Jin, M.; Zhu, L.; Li, L.; Jin, C.; An, Y. Downregulation of miR-493 promoted melanoma proliferation by suppressing IRS4 expression. Tumour Biol. 2017, 39, 1010428317701640. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Y.; Xu, D.; Cao, L. hsa_circ_0023409 Accelerates Gastric Cancer Cell Growth and Metastasis through Regulating the IRS4/PI3K/AKT Pathway. Cell Transplant. 2021, 30, 963689720975390. [Google Scholar] [CrossRef]
- Li, X.; Zhong, L.; Wang, Z.; Chen, H.; Liao, D.; Zhang, R.; Zhang, H.; Kang, T. Phosphorylation of IRS4 by CK1γ2 promotes its degradation by CHIP through the ubiquitin/lysosome pathway. Theranostics 2018, 8, 3643–3653. [Google Scholar] [CrossRef]
- Li, S.S.; Dong, Y.H.; Liu, Z.P. Recent Advances in the Development of Casein Kinase 1 Inhibitors. Curr. Med. Chem. 2021, 28, 1585–1604. [Google Scholar] [CrossRef] [PubMed]
- Mihindukulasuriya, K.A.; Zhou, G.; Qin, J.; Tan, T.H. Protein phosphatase 4 interacts with and down-regulates insulin receptor substrate 4 following tumor necrosis factor-alpha stimulation. J. Biol. Chem. 2004, 279, 46588–46594. [Google Scholar] [CrossRef]
- Cuevas, E.P.; Escribano, O.; Chiloeches, A.; Ramirez Rubio, S.; Román, I.D.; Fernández-Moreno, M.D.; Guijarro, L.G. Role of insulin receptor substrate-4 in IGF-I-stimulated HEPG2 proliferation. J. Hepatol. 2007, 46, 1089–1098. [Google Scholar] [CrossRef]
- Ursø, B.; Ilondo, M.M.; Holst, P.A.; Christoffersen, C.T.; Ouwens, M.; Giorgetti, S.; Van Obberghen, E.; Naor, D.; Tornqvist, H.; De Meyts, P. IRS-4 mediated mitogenic signalling by insulin and growth hormone in LB cells, a murine T-cell lymphoma devoid of IGF-I receptors. Cell Signal. 2003, 15, 385–394. [Google Scholar] [CrossRef]
- Gahr, S.; Merger, M.; Bollheimer, L.C.; Hammerschmied, C.G.; Schölmerich, J.; Hügl, S.R. Hepatocyte growth factor stimulates proliferation of pancreatic beta-cells particularly in the presence of subphysiological glucose concentrations. J. Mol. Endocrinol. 2002, 28, 99–110. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wauman, J.; De Smet, A.S.; Catteeuw, D.; Belsham, D.; Tavernier, J. Insulin receptor substrate 4 couples the leptin receptor to multiple signaling pathways. Mol. Endocrinol. 2008, 22, 965–977. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fantin, V.R.; Keller, S.R.; Lienhard, G.E.; Wang, L.M. Insulin receptor substrate 4 supports insulin- and interleukin 4-stimulated proliferation of hematopoietic cells. Biochem. Biophys. Res. Commun. 1999, 260, 718–723. [Google Scholar] [CrossRef]
- Sanmartín-Salinas, P.; Lobo, M.D.V.T.; Noguerales-Fraguas, F.; Londoño, M.T.; Jiménez-Ruiz, A.; Guijarro, L.G. Insulin receptor substrate-4 is overexpressed in colorectal cancer and promotes retinoblastoma-cyclin-dependent kinase activation. J. Gastroenterol. 2018, 53, 932–944. [Google Scholar] [CrossRef]
- Escribano, O.; Fernández-Moreno, M.D.; Zueco, J.A.; Menor, C.; Fueyo, J.; Ropero, R.M.; Diaz-Laviada, I.; Román, I.D.; Guijarro, L.G. Insulin receptor substrate-4 signaling in quiescent rat hepatocytes and in regenerating rat liver. Hepatology 2003, 37, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Myers, M.G., Jr.; White, M.F. IRS-4 mediates protein kinase B signaling during insulin stimulation without promoting antiapoptosis. Mol. Cell. Biol. 2000, 20, 126–138. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lingohr, M.K.; Dickson, L.M.; Wrede, C.E.; Briaud, I.; McCuaig, J.F.; Myers, M.G., Jr.; Rhodes, C.J. Decreasing IRS-2 expression in pancreatic beta-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol. Cell. Endocrinol. 2003, 209, 17–31. [Google Scholar] [CrossRef]
- Guijarro, L.G.; Sanmartin-Salinas, P.; Pérez-Cuevas, E.; Toledo-Lobo, M.V.; Monserrat, J.; Zoullas, S.; Sáez, M.A.; Álvarez-Mon, M.A.; Bujan, J.; Noguerales-Fraguas, F.; et al. Actinomycin D Arrests Cell Cycle of Hepatocellular Carcinoma Cell Lines and Induces p53-Dependent Cell Death: A Study of the Molecular Mechanism Involved in the Protective Effect of IRS-4. Pharmaceuticals 2021, 14, 845. [Google Scholar] [CrossRef] [PubMed]
- Homma, Y.; Kanno, S.I.; Sasaki, K.; Nishita, M.; Yasui, A.; Asano, T.; Ohashi, K.; Mizuno, K. Insulin receptor substrate-4 binds to Slingshot-1 phosphatase and promotes cofilin dephosphorylation. J. Biol. Chem. 2014, 289, 26302–26313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, F.; Fonseca, N.A.; He, Y.; Fujita, M.; Nakagawa, H.; Zhang, Z.; Brazma, A.; Amin, S.B.; Awadalla, P.; et al. High-Coverage Whole-Genome Analysis of 1220 Cancers Reveals Hundreds of Genes Deregulated by Rearrangement-Mediated Cis-Regulatory Alterations. Nat. Commun. 2020, 11, 736. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Eng, C.; Nieman, L.Z.; Kapadia, A.S.; Du, X.L. Trends in Colorectal Cancer Incidence by Anatomic Site and Disease Stage in the United States from 1976 to 2005. Am. J. Clin. Oncol. 2011, 34, 573–580. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Grady, W.M.; Pritchard, C.C. Molecular Alterations and Biomarkers in Colorectal Cancer. Toxicol. Pathol. 2014, 42, 124–139. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Tsang, A.H.F.; Cheng, K.H.; Wong, A.S.P.; Ng, S.S.M.; Ma, B.B.Y.; Chan, C.M.L.; Tsui, N.B.Y.; Chan, L.W.C.; Yung, B.Y.M.; Wong, S.C.C. Current and Future Molecular Diagnostics in Colorectal Cancer and Colorectal Adenoma. World J. Gastroenterol. 2014, 20, 3847–3857. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic Instability in Colorectal Cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Duong, H.Q. The Molecular Characteristics of Colorectal Cancer: Implications for Diagnosis and Therapy. Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef]
- Sanmartín-Salinas, P.; Toledo-Lobo, M.V.; Noguerales-Fraguas, F.; Fernández-Contreras, M.E.; Guijarro, L.G. Overexpression of Insulin Receptor Substrate-4 Is Correlated with Clinical Staging in Colorectal Cancer Patients. J. Mol. Histol. 2018, 49, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, M.W.; Peh, J.; Hergenrother, P.J. Procaspase-3 Overexpression in Cancer: A Paradoxical Observation with Therapeutic Potential. ACS Chem. Biol. 2019, 14, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, L.; Meyer, M.; Fay, J.; Curry, S.; Bacon, O.; Duessmann, H.; John, K.; Boland, K.C.; McNamara, D.A.; Kay, E.W.; et al. Low Levels of Caspase-3 Predict Favourable Response to 5FU-Based Chemotherapy in Advanced Colorectal Cancer: Caspase-3 Inhibition as a Therapeutic Approach. Cell Death Dis. 2016, 7, e2087. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.M.; Rank, K.B.; Zeng, Y.; Capen, A.; Yadav, V.; Manro, J.R.; Engler, T.A.; Chedid, M. Activating the Wnt/β-Catenin Pathway for the Treatment of Melanoma--Application of LY2090314, a Novel Selective Inhibitor of Glycogen Synthase Kinase-3. PLoS ONE 2015, 10, e0125028. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-Catenin Signaling Pathway in Cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Sitarz, R.; Skierucha, M.; Mielko, J.; Offerhaus, G.J.A.; Maciejewski, R.; Polkowski, W.P. Gastric Cancer: Epidemiology, Prevention, Classification, and Treatment. Cancer Manag. Res. 2018, 10, 239. [Google Scholar] [CrossRef]
- Liang, Y.X.; Deng, J.Y.; Guo, H.H.; Ding, X.W.; Wang, X.N.; Wang, B.G.; Zhang, L.; Liang, H. Characteristics and Prognosis of Gastric Cancer in Patients Aged ≥ 70 Years. World J. Gastroenterol. 2013, 19, 6568–6578. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, J.; Hou, L.; Wang, G.; Liu, H.; Zhang, R.; Chen, X.; Zhu, J. Correction to: LncRNA AK023391 Promotes Tumorigenesis and Invasion of Gastric Cancer through Activation of the PI3K/Akt Signaling Pathway. J. Exp. Clin. Cancer Res. 2020, 39, 155. [Google Scholar] [CrossRef]
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular Carcinoma (HCC): Epidemiology, Etiology and Molecular Classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Cantarini, M.C.; de la Monte, S.M.; Pang, M.; Tong, M.; D’Errico, A.; Trevisani, F.; Wands, J.R. Aspartyl-asparagyl beta hydroxylase over-expression in human hepatoma is linked to activation of insulin-like growth factor and notch signaling mechanisms. Hepatology 2006, 44, 446–457. [Google Scholar] [CrossRef]
- Ganapathy, E.; Khachatoorian, R.; French, S.W. Glypican 3 increases IRS4 phosphorylation and stimulates Hepatocellular Carcinoma cell proliferation. FASEB J. 2013, 27, 872.11. [Google Scholar] [CrossRef]
- Klein, A.P. Pancreatic Cancer Epidemiology: Understanding the Role of Lifestyle and Inherited Risk Factors HHS Public Access. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Pekarek, L.; Fraile-Martinez, O.; Garcia-Montero, C.; Alvarez-Mon, M.A.; Acero, J.; Ruiz-Llorente, L.; García-Honduvilla, N.; Albillos, A.; Buján, J.; Alvarez-Mon, M.; et al. Towards an updated view on the clinical management of pancreatic adenocarcinoma: Current and future perspectives. Oncol. Lett. 2021, 22, 809. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Kommalapati, A.; Tella, S.H.; Goyal, G.; Ma, W.W.; Mahipal, A. Contemporary Management of Localized Resectable Pancreatic Cancer. Cancers 2018, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Pekarek, L.; Fraile-Martinez, O.; Garcia-Montero, C.; Saez, M.A.; Barquero-Pozanco, I.; Del Hierro-Marlasca, L.; de Castro Martinez, P.; Romero-Bazán, A.; Alvarez-Mon, M.A.; Monserrat, J.; et al. Clinical Applications of Classical and Novel Biological Markers of Pancreatic Cancer. Cancers 2022, 14, 1866. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Pekarek, L.; Garcia-Montero, C.; Fraile-Martinez, O.; Saez, M.A.; Asúnsolo, A.; Alvarez-Mon, M.A.; Monserrat, J.; Coca, S.; Toledo-Lobo, M.V.; et al. Prognostic Role of IRS-4 in the Survival of Patients with Pancreatic Cancer. Histol. Histopathol. 2022, 37, 449–459. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Buján, J.; García-Honduvilla, N.; Coca, S. Signal Transduction Pathways in Breast Cancer: The Important Role of PI3K/Akt/MTOR. J. Oncol. 2020, 2020, 9258396. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Hosseini, M.; Shahidsales, S.; Maftouh, M.; Ferns, G.A.; Ghayour-Mobarhan, M.; Hassanian, S.M.; Avan, A. Targeting the Akt/PI3K Signaling Pathway as a Potential Therapeutic Strategy for the Treatment of Pancreatic Cancer. Curr. Med. Chem. 2017, 24, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Fraile-Martínez, O.; García-Montero, C.; Pekarek, L.; Guijarro, L.G.; Castellanos, A.J.; Sanchez-Trujillo, L.; García-Honduvilla, N.; Álvarez-Mon, M.; Buján, J.; et al. Physical Activity as an Imperative Support in Breast Cancer Management. Cancers 2021, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.K.; Guan, J.L. Breast Cancer: Multiple Subtypes within a Tumor? Trends Cancer 2017, 3, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef]
- Ikink, G.J.; Hilkens, J. Insulin Receptor Substrate 4(IRS4) Is a Constitutive Active Oncogenic Driver Collaborating with HER2 and Causing Therapeutic Resistance. Mol. Cell. Oncol. 2017, 4, e1279722. [Google Scholar] [CrossRef]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef]
- Webb, P.M.; Jordan, S.J. Epidemiology of Epithelial Ovarian Cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 41, 3–14. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Dvorská, D.; Braný, D.; Danková, Z.; Halašová, E.; Višňovský, J. Molecular and Clinical Attributes of Uterine Leiomyomas. Tumour Biol. 2017, 39, 1010428317710226. [Google Scholar] [CrossRef] [PubMed]
- Mehine, M.; Kaasinen, E.; Mäkinen, N.; Katainen, R.; Kämpjärvi, K.; Pitkänen, E.; Heinonen, H.-R.; Bützow, R.; Kilpivaara, O.; Kuosmanen, A.; et al. Characterization of Uterine Leiomyomas by Whole-Genome Sequencing. N. Engl. J. Med. 2013, 369, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Mehine, M.; Mäkinen, N.; Heinonen, H.R.; Aaltonen, L.A.; Vahteristo, P. Genomics of Uterine Leiomyomas: Insights from High-Throughput Sequencing. Fertil. Steril. 2014, 102, 621–629. [Google Scholar] [CrossRef]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Chansky, K.; Detterbeck, F.C.; Nicholson, A.G.; Rusch, V.W.; Vallières, E.; Groome, P.; Kennedy, C.; Krasnik, M.; Peake, M.; Shemanski, L.; et al. The IASLC Lung Cancer Staging Project: External Validation of the Revision of the TNM Stage Groupings in the Eighth Edition of the TNM Classification of Lung Cancer. J. Thorac. Oncol. 2017, 12, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wang, H.; Li, C. Signal Pathways of Melanoma and Targeted Therapy. Signal Transduct. Target. Ther. 2021, 6, 424. [Google Scholar] [CrossRef]
- Shull, A.Y.; Latham-Schwark, A.; Ramasamy, P.; Leskoske, K.; Oroian, D.; Birtwistle, M.R.; Buckhaults, P.J. Novel Somatic Mutations to PI3K Pathway Genes in Metastatic Melanoma. PLoS ONE 2012, 7, e0043369. [Google Scholar] [CrossRef]
- Bardelli, V.; Arniani, S.; Pierini, V.; Di Giacomo, D.; Pierini, T.; Gorello, P.; Mecucci, C.; La Starza, R. T-Cell Acute Lymphoblastic Leukemia: Biomarkers and Their Clinical Usefulness. Genes 2021, 12, 1118. [Google Scholar] [CrossRef]
- Karrman, K.; Kjeldsen, E.; Lassen, C.; Isaksson, M.; Davidsson, J.; Andersson, A.; Hasle, H.; Fioretos, T.; Johansson, B. The t(X;7)(Q22;Q34) in Paediatric T-Cell Acute Lymphoblastic Leukaemia Results in Overexpression of the Insulin Receptor Substrate 4 Gene through Illegitimate Recombination with the T-Cell Receptor Beta Locus. Br. J. Haematol. 2009, 144, 546–551. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational Loss of PTEN Induces Resistance to NOTCH1 Inhibition in T-Cell Leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef]
- DaCambra, M.P.; Gupta, S.K.; Ferri-De-Barros, F. Subungual Exostosis of the Toes: A Systematic Review. Clin. Orthop. Relat. Res. 2014, 472, 1251–1259. [Google Scholar] [CrossRef]
- Mertens, F.; Möller, E.; Mandahl, N.; Picci, P.; Perez-Atayde, A.R.; Samson, I.; Sciot, R.; Debiec-Rychter, M. The t(X;6) in Subungual Exostosis Results in Transcriptional Deregulation of the Gene for Insulin Receptor Substrate 4. Int. J. Cancer 2011, 128, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Buerki, R.A.; Horbinski, C.M.; Kruser, T.; Horowitz, P.M.; James, C.D.; Lukas, R.V. An Overview of Meningiomas. Future Oncol. 2018, 14, 2161. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Franceschi, E.; Tosoni, A.; Di Nunno, V.; Gatto, L.; Lodi, R.; Brandes, A.A. Meningioma: Not Always a Benign Tumor. A Review of Advances in the Treatment of Meningiomas. CNS Oncol. 2021, 10, CNS72. [Google Scholar] [CrossRef] [PubMed]
- Torres-Martin, M.; Lassaletta, L.; Isla, A.; De Campos, J.M.; Pinto, G.R.; Burbano, R.R.; Castresana, J.S.; Melendez, B.; Rey, J.A. Global Expression Profile in Low Grade Meningiomas and Schwannomas Shows Upregulation of PDGFD, CDH1 and SLIT2 Compared to Their Healthy Tissue. Oncol. Rep. 2014, 32, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Stelcer, E.; Jopek, K.; Blatkiewicz, M.; Olechnowicz, A.; Kamiński, K.; Szyszka, M.; Suchorska, W.M.; Ruciński, M. Gene expression profile of hiPSC-derived cells differentiated with growth factors, forskolin and conditioned medium from human adrenocortical cell line. Adv. Clin. Exp. Med. 2023. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Tumor | Expression | Mechanism of Dysregulation | Carcinogenic Role | References |
|---|---|---|---|---|
| Colorectal cancer | Overexpressed | Stimulated G1 checkpoint cell cycle | IRS4/BRK/pIGF-1R complex (Tumor development and survival) | [47,67] |
| Gastric cancer | Overexpressed | Non-coding microRNAs | has_circ_0023409 (restrict cell survival and proliferative potential) | [58] |
| Hepatocellular carcinoma | Overexpressed | Not specified | ERK hyperactivation | [46,62] |
| Pancreatic cancer | Overexpressed | Not specified | PI3K/Akt hyperactivation | [99] |
| Breast cancer | Overexpressed | Not specified | insulin/IGF1 and AKT activation (tumor cell proliferation and invasiveness) | [107] |
| Ovarian cancer | Overexpressed | Tyr779 enables IRS4 to attract p85 | FER tyrosine kinase | [37] |
| Leiomyomas | Overexpressed | Mutations in Xq22 | CRR (Chromosome rearrangements) | [112,113] |
| Lung cancer | Overexpressed | Not specified | PI3K/Akt and Ras/MAPK pathways | [36,55] |
| Melanoma | Overexpressed | Mutations | miR-493 (suppress tumor growth) | [57] |
| T-cell acute lymphoblastic leukemia | Overexpressed | t(X;7) | CRR (Chromosome rearrangements) | [119] |
| Subungual exostosis | - | t(X;6) | CRR (Chromosome rearrangements) | [122] |
| Meningiomas | - | Mutations | - | [125] |
| Adrenocortical carcinoma | Overexpressed | Induction by growth factors | Endocrine differentiation | [126] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guijarro, L.G.; Justo Bermejo, F.J.; Boaru, D.L.; De Castro-Martinez, P.; De Leon-Oliva, D.; Fraile-Martínez, O.; Garcia-Montero, C.; Alvarez-Mon, M.; Toledo-Lobo, M.d.V.; Ortega, M.A. Is Insulin Receptor Substrate4 (IRS4) a Platform Involved in the Activation of Several Oncogenes? Cancers 2023, 15, 4651. https://doi.org/10.3390/cancers15184651
Guijarro LG, Justo Bermejo FJ, Boaru DL, De Castro-Martinez P, De Leon-Oliva D, Fraile-Martínez O, Garcia-Montero C, Alvarez-Mon M, Toledo-Lobo MdV, Ortega MA. Is Insulin Receptor Substrate4 (IRS4) a Platform Involved in the Activation of Several Oncogenes? Cancers. 2023; 15(18):4651. https://doi.org/10.3390/cancers15184651
Chicago/Turabian StyleGuijarro, Luis G., Francisco Javier Justo Bermejo, Diego Liviu Boaru, Patricia De Castro-Martinez, Diego De Leon-Oliva, Oscar Fraile-Martínez, Cielo Garcia-Montero, Melchor Alvarez-Mon, María del Val Toledo-Lobo, and Miguel A. Ortega. 2023. "Is Insulin Receptor Substrate4 (IRS4) a Platform Involved in the Activation of Several Oncogenes?" Cancers 15, no. 18: 4651. https://doi.org/10.3390/cancers15184651
APA StyleGuijarro, L. G., Justo Bermejo, F. J., Boaru, D. L., De Castro-Martinez, P., De Leon-Oliva, D., Fraile-Martínez, O., Garcia-Montero, C., Alvarez-Mon, M., Toledo-Lobo, M. d. V., & Ortega, M. A. (2023). Is Insulin Receptor Substrate4 (IRS4) a Platform Involved in the Activation of Several Oncogenes? Cancers, 15(18), 4651. https://doi.org/10.3390/cancers15184651