
A TLR4 Agonist Induces Osteosarcoma Regression by Inducing an Antitumor Immune Response and Reprogramming M2 Macrophages to M1 Macrophages

 , , ,
, , ,  and
and 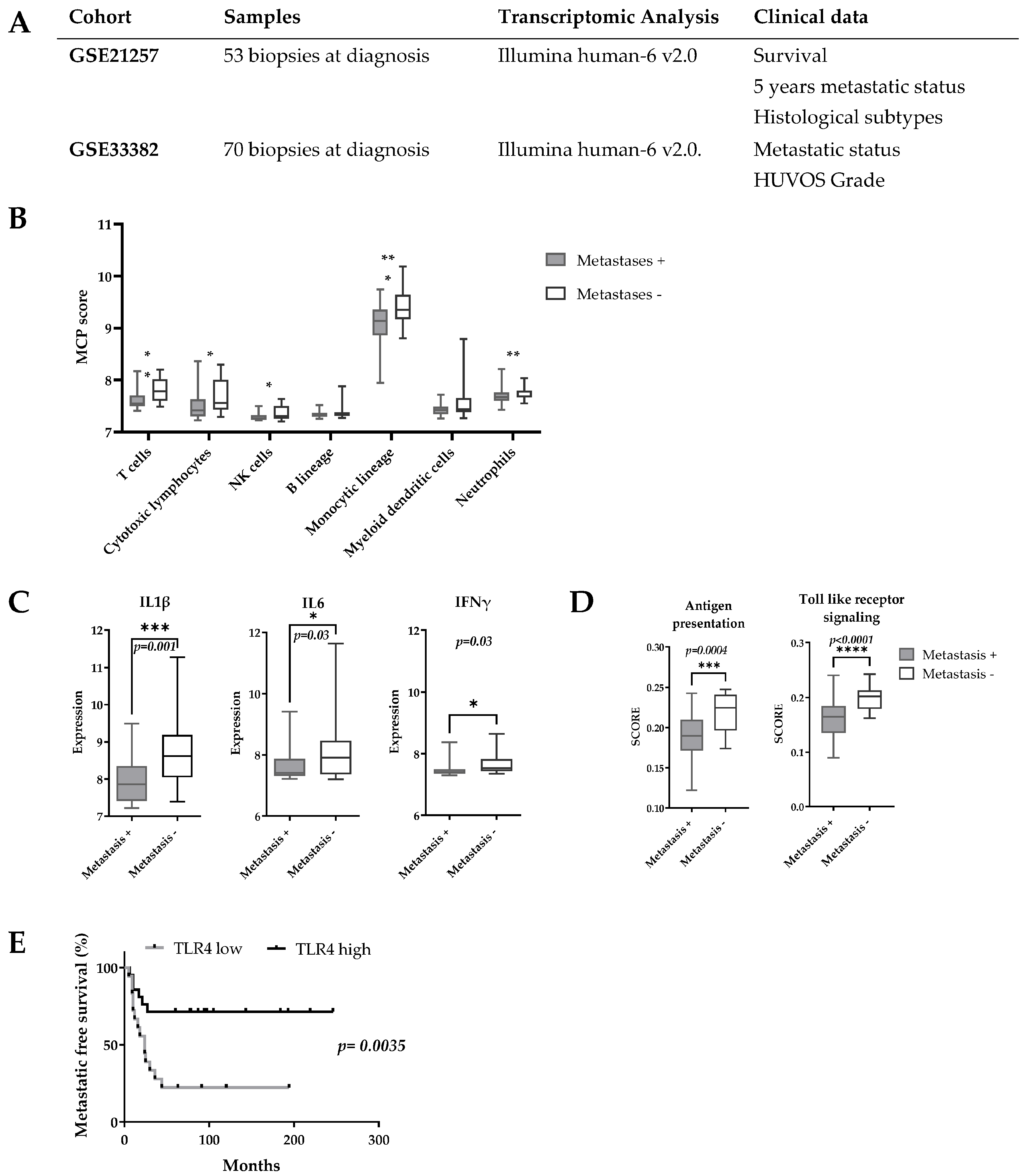{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analyses
2.2. In Vivo Experimental Model and TLR4 Agonist (Lipo-MP-LPS) Formulation
- Rat osteosarcoma model
- Modified TLR4 agonist (Lipo-MP-LPS)
2.3. Lipo-MP-LPS Efficacy Study
2.4. Immune Infiltrates Analysis at Early Stage of Antitumor Response
2.5. Flow Cytometry
- Single cell suspension preparation and staining
- FACS analyses
2.6. ELISpot Analysis
2.7. Bone Marrow Derived Macrophage (BMDM) Generation and Differentiation toward M1 and M2 Phenotype
2.8. Functional Analyses of Lipo-MP-LPS Treated Macrophages
- Phagocytosis assay
- Cytokines profile assay
2.9. Immunohistochemistry
2.10. Statistical Analyses
3. Results
3.1. A transcriptomic Pro-Inflammatory Immune Profile Is Associated with Delayed Development of Metastases in OsA
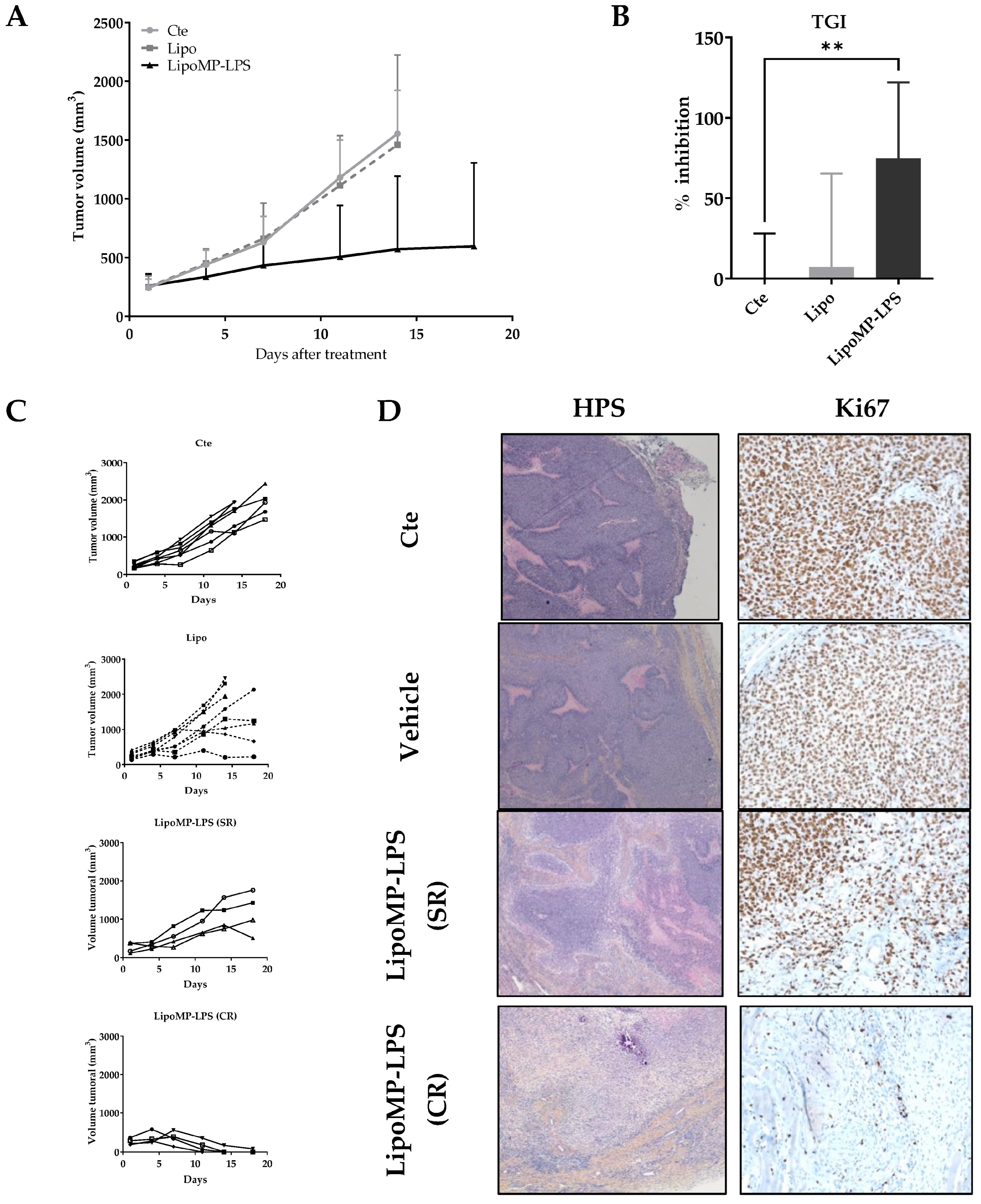
3.2. Lipo-MP-LPS Induced Regression of Primary OsA
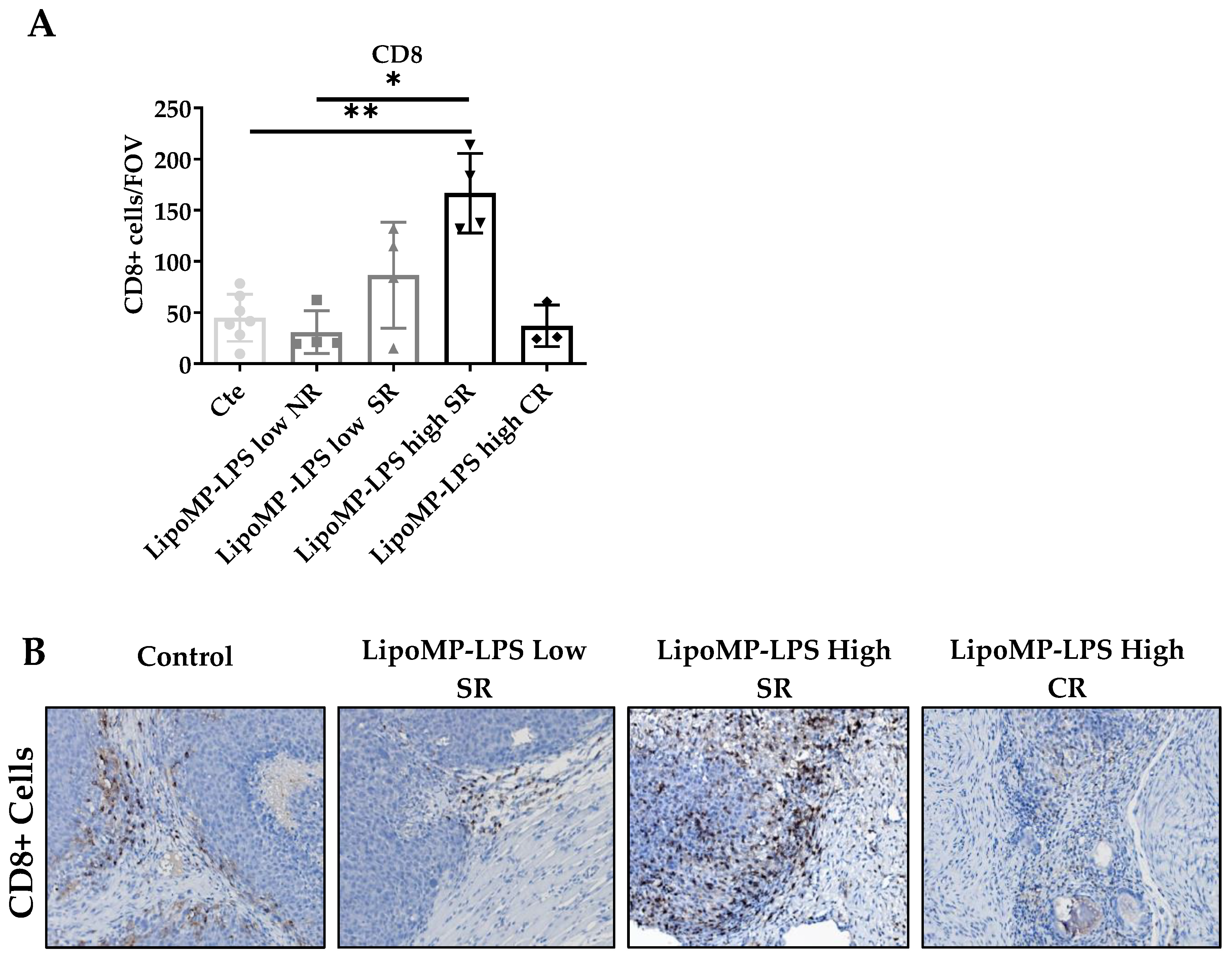
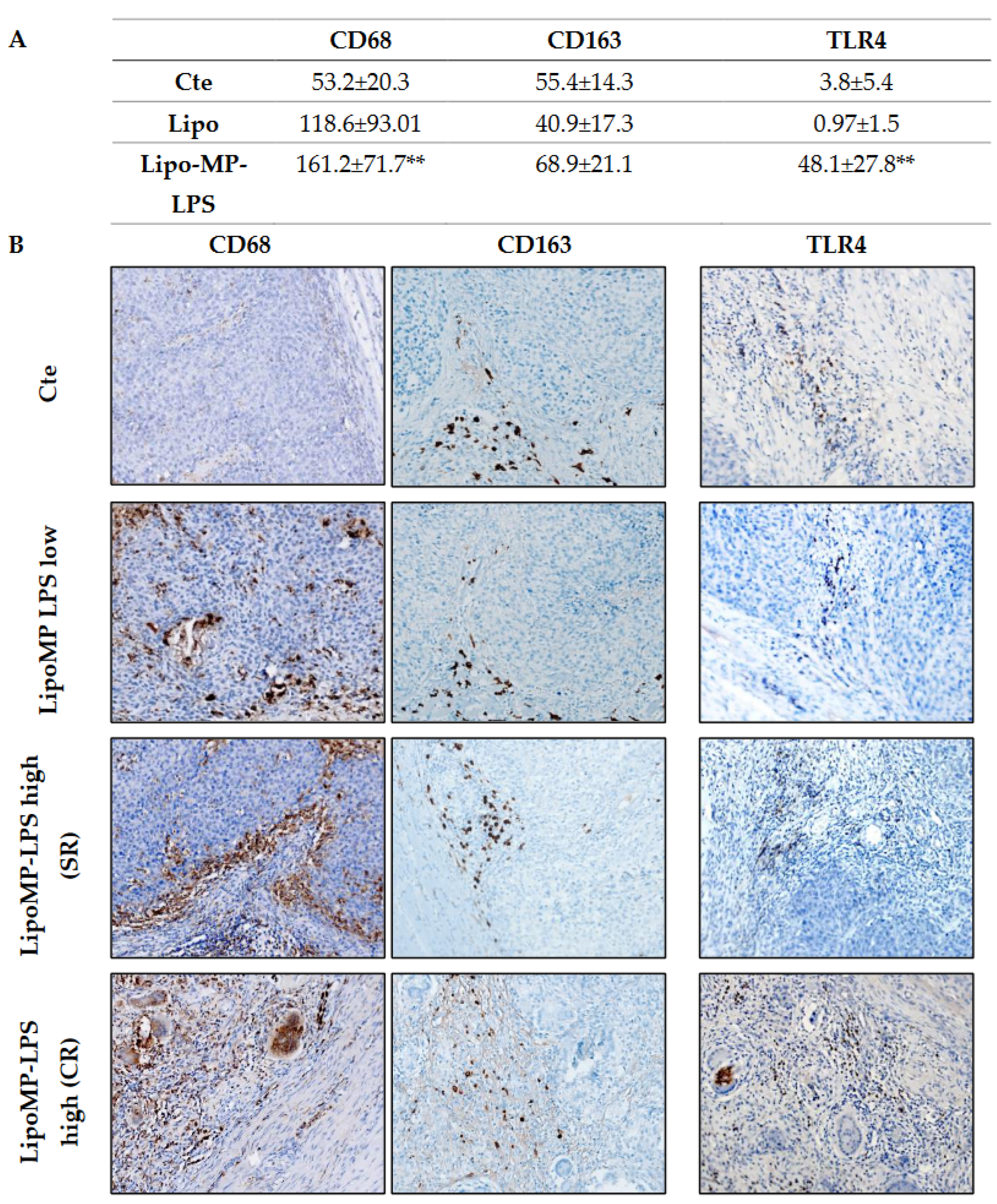
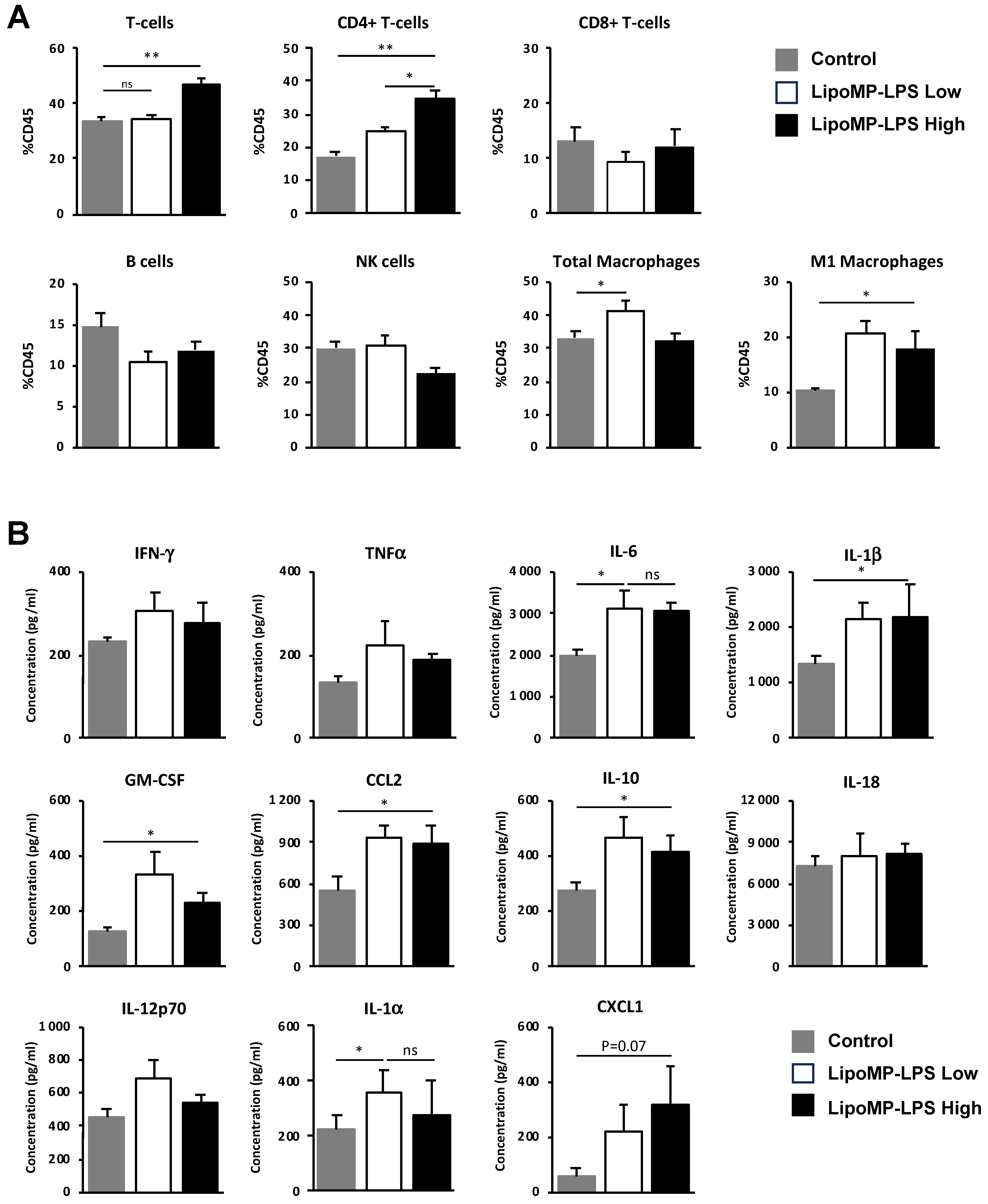
3.3. Lipo-MP-LPS Changed the Composition of OsA Immune Microenvironment
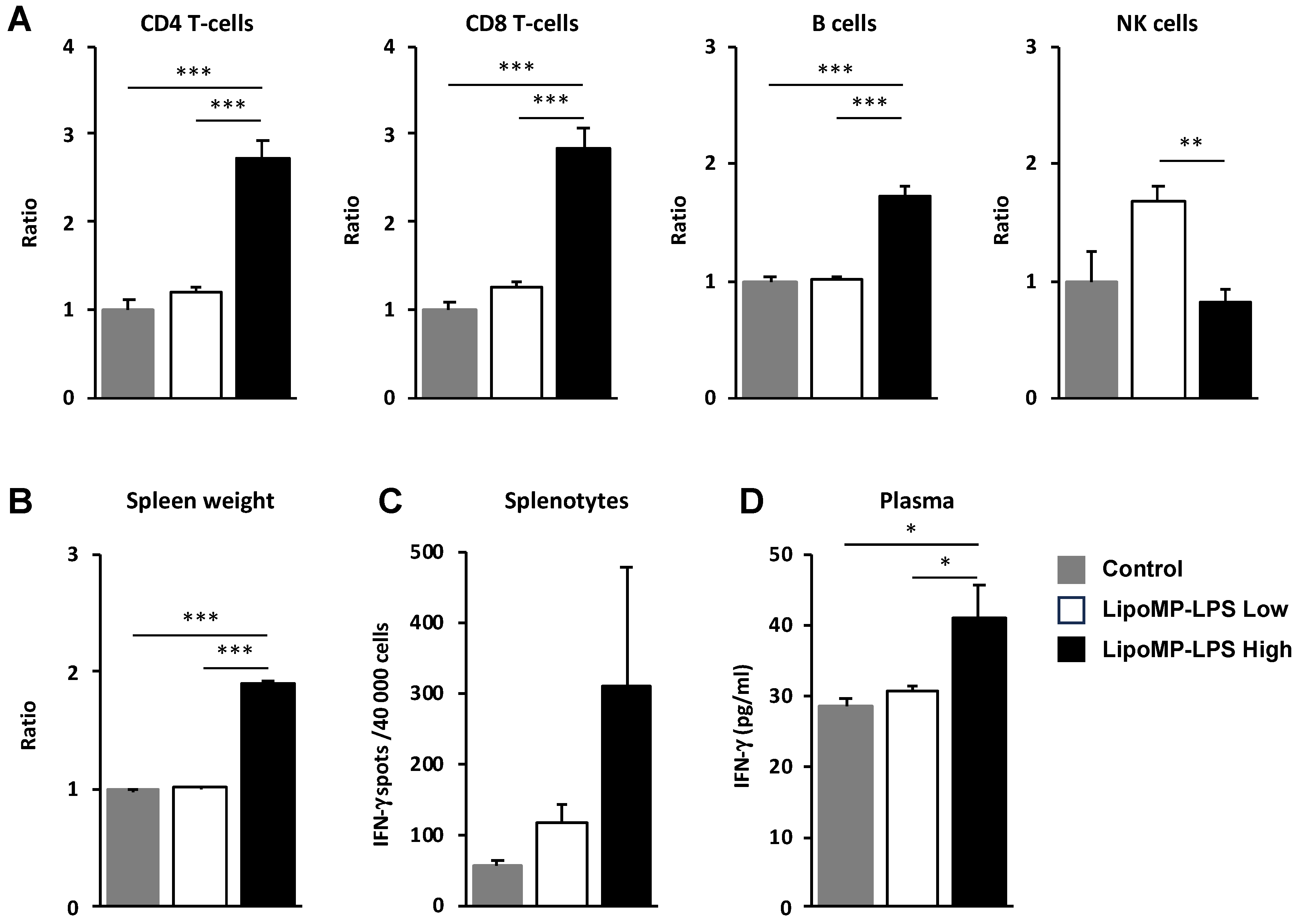
3.4. Lipo-MP-LPS Induced a Systemic Inflammatory Response
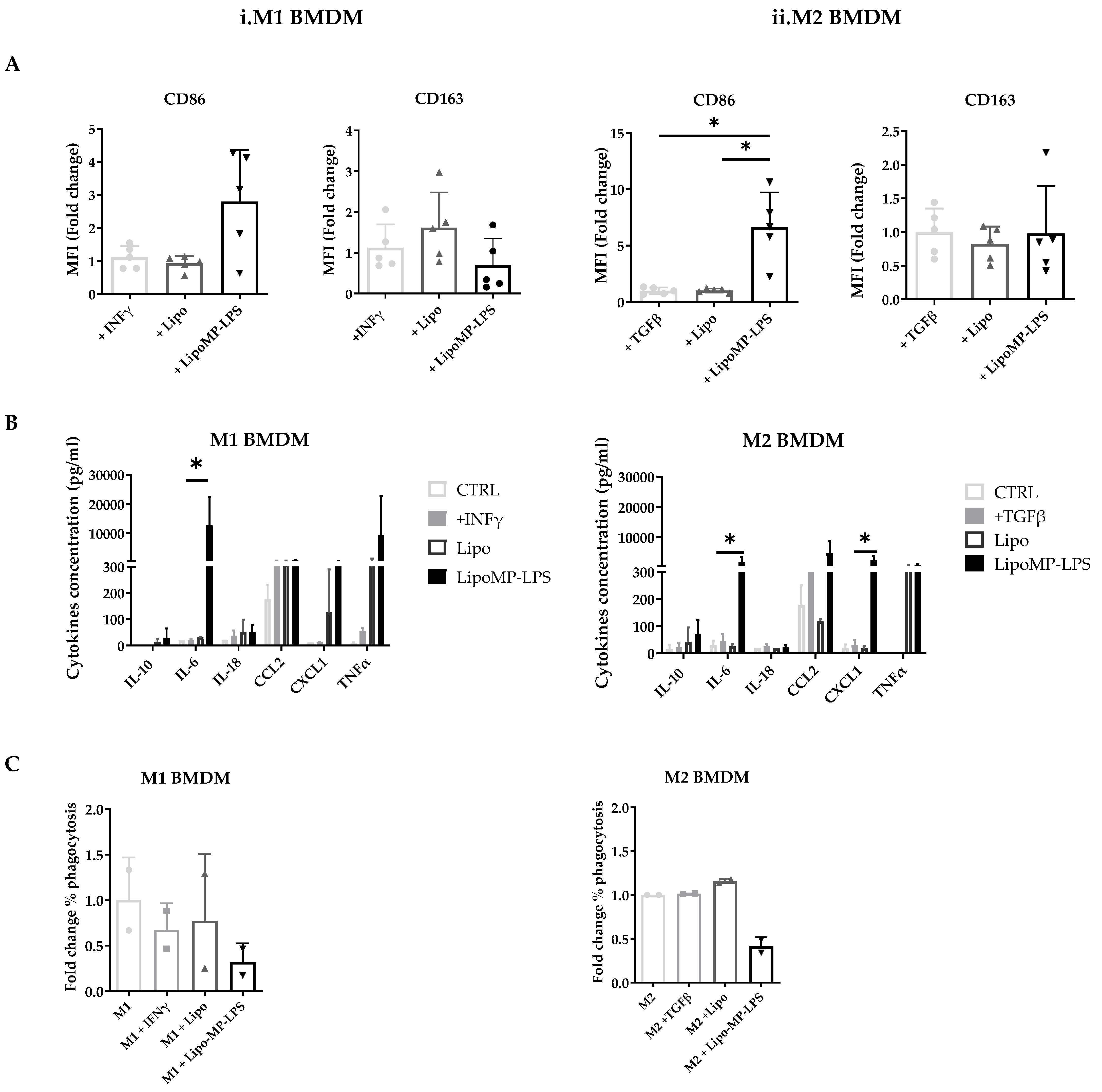
3.5. Lipo-MP-LPS Directs Macrophages toward a Pro-Inflammatory Phenotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, J.; Sun, H.; Li, J.; Guo, Y.; Zhang, K.; Lang, C.; Zou, C.; Ma, H. Increased Survival of Patients Aged 0–29 Years with Osteosarcoma: A Period Analysis, 1984–2013. Cancer Med. 2018, 7, 3652–3661. [Google Scholar] [CrossRef] [PubMed]
- Marko, T.A.; Diessner, B.J.; Spector, L.G. Prevalence of Metastasis at Diagnosis of Osteosarcoma: An International Comparison. Pediatr. Blood Cancer 2016, 63, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Aljubran, A.H.; Griffin, A.; Pintilie, M.; Blackstein, M. Osteosarcoma in Adolescents and Adults: Survival Analysis with and without Lung Metastases. Ann. Oncol. 2009, 20, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Smeland, S.; Bielack, S.S.; Whelan, J.; Bernstein, M.; Hogendoorn, P.; Krailo, M.D.; Gorlick, R.; Janeway, K.A.; Ingleby, F.C.; Anninga, J.; et al. Survival and Prognosis with Osteosarcoma: Outcomes in More than 2000 Patients in the EURAMOS-1 (European and American Osteosarcoma Study) Cohort. Eur. J. Cancer 2019, 109, 36–50. [Google Scholar] [CrossRef]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary Genomic Approaches Highlight the PI3K/MTOR Pathway as a Common Vulnerability in Osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent Mutation of IGF Signalling Genes and Distinct Patterns of Genomic Rearrangement in Osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef]
- Chawla, S.P.; Staddon, A.P.; Baker, L.H.; Schuetze, S.M.; Tolcher, A.W.; D’Amato, G.Z.; Blay, J.-Y.; Mita, M.M.; Sankhala, K.K.; Berk, L.; et al. Phase II Study of the Mammalian Target of Rapamycin Inhibitor Ridaforolimus in Patients with Advanced Bone and Soft Tissue Sarcomas. J. Clin. Oncol. 2012, 30, 78–84. [Google Scholar] [CrossRef]
- Isakoff, M.S.; Goldsby, R.; Villaluna, D.; Krailo, M.D.; Hingorani, P.; Collier, A.; Morris, C.D.; Kolb, E.A.; Doski, J.J.; Womer, R.B.; et al. A Phase II Study of Eribulin in Recurrent or Refractory Osteosarcoma: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2019, 66, e27524. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Le Deley, M.-C.; Rédini, F.; Pacquement, H.; Marec-Bérard, P.; Petit, P.; Brisse, H.; Lervat, C.; Gentet, J.-C.; Entz-Werlé, N.; et al. Zoledronate in Combination with Chemotherapy and Surgery to Treat Osteosarcoma (OS2006): A Randomised, Multicentre, Open-Label, Phase 3 Trial. Lancet Oncol. 2016, 17, 1070–1080. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-Cell RNA Landscape of Intratumoral Heterogeneity and Immunosuppressive Microenvironment in Advanced Osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef]
- Casanova, J.M.; Almeida, J.-S.; Reith, J.D.; Sousa, L.M.; Fonseca, R.; Freitas-Tavares, P.; Santos-Rosa, M.; Rodrigues-Santos, P. Tumor-Infiltrating Lymphocytes and Cancer Markers in Osteosarcoma: Influence on Patient Survival. Cancers 2021, 13, 6075. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. Int. J. Mol. Sci. 2020, 21, 5207. [Google Scholar] [CrossRef] [PubMed]
- Ligon, J.A.; Choi, W.; Cojocaru, G.; Fu, W.; Hsiue, E.H.-C.; Oke, T.F.; Siegel, N.; Fong, M.H.; Ladle, B.; Pratilas, C.A.; et al. Pathways of Immune Exclusion in Metastatic Osteosarcoma Are Associated with Inferior Patient Outcomes. J. Immunother. Cancer 2021, 9, e001772. [Google Scholar] [CrossRef] [PubMed]
- Marchais, A.; Marques da Costa, M.E.; Job, B.; Abbas, R.; Drubay, D.; Piperno-Neumann, S.; Fromigué, O.; Gomez-Brouchet, A.; Redini, F.; Droit, R.; et al. Immune Infiltrate and Tumor Microenvironment Transcriptional Programs Stratify Pediatric Osteosarcoma into Prognostic Groups at Diagnosis. Cancer Res. 2022, 82, 974–985. [Google Scholar] [CrossRef]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: The Addition of Muramyl Tripeptide to Chemotherapy Improves Overall Survival--a Report from the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to Anti-PD1 Therapy with Nivolumab in Metastatic Sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in Advanced Soft Tissue and Bone Sarcomas: Results of SARC028, A Multicentre, Single Arm, Phase 2 Trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Shen, H.; Tesar, B.M.; Walker, W.E.; Goldstein, D.R. Dual Signaling of MyD88 and TRIF Is Critical for Maximal TLR4-Induced Dendritic Cell Maturation. J. Immunol. 2008, 181, 1849–1858. [Google Scholar] [CrossRef]
- Berendt, M.J.; North, R.J.; Kirstein, D.P. The Immunological Basis of Endotoxin-Induced Tumor Regression. Requirement for a Pre-Existing State of Concomitant Anti-Tumor Immunity. J. Exp. Med. 1978, 148, 1560–1569. [Google Scholar] [CrossRef]
- McCarthy, E.F. The Toxins of William B. Coley and the Treatment of Bone and Soft-Tissue Sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Lascelles, B.D.X.; Dernell, W.S.; Correa, M.T.; Lafferty, M.; Devitt, C.M.; Kuntz, C.A.; Straw, R.C.; Withrow, S.J. Improved Survival Associated with Postoperative Wound Infection in Dogs Treated with Limb-Salvage Surgery for Osteosarcoma. Ann. Surg. Oncol. 2005, 12, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.U.; Xu, S.-F.; Xu, M.; Yu, X.-C. Postoperative Infection and Survival in Osteosarcoma Patients: Reconsideration of Immunotherapy for Osteosarcoma. Mol. Clin. Oncol. 2015, 3, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Jeys, L.M.; Grimer, R.J.; Carter, S.R.; Tillman, R.M.; Abudu, A. Post Operative Infection and Increased Survival in Osteosarcoma Patients: Are They Associated? Ann. Surg. Oncol. 2007, 14, 2887–2895. [Google Scholar] [CrossRef] [PubMed]
- Tuohy, J.L.; Somarelli, J.A.; Borst, L.B.; Eward, W.C.; Lascelles, B.D.X.; Fogle, J.E. Immune Dysregulation and Osteosarcoma: Staphylococcus Aureus Downregulates TGF-β and Heightens the Inflammatory Signature in Human and Canine Macrophages Suppressed by Osteosarcoma. Vet. Comp. Oncol. 2020, 18, 64–75. [Google Scholar] [CrossRef]
- Yahiro, K.; Matsumoto, Y.; Yamada, H.; Endo, M.; Setsu, N.; Fujiwara, T.; Nakagawa, M.; Kimura, A.; Shimada, E.; Okada, S.; et al. Activation of TLR4 Signaling Inhibits Progression of Osteosarcoma by Stimulating CD8-Positive Cytotoxic Lymphocytes. Cancer Immunol. Immunother. 2020, 69, 745–758. [Google Scholar] [CrossRef]
- Engelhardt, R.; Mackensen, A.; Galanos, C. Phase I Trial of Intravenously Administered Endotoxin (Salmonella Abortus Equi) in Cancer Patients. Cancer Res. 1991, 51, 2524–2530. [Google Scholar]
- Paavonen, J.; Jenkins, D.; Bosch, F.X.; Naud, P.; Salmerón, J.; Wheeler, C.M.; Chow, S.-N.; Apter, D.L.; Kitchener, H.C.; Castellsague, X.; et al. Efficacy of a Prophylactic Adjuvanted Bivalent L1 Virus-like-Particle Vaccine against Infection with Human Papillomavirus Types 16 and 18 in Young Women: An Interim Analysis of a Phase III Double-Blind, Randomised Controlled Trial. Lancet 2007, 369, 2161–2170. [Google Scholar] [CrossRef]
- Shetab Boushehri, M.A.; Lamprecht, A. TLR4-Based Immunotherapeutics in Cancer: A Review of the Achievements and Shortcomings. Mol. Pharm. 2018, 15, 4777–4800. [Google Scholar] [CrossRef]
- Romerio, A.; Peri, F. Increasing the Chemical Variety of Small-Molecule-Based TLR4 Modulators: An Overview. Front. Immunol. 2020, 11, 1210. [Google Scholar] [CrossRef]
- Villani, A.; Fabbrocini, G.; Costa, C.; Carmela Annunziata, M.; Scalvenzi, M. Merkel Cell Carcinoma: Therapeutic Update and Emerging Therapies. Dermatol. Ther. 2019, 9, 209–222. [Google Scholar] [CrossRef]
- Flowers, C.; Panizo, C.; Isufi, I.; Herrera, A.F.; Okada, C.; Cull, E.H.; Kis, B.; Chaves, J.M.; Bartlett, N.L.; Ai, W.; et al. Intratumoral G100 Induces Systemic Immunity and Abscopal Tumor Regression in Patients with Follicular Lymphoma: Results of a Phase 1/ 2 Study Examining G100 Alone and in Combination with Pembrolizumab. Blood 2017, 130, 2771. [Google Scholar] [CrossRef]
- Hug, B.A.; Matheny, C.J.; Burns, O.; Struemper, H.; Wang, X.; Washburn, M.L. Safety, Pharmacokinetics, and Pharmacodynamics of the TLR4 Agonist GSK1795091 in Healthy Individuals: Results from a Randomized, Double-Blind, Placebo-Controlled, Ascending Dose Study. Clin. Ther. 2020, 42, 1519–1534. [Google Scholar] [CrossRef] [PubMed]
- Chettab, K.; Fitzsimmons, C.; Novikov, A.; Denis, M.; Phelip, C.; Mathé, D.; Choffour, P.A.; Beaumel, S.; Fourmaux, E.; Norca, P.; et al. A Systemically Administered Detoxified TLR4 Agonist Displays Potent Antitumor Activity and an Acceptable Tolerance Profile in Preclinical Models. Front. Immunol. 2023, 14, 1066402. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the Population Abundance of Tissue-Infiltrating Immune and Stromal Cell Populations Using Gene Expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA Interference Reveals That Oncogenic KRAS-Driven Cancers Require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Dutour, A.; Josserand, V.; Jury, D.; Guillermet, S.; Decouvelaere, A.V.; Chotel, F.; Pointecouteau, T.; Rizo, P.; Coll, J.L.; Blay, J.Y. Targeted Imaging of Avβ3 Expressing Sarcoma Tumor Cells in Vivo in Pre-Operative Setting Using near Infrared: A Potential Tool to Reduce Incomplete Surgical Resection. Bone 2014, 62, 71–78. [Google Scholar] [CrossRef]
- Dutour, A.; Decouvelaere, A.-V.; Monteil, J.; Duclos, M.-E.; Roualdes, O.; Rousseau, R.; Marec-Bérard, P. 18F-FDG PET SUVmax Correlates with Osteosarcoma Histologic Response to Neoadjuvant Chemotherapy: Preclinical Evaluation in an Orthotopic Rat Model. J. Nucl. Med. 2009, 50, 1533–1540. [Google Scholar] [CrossRef][Green Version]
- Caroff, M.; Novikov, A. Micromethods for Lipid A Isolation and Structural Characterization. Methods Mol. Biol. 2011, 739, 135–146. [Google Scholar] [CrossRef]
- Barrett, J.P.; Costello, D.A.; O’Sullivan, J.; Cowley, T.R.; Lynch, M.A. Bone Marrow-Derived Macrophages from Aged Rats Are More Responsive to Inflammatory Stimuli. J. Neuroinflamm. 2015, 12, 67. [Google Scholar] [CrossRef]
- Mia, S.; Warnecke, A.; Zhang, X.-M.; Malmström, V.; Harris, R.A. An Optimized Protocol for Human M2 Macrophages Using M-CSF and IL-4/IL-10/TGF-β Yields a Dominant Immunosuppressive Phenotype. Scand. J. Immunol. 2014, 79, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wang, X.; Zhou, Y.; Xiao, Y. RANKL-Induced M1 Macrophages Are Involved in Bone Formation. Bone Res. 2017, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cai, X.; Ren, F.; Ye, Y.; Wang, F.; Zheng, C.; Qian, Y.; Zhang, M. The Macrophage-Osteoclast Axis in Osteoimmunity and Osteo-Related Diseases. Front. Immunol. 2021, 12, 664871. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.-M.; Marie, B.; Larousserie, F.; Entz-Werlé, N.; de Pinieux, G.; et al. CD163-Positive Tumor-Associated Macrophages and CD8-Positive Cytotoxic Lymphocytes Are Powerful Diagnostic Markers for the Therapeutic Stratification of Osteosarcoma Patients: An Immunohistochemical Analysis of the Biopsies Fromthe French OS2006 Phase 3 Trial. Oncoimmunology 2017, 6, e1331193. [Google Scholar] [CrossRef]
- Richert, I.; Dutour, A. The Immune Environment of Bone Sarcomas. In Bone Cancer; Elsevier: Amsterdam, The Netherlands, 2022; pp. 189–201. [Google Scholar]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune Infiltration and PD-L1 Expression in the Tumor Microenvironment Are Prognostic in Osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef]
- Biteau, K.; Guiho, R.; Chatelais, M.; Taurelle, J.; Chesneau, J.; Corradini, N.; Heymann, D.; Redini, F. L-MTP-PE and Zoledronic Acid Combination in Osteosarcoma: Preclinical Evidence of Positive Therapeutic Combination for Clinical Transfer. Am. J. Cancer Res. 2016, 6, 677–689. [Google Scholar]
- Gilabert-Oriol, R.; Ryan, G.M.; Leung, A.W.Y.; Firmino, N.S.; Bennewith, K.L.; Bally, M.B. Liposomal Formulations to Modulate the Tumour Microenvironment and Antitumour Immune Response. Int. J. Mol. Sci. 2018, 19, 2922. [Google Scholar] [CrossRef]
- Bohannon, J.K.; Hernandez, A.; Enkhbaatar, P.; Adams, W.L.; Sherwood, E.R. The Immunobiology of Toll-like Receptor 4 Agonists: From Endotoxin Tolerance to Immunoadjuvants. Shock 2013, 40, 451–462. [Google Scholar] [CrossRef]
- Pahl, J.H.W.; Kwappenberg, K.M.C.; Varypataki, E.M.; Santos, S.J.; Kuijjer, M.L.; Mohamed, S.; Wijnen, J.T.; van Tol, M.J.D.; Cleton-Jansen, A.-M.; Egeler, R.M.; et al. Macrophages Inhibit Human Osteosarcoma Cell Growth after Activation with the Bacterial Cell Wall Derivative Liposomal Muramyl Tripeptide in Combination with Interferon-γ. J. Exp. Clin. Cancer Res. 2014, 33, 27. [Google Scholar] [CrossRef]
- Asano, T.; McWatters, A.; An, T.; Matsushima, K.; Kleinerman, E.S. Liposomal Muramyl Tripeptide Up-Regulates Interleukin-1 Alpha, Interleukin-1 Beta, Tumor Necrosis Factor-Alpha, Interleukin-6 and Interleukin-8 Gene Expression in Human Monocytes. J. Pharmacol. Exp. Ther. 1994, 268, 1032–1039. [Google Scholar]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (Alternative) Macrophage Polarization Are Related Processes Orchestrated by P50 Nuclear Factor KappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef]
- Garrido-Martin, E.M.; Mellows, T.W.P.; Clarke, J.; Ganesan, A.-P.; Wood, O.; Cazaly, A.; Seumois, G.; Chee, S.J.; Alzetani, A.; King, E.V.; et al. M1hot Tumor-Associated Macrophages Boost Tissue-Resident Memory T Cells Infiltration and Survival in Human Lung Cancer. J. Immunother. Cancer 2020, 8, e000778. [Google Scholar] [CrossRef]
- Gomez-Brouchet, A.; Gilhodes, J.; Acker, N.V.; Brion, R.; Bouvier, C.; Assemat, P.; Gaspar, N.; Aubert, S.; Guinebretiere, J.-M.; Marie, B.; et al. Characterization of Macrophages and Osteoclasts in the Osteosarcoma Tumor Microenvironment at Diagnosis: New Perspective for Osteosarcoma Treatment? Cancers 2021, 13, 423. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.-Q.; Guo, C.; Song, G.-H.; Fang, N.; Fan, W.-J.; Chen, X.-D.; Yuan, L.; Wang, Z.-Q. Lipopolysaccharide (LPS) Promotes Osteoclast Differentiation and Activation by Enhancing the MAPK Pathway and COX-2 Expression in RAW264.7 Cells. Int. J. Mol. Med. 2013, 32, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, S.; Fiorino, C.; Naik, U.; Sun, H.S.; Harrison, R.E. Modulation of Osteoclastogenesis with Macrophage M1- and M2-Inducing Stimuli. PLoS ONE 2014, 9, e104498. [Google Scholar] [CrossRef]
- Strålberg, F.; Kassem, A.; Kasprzykowski, F.; Abrahamson, M.; Grubb, A.; Lindholm, C.; Lerner, U.H. Inhibition of Lipopolysaccharide-Induced Osteoclast Formation and Bone Resorption in Vitro and in Vivo by Cysteine Proteinase Inhibitors. J. Leukoc. Biol. 2017, 101, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Palomero, J.; Grabowska, J.; Wang, L.; de Rink, I.; van Helvert, L.; Borst, J. Macrophages and Osteoclasts Stem from a Bipotent Progenitor Downstream of a Macrophage/Osteoclast/Dendritic Cell Progenitor. Blood Adv. 2017, 1, 1993–2006. [Google Scholar] [CrossRef]
- Fritzsching, B.; Fellenberg, J.; Moskovszky, L.; Sápi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendrõi, M.; Bosch, A.L.; et al. CD8+/FOXP3+-Ratio in Osteosarcoma Microenvironment Separates Survivors from Non-Survivors: A Multicenter Validated Retrospective Study. Oncoimmunology 2015, 4, e990800. [Google Scholar] [CrossRef]
- Simard, F.A.; Richert, I.; Vandermoeten, A.; Decouvelaere, A.-V.; Michot, J.-P.; Caux, C.; Blay, J.-Y.; Dutour, A. Description of the Immune Microenvironment of Chondrosarcoma and Contribution to Progression. OncoImmunology 2017, 6, e1265716. [Google Scholar] [CrossRef] [PubMed]
- Viitala, M.K.; Virtakoivu, R.; Tadayon, S.; Rannikko, J.; Jalkanen, S.; Hollmén, M. Immunotherapeutic Blockade of Macrophage Clever-1 Reactivates the CD8+ T Cell Response Against Immunosuppressive Tumors. Clin. Cancer Res. 2019, 25, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Nestvold, J.; Wang, M.-Y.; Camilio, K.A.; Zinöcker, S.; Tjelle, T.E.; Lindberg, A.; Haug, B.E.; Kvalheim, G.; Sveinbjørnsson, B.; Rekdal, Ø. Oncolytic Peptide LTX-315 Induces an Immune-Mediated Abscopal Effect in a Rat Sarcoma Model. Oncoimmunology 2017, 6. [Google Scholar] [CrossRef]
- Markel, J.E.; Noore, J.; Emery, E.J.; Bobnar, H.J.; Kleinerman, E.S.; Lindsey, B.A. Using the Spleen as an In Vivo Systemic Immune Barometer Alongside Osteosarcoma Disease Progression and Immunotherapy with α-PD-L1. Sarcoma 2018, 2018, 8694397. [Google Scholar] [CrossRef]
- Pantel, A.; Cheong, C.; Dandamudi, D.; Shrestha, E.; Mehandru, S.; Brane, L.; Ruane, D.; Teixeira, A.; Bozzacco, L.; Steinman, R.M.; et al. A New Synthetic TLR4 Agonist, GLA, Allows Dendritic Cells Targeted with Antigen to Elicit Th1 T-Cell Immunity in Vivo. Eur. J. Immunol. 2012, 42, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Farrell, C.J.; Zaupa, C.; Barnard, Z.; Maley, J.; Martuza, R.L.; Rabkin, S.D.; Curry, W.T. Combination Immunotherapy for Tumors via Sequential Intratumoral Injections of Oncolytic Herpes Simplex Virus 1 and Immature Dendritic Cells. Clin. Cancer Res. 2008, 14, 7711–7716. [Google Scholar] [CrossRef]
- Huang, J.-H.; Zhang, S.-N.; Choi, K.-J.; Choi, I.-K.; Kim, J.-H.; Lee, M.; Kim, H.; Yun, C.-O. Therapeutic and Tumor-Specific Immunity Induced by Combination of Dendritic Cells and Oncolytic Adenovirus Expressing IL-12 and 4-1BBL. Mol. Ther. 2010, 18, 264–274. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, X.; Liu, T.; Zhang, X. Strategies and Developments of Immunotherapies in Osteosarcoma. Oncol. Lett. 2016, 11, 511–520. [Google Scholar] [CrossRef]
- Punzo, F.; Bellini, G.; Tortora, C.; Pinto, D.D.; Argenziano, M.; Pota, E.; Paola, A.D.; Martino, M.D.; Rossi, F. Mifamurtide and TAM-like Macrophages: Effect on Proliferation, Migration and Differentiation of Osteosarcoma Cells. Oncotarget 2020, 11, 687–698. [Google Scholar] [CrossRef]
- Cascini, C.; Ratti, C.; Botti, L.; Parma, B.; Cancila, V.; Salvaggio, A.; Meazza, C.; Tripodo, C.; Colombo, M.P.; Chiodoni, C. Rewiring Innate and Adaptive Immunity with TLR9 Agonist to Treat Osteosarcoma. J. Exp. Clin. Cancer Res. 2023, 42, 154. [Google Scholar] [CrossRef]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-Cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richert, I.; Berchard, P.; Abbes, L.; Novikov, A.; Chettab, K.; Vandermoeten, A.; Dumontet, C.; Karanian, M.; Kerzerho, J.; Caroff, M.; et al. A TLR4 Agonist Induces Osteosarcoma Regression by Inducing an Antitumor Immune Response and Reprogramming M2 Macrophages to M1 Macrophages. Cancers 2023, 15, 4635. https://doi.org/10.3390/cancers15184635
Richert I, Berchard P, Abbes L, Novikov A, Chettab K, Vandermoeten A, Dumontet C, Karanian M, Kerzerho J, Caroff M, et al. A TLR4 Agonist Induces Osteosarcoma Regression by Inducing an Antitumor Immune Response and Reprogramming M2 Macrophages to M1 Macrophages. Cancers. 2023; 15(18):4635. https://doi.org/10.3390/cancers15184635
Chicago/Turabian StyleRichert, Iseulys, Paul Berchard, Lhorra Abbes, Alexey Novikov, Kamel Chettab, Alexandra Vandermoeten, Charles Dumontet, Marie Karanian, Jerome Kerzerho, Martine Caroff, and et al. 2023. "A TLR4 Agonist Induces Osteosarcoma Regression by Inducing an Antitumor Immune Response and Reprogramming M2 Macrophages to M1 Macrophages" Cancers 15, no. 18: 4635. https://doi.org/10.3390/cancers15184635
APA StyleRichert, I., Berchard, P., Abbes, L., Novikov, A., Chettab, K., Vandermoeten, A., Dumontet, C., Karanian, M., Kerzerho, J., Caroff, M., Blay, J.-Y., & Dutour, A. (2023). A TLR4 Agonist Induces Osteosarcoma Regression by Inducing an Antitumor Immune Response and Reprogramming M2 Macrophages to M1 Macrophages. Cancers, 15(18), 4635. https://doi.org/10.3390/cancers15184635