
Repurposing Clemastine to Target Glioblastoma Cell Stemness
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Chemicals, Other Reagents and Plasmids
2.3. Plasmid Electroporation
2.4. BTIC Proliferation Assay, Cell Cycle Analysis and ELDA
2.5. In Vivo Drug Treatment
2.6. Reverse Transcription and Real-Time Quantitative PCR (RT-qPCR)
2.7. Protein Extraction and Immunoblotting
2.8. Immunofluorescence Staining
2.9. RNA-Seq Data and Pathway Analysis
2.10. Statistical Analysis
3. Results
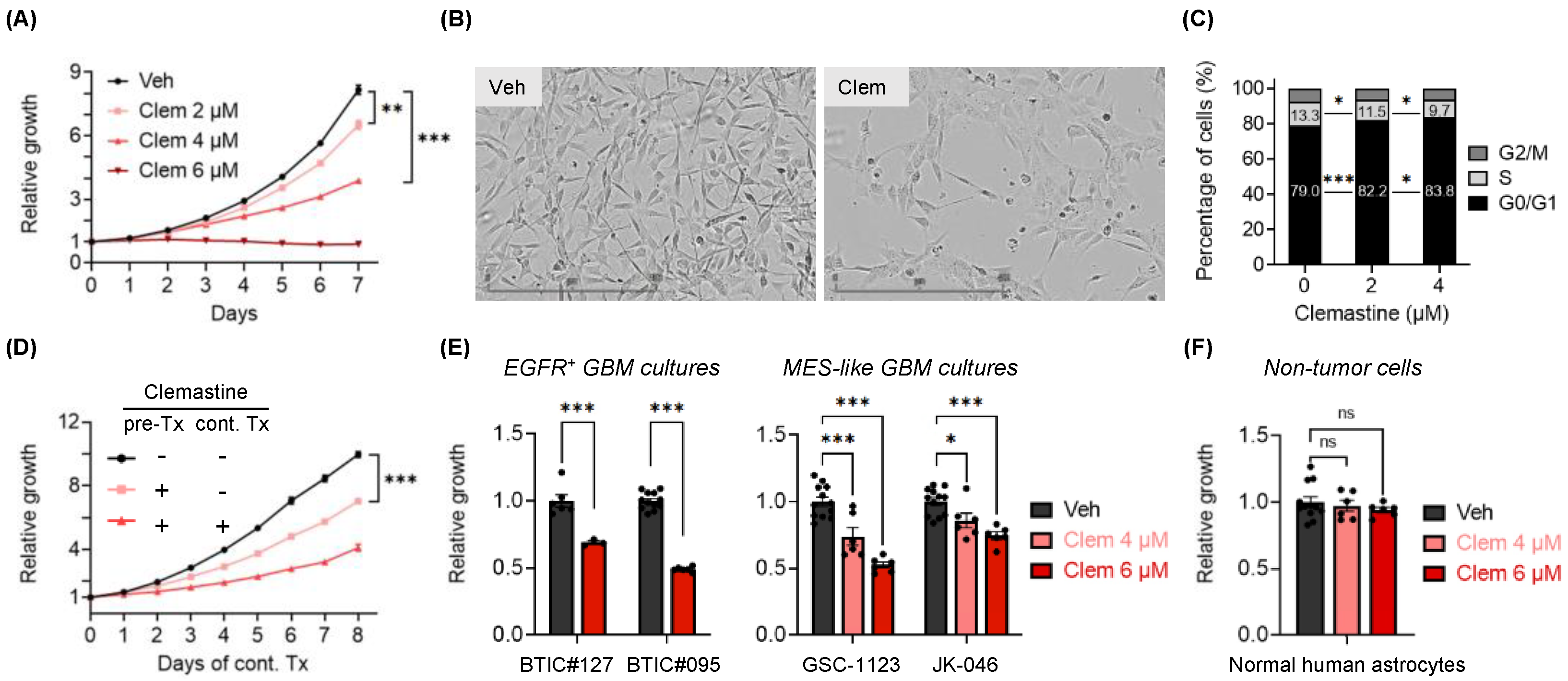
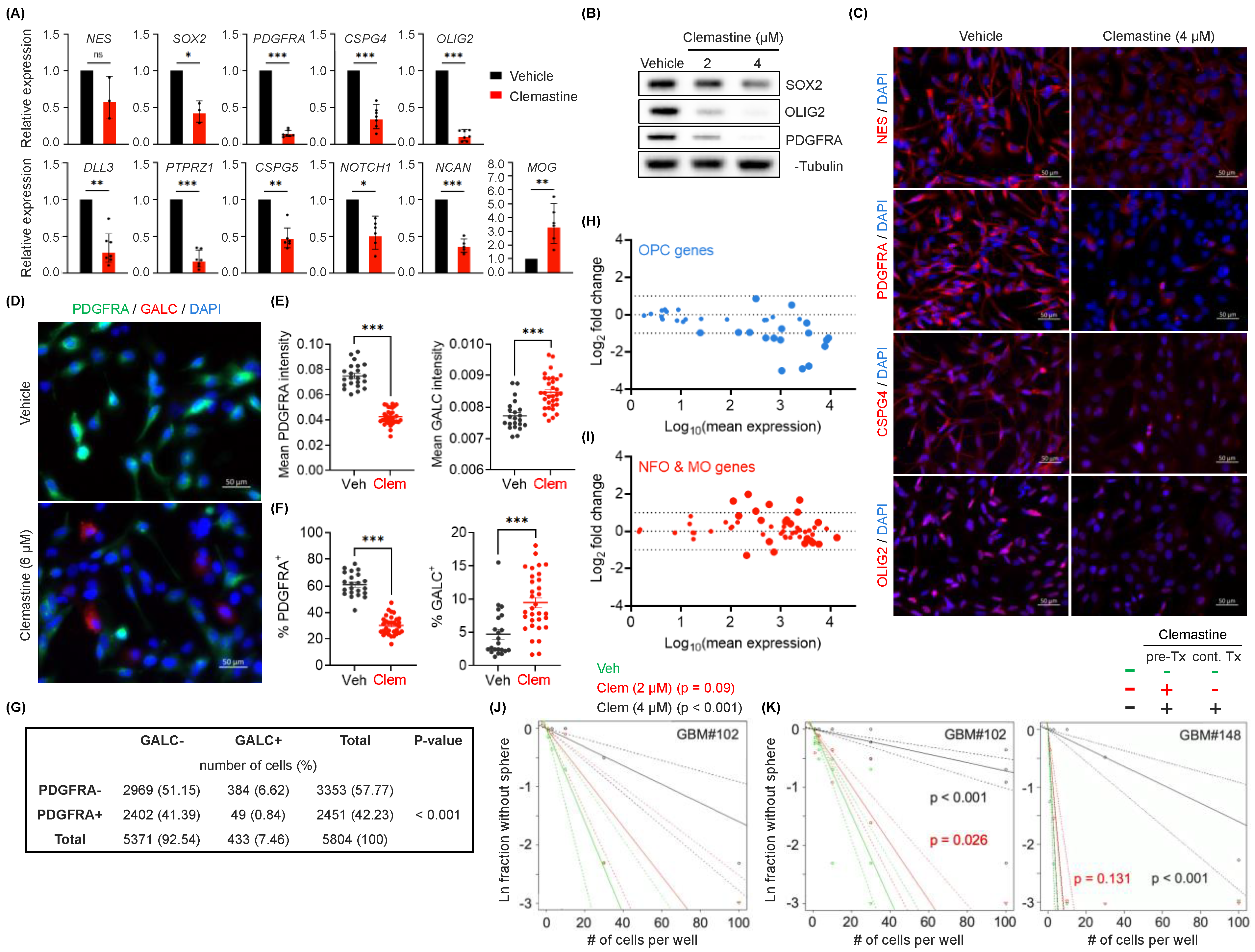
3.1. Clemastine Suppresses the Propagation of Patient-Derived BTIC Cultures
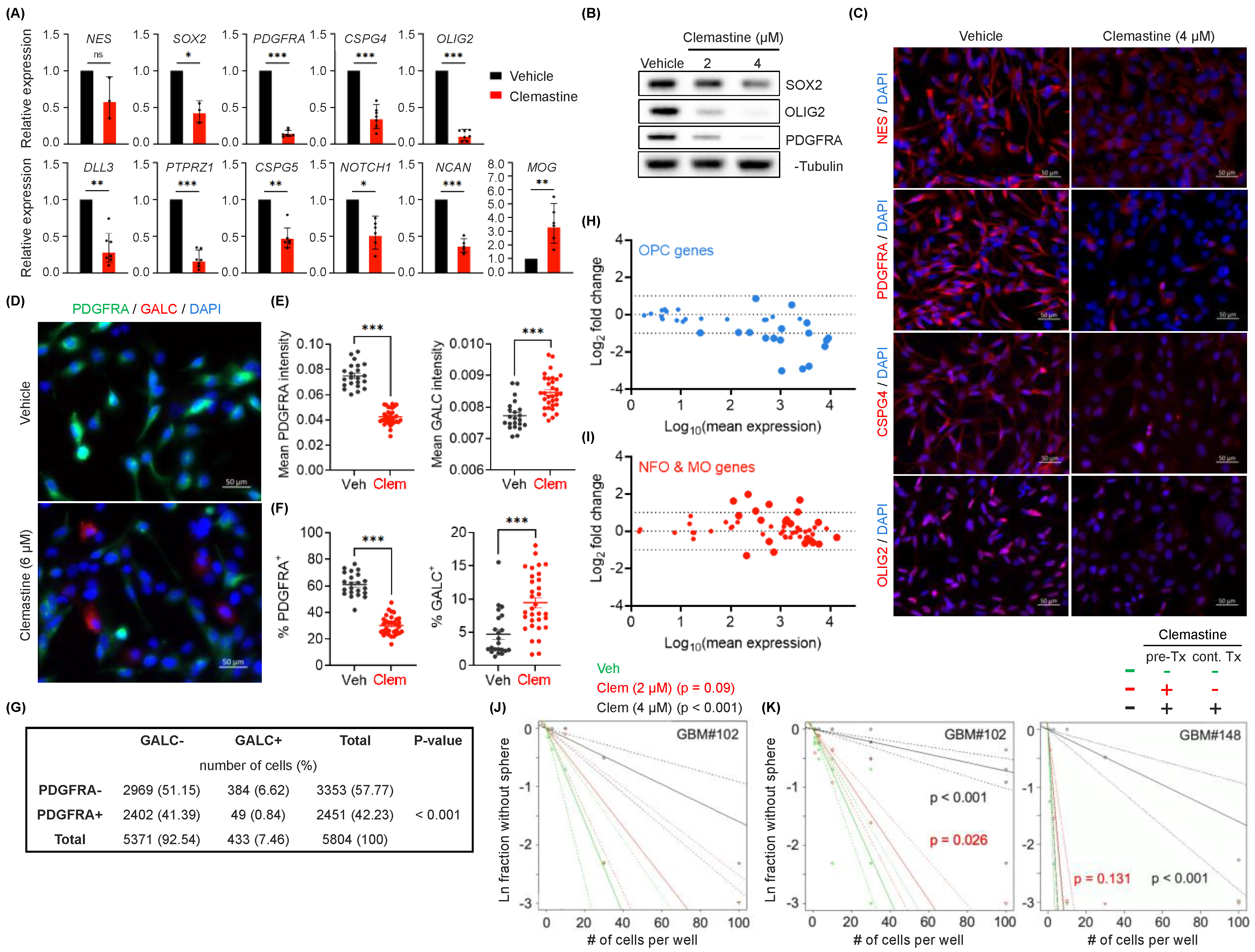
3.2. Clemastine Attenuates the Stemness and Progenitor Cell Features of PDGFRA+ BTICs
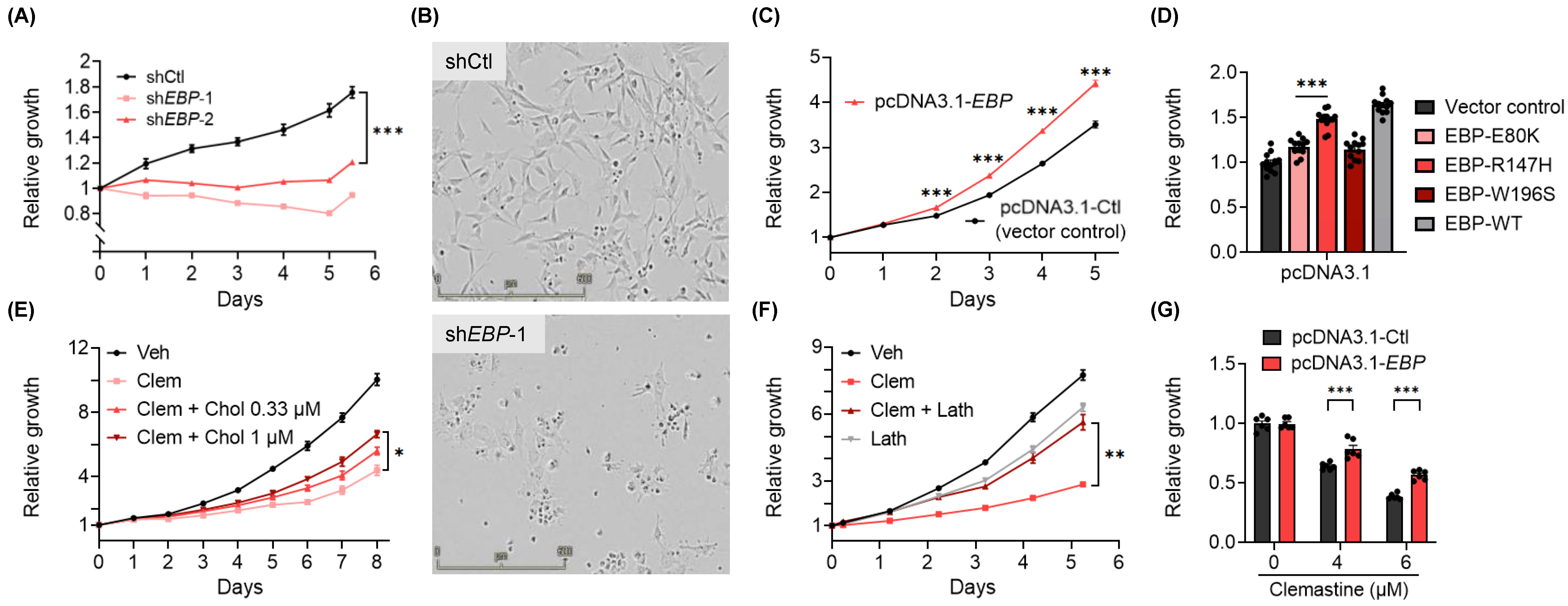
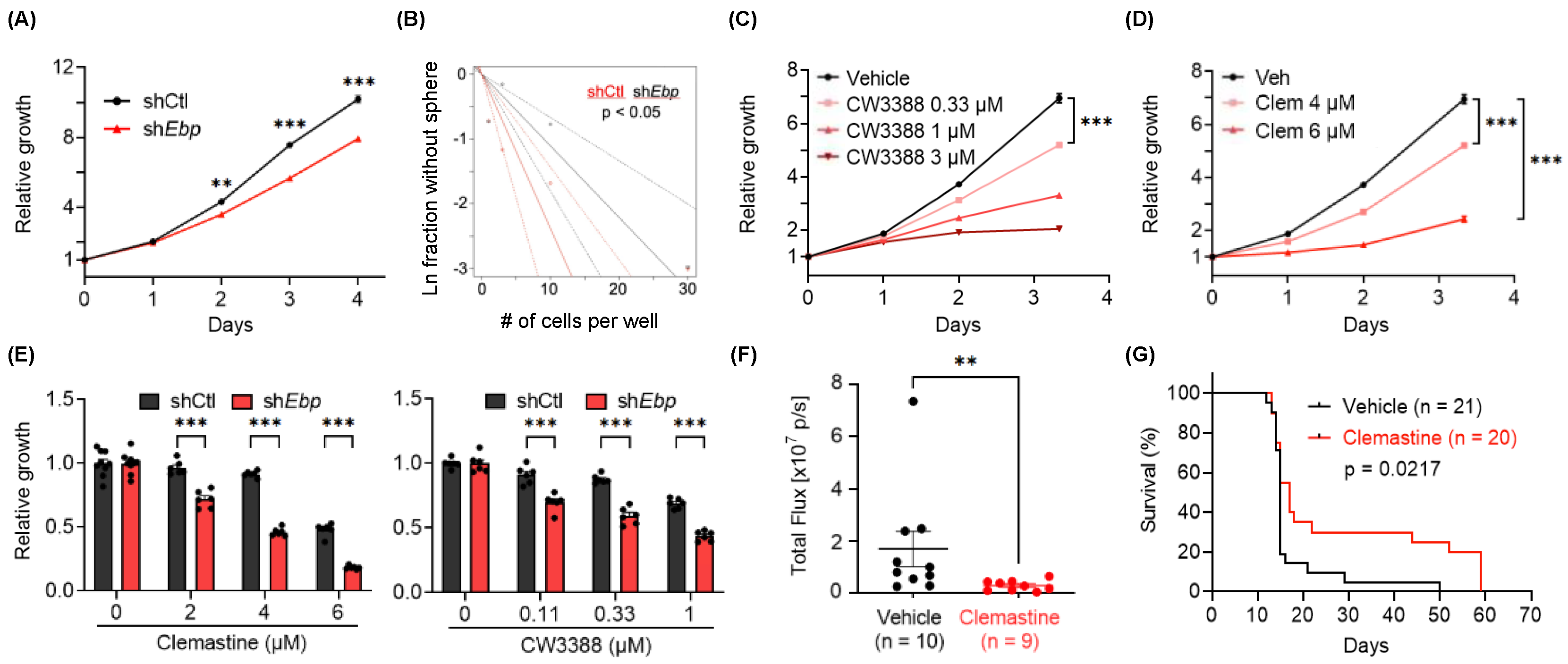
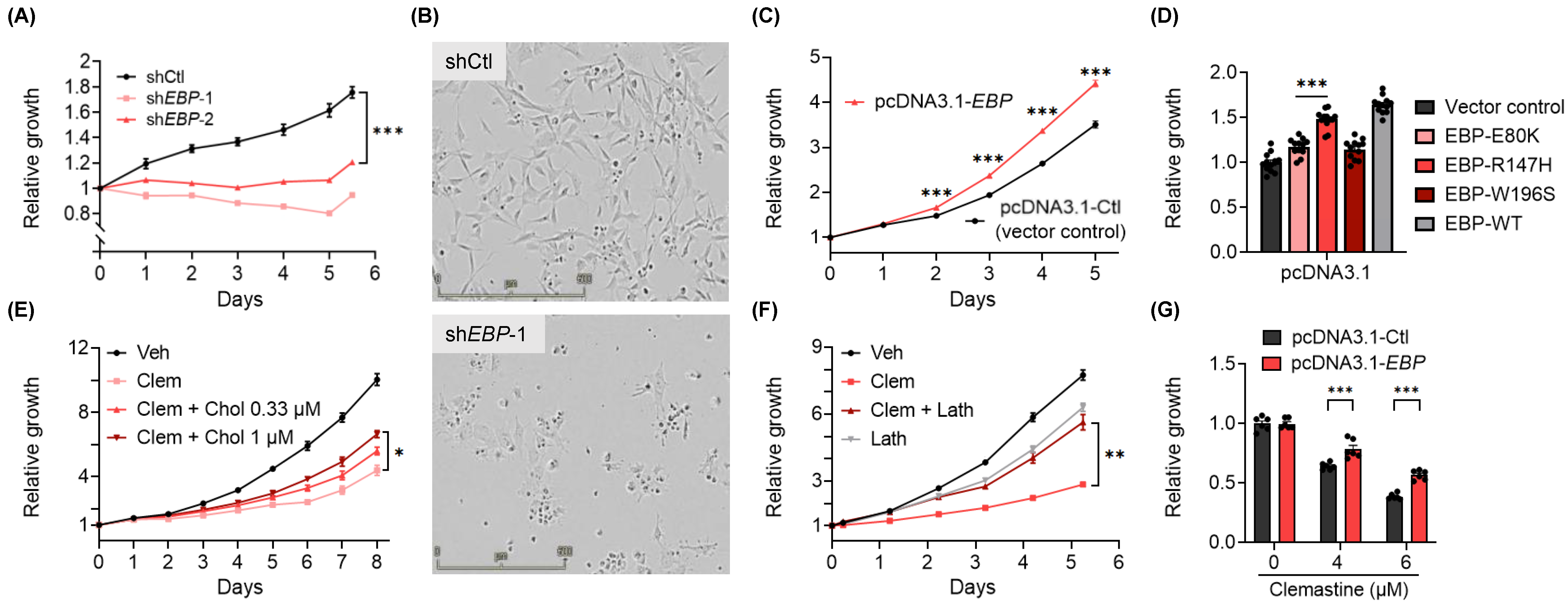
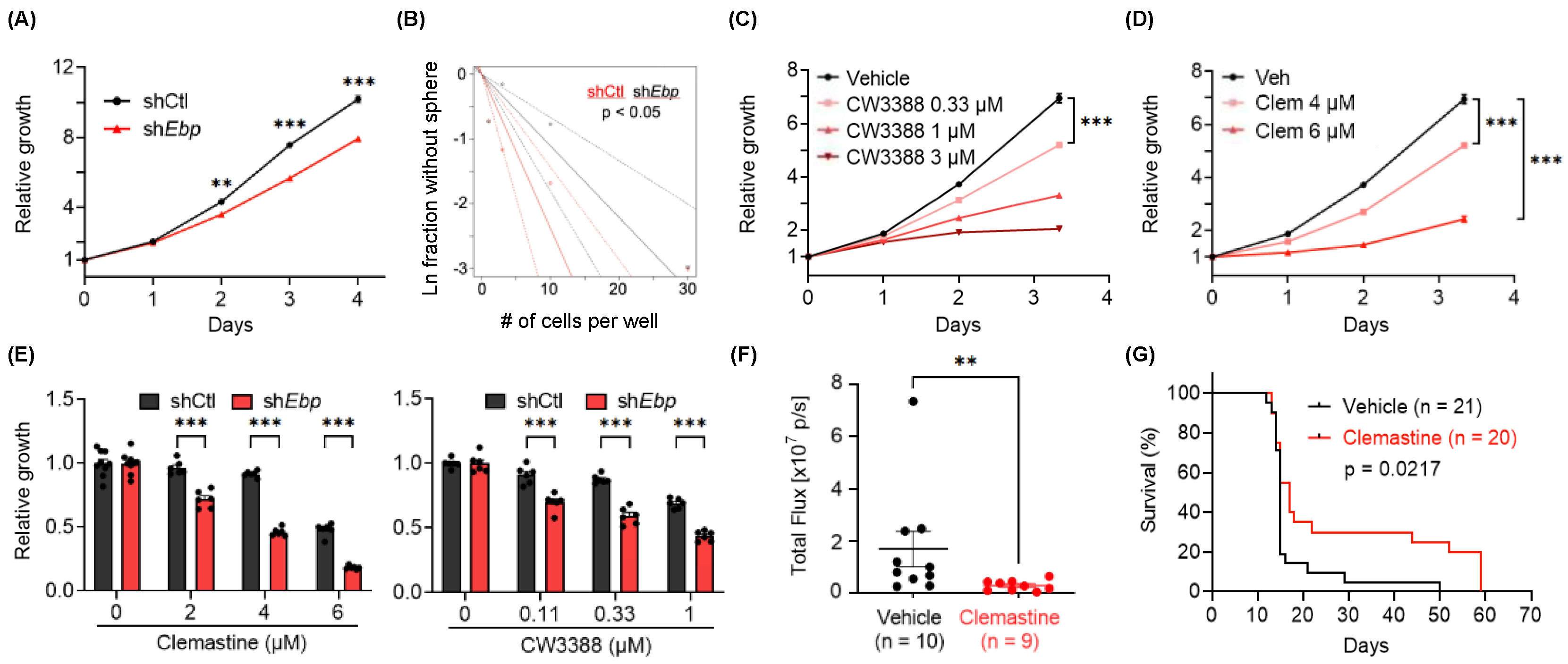
3.3. EBP, A Pharmacological Target of Clemastine, Is Essential for BTIC Propagation
3.4. Clemastine Suppresses Tumorigenicity in A Mouse Glioma Model Representative of PN GBMs
3.5. Clemastine Treatment Broadly Alters Multiple Signaling Pathways in Glioma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Marziali, G.; Signore, M.; Buccarelli, M.; Grande, S.; Palma, A.; Biffoni, M.; Rosi, A.; D’Alessandris, Q.G.; Martini, M.; Larocca, L.M.; et al. Metabolic/Proteomic Signature Defines Two Glioblastoma Subtypes With Different Clinical Outcome. Sci. Rep. 2016, 6, 21557. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef] [PubMed]
- Colman, H.; Zhang, L.; Sulman, E.P.; McDonald, J.M.; Shooshtari, N.L.; Rivera, A.; Popoff, S.; Nutt, C.L.; Louis, D.N.; Cairncross, J.G.; et al. A multigene predictor of outcome in glioblastoma. Neuro-Oncol. 2010, 12, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergstrom, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sonmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Chen, Y.; Zhao, C.; Wang, H.; He, D.; Xu, L.; Wang, J.; He, X.; Deng, Y.; Lu, E.E.; et al. Olig2-Dependent Reciprocal Shift in PDGF and EGF Receptor Signaling Regulates Tumor Phenotype and Mitotic Growth in Malignant Glioma. Cancer Cell 2016, 29, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Riester, M.; Cheng, Y.K.; Huse, J.T.; Squatrito, M.; Helmy, K.; Charles, N.; Michor, F.; Holland, E.C. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 2014, 26, 288–300. [Google Scholar] [CrossRef]
- Fang, X.; Huang, Z.; Zhou, W.; Wu, Q.; Sloan, A.E.; Ouyang, G.; McLendon, R.E.; Yu, J.S.; Rich, J.N.; Bao, S. The zinc finger transcription factor ZFX is required for maintaining the tumorigenic potential of glioblastoma stem cells. Stem Cells 2014, 32, 2033–2047. [Google Scholar] [CrossRef]
- Fang, X.; Zhou, W.; Wu, Q.; Huang, Z.; Shi, Y.; Yang, K.; Chen, C.; Xie, Q.; Mack, S.C.; Wang, X.; et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J. Exp. Med. 2017, 214, 245–267. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef]
- Tao, W.; Chu, C.; Zhou, W.; Huang, Z.; Zhai, K.; Fang, X.; Huang, Q.; Zhang, A.; Wang, X.; Yu, X.; et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat. Commun. 2020, 11, 3015. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Li, Z.; Sathornsumetee, S.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008, 68, 6043–6048. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.Y.; Morton, A.R.; et al. Targeting Glioblastoma Stem Cells through Disruption of the Circadian Clock. Cancer Discov. 2019, 9, 1556–1573. [Google Scholar] [CrossRef]
- Jin, X.; Kim, L.J.Y.; Wu, Q.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Wang, X.; Mack, S.C.; Miller, T.E.; et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat. Med. 2017, 23, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Guryanova, O.A.; Zhou, W.; Liu, C.; Huang, Z.; Fang, X.; Wang, X.; Chen, C.; Wu, Q.; He, Z.; et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci. Transl. Med. 2018, 10, eaah6816. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Wickman, S.; Eyler, C.; Heddleston, J.; Shi, Q.; Lathia, J.D.; Macswords, J.; Lee, J.; McLendon, R.E.; et al. Targeting A20 decreases glioma stem cell survival and tumor growth. PLoS Biol. 2010, 8, e1000319. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- De The, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Storm, E.E.; Durinck, S.; de Sousa e Melo, F.; Tremayne, J.; Kljavin, N.; Tan, C.; Ye, X.; Chiu, C.; Pham, T.; Hongo, J.A.; et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature 2016, 529, 97–100. [Google Scholar] [CrossRef]
- Yan, M.; Zhang, Y.; He, B.; Xiang, J.; Wang, Z.F.; Zheng, F.M.; Xu, J.; Chen, M.Y.; Zhu, Y.L.; Wen, H.J.; et al. IKKalpha restoration via EZH2 suppression induces nasopharyngeal carcinoma differentiation. Nat. Commun. 2014, 5, 3661. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Weinberg, R.A. Targeting the epithelial-to-mesenchymal transition: The case for differentiation-based therapy. Cold Spring Harb. Symp. Quant. Biol. 2017, 81, 11–19. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef]
- Li, Y.; Yin, W.; Wang, X.; Zhu, W.; Huang, Y.; Yan, G. Cholera toxin induces malignant glioma cell differentiation via the PKA/CREB pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 13438–13443. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Luan, Y.; Cai, J.; Wu, S.; Mai, J.; Gu, J.; Zhang, H.; Li, K.; Lin, Y.; Xiao, X.; et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1alpha Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017, 18, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef]
- Chen, J.Y.; Lai, Y.S.; Tsai, H.J.; Kuo, C.C.; Yen, B.L.; Yeh, S.P.; Sun, H.S.; Hung, W.C. The oncometabolite R-2-hydroxyglutarate activates NF-kappaB-dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci. Rep. 2016, 6, 32428. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Fancy, S.P.J.; Shen, Y.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dupree, J.L.; Gacias, M.; Frawley, R.; Sikder, T.; Naik, P.; Casaccia, P. Clemastine Enhances Myelination in the Prefrontal Cortex and Rescues Behavioral Changes in Socially Isolated Mice. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 957–962. [Google Scholar] [CrossRef]
- Hansen, L.J.; Sun, R.; Yang, R.; Singh, S.X.; Chen, L.H.; Pirozzi, C.J.; Moure, C.J.; Hemphill, C.; Carpenter, A.B.; Healy, P.; et al. MTAP Loss Promotes Stemness in Glioblastoma and Confers Unique Susceptibility to Purine Starvation. Cancer Res. 2019, 79, 3383–3394. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.X.; Yang, R.; Roso, K.; Hansen, L.J.; Du, C.; Chen, L.H.; Greer, P.K.; Pirozzi, C.J.; He, Y. Purine Synthesis Inhibitor L-Alanosine Impairs Mitochondrial Function and Stemness of Brain Tumor Initiating Cells. Biomedicines 2022, 10, 751. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, M.; Das, S.; Berns, E.J.; Kim, J.; Stupp, S.I.; Kessler, J.A. Nanofiber-mediated inhibition of focal adhesion kinase sensitizes glioma stemlike cells to epidermal growth factor receptor inhibition. Neuro-Oncol. 2013, 15, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, M.R.; Sabatini, D.M.; Carpenter, A.E. CellProfiler: Free, versatile software for automated biological image analysis. Biotechniques 2007, 42, 71–75. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Hubler, Z.; Allimuthu, D.; Bederman, I.; Elitt, M.S.; Madhavan, M.; Allan, K.C.; Shick, H.E.; Garrison, E.; Karl, M.T.; Factor, D.C.; et al. Accumulation of 8,9-unsaturated sterols drives oligodendrocyte formation and remyelination. Nature 2018, 560, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Moghaddasi, M.; Nabovvati, M.; Koushki, A.; Soltansanjari, M.; Sardarinia, M.; Mohebi, N.; Rabani, S. Randomized control trial of evaluation of Clemastine effects on visual evoked potential, nerve fiber layer and ganglion cell layer complex in patients with optic neuritis. Clin. Neurol. Neurosurg. 2020, 193, 105741. [Google Scholar] [CrossRef]
- Allimuthu, D.; Hubler, Z.; Najm, F.J.; Tang, H.; Bederman, I.; Seibel, W.; Tesar, P.J.; Adams, D.J. Diverse Chemical Scaffolds Enhance Oligodendrocyte Formation by Inhibiting CYP51, TM7SF2, or EBP. Cell Chem. Biol. 2019, 26, 593–599.e594. [Google Scholar] [CrossRef]
- Wiese, C.; Rolletschek, A.; Kania, G.; Blyszczuk, P.; Tarasov, K.V.; Tarasova, Y.; Wersto, R.P.; Boheler, K.R.; Wobus, A.M. Nestin expression—A property of multi-lineage progenitor cells? Cell. Mol. Life Sci. 2004, 61, 2510–2522. [Google Scholar] [CrossRef]
- Ellis, P.; Fagan, B.M.; Magness, S.T.; Hutton, S.; Taranova, O.; Hayashi, S.; McMahon, A.; Rao, M.; Pevny, L. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev. Neurosci. 2004, 26, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, S.; Anderson, D.J. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 2000, 25, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.L.; Garcia-Verdugo, J.M.; Gil-Perotin, S.; Roy, M.; Quinones-Hinojosa, A.; VandenBerg, S.; Alvarez-Buylla, A. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 2006, 51, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Nishiyama, A.; Peterson, J.; Prineas, J.; Trapp, B.D. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 6404–6412. [Google Scholar] [CrossRef] [PubMed]
- Plemel, J.R.; Liu, W.-Q.; Yong, V.W. Remyelination therapies: A new direction and challenge in multiple sclerosis. Nat. Rev. Drug Discov. 2017, 16, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Zhong, S.; Zhang, S.; Fan, X.; Wu, Q.; Yan, L.; Dong, J.; Zhang, H.; Li, L.; Sun, L.; Pan, N.; et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 2018, 555, 524–528. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nature reviews. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.J.; Papadopoli, D.; Jewer, M.; Del Rincon, S.; Topisirovic, I.; Lawrence, M.G.; Postovit, L.M. Cancer Plasticity: The Role of mRNA Translation. Trends Cancer 2021, 7, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Shirakawa, O.; Kuno, T.; Tanaka, C. Antimuscarinic effects of antihistamines: Quantitative evaluation by receptor-binding assay. Jpn. J. Pharmacol. 1987, 43, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Niu, J.; Hoi, K.K.; Zhao, C.; Caganap, S.D.; Henry, R.G.; Dao, D.Q.; Zollinger, D.R.; Mei, F.; Shen, Y.A.; et al. Clemastine rescues myelination defects and promotes functional recovery in hypoxic brain injury. Brain 2018, 141, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Welliver, R.R.; Polanco, J.J.; Seidman, R.A.; Sinha, A.K.; O’Bara, M.A.; Khaku, Z.M.; Santiago Gonzalez, D.A.; Nishiyama, A.; Wess, J.; Feltri, M.L.; et al. Muscarinic Receptor M3R Signaling Prevents Efficient Remyelination by Human and Mouse Oligodendrocyte Progenitor Cells. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 6921–6932. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, M.; Sakurada, Y.; Iwabuchi, K.; Mochizuki, H.; Kato, M.; Aoki, M.; Funaki, Y.; Itoh, M.; Iwata, R.; Wong, D.F.; et al. Central effects of fexofenadine and cetirizine: Measurement of psychomotor performance, subjective sleepiness, and brain histamine H1-receptor occupancy using 11C-doxepin positron emission tomography. J. Clin. Pharmacol. 2004, 44, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef]
- Braverman, N.; Lin, P.; Moebius, F.F.; Obie, C.; Moser, A.; Glossmann, H.; Wilcox, W.R.; Rimoin, D.L.; Smith, M.; Kratz, L.; et al. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat. Genet. 1999, 22, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Laggner, C.; Schieferer, C.; Fiechtner, B.; Poles, G.; Hoffmann, R.D.; Glossmann, H.; Langer, T.; Moebius, F.F. Discovery of high-affinity ligands of sigma1 receptor, ERG2, and emopamil binding protein by pharmacophore modeling and virtual screening. J. Med. Chem. 2005, 48, 4754–4764. [Google Scholar] [CrossRef]
- Moebius, F.F.; Reiter, R.J.; Bermoser, K.; Glossmann, H.; Cho, S.Y.; Paik, Y.K. Pharmacological analysis of sterol delta8-delta7 isomerase proteins with [3H]ifenprodil. Mol. Pharmacol. 1998, 54, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Theodoropoulos, P.C.; Eskiocak, U.; Wang, W.; Moon, Y.A.; Posner, B.; Williams, N.S.; Wright, W.E.; Kim, S.B.; Nijhawan, D.; et al. Selective targeting of mutant adenomatous polyposis coli (APC) in colorectal cancer. Sci. Transl. Med. 2016, 8, 361ra140. [Google Scholar] [CrossRef]
- Moebius, F.F.; Soellner, K.E.; Fiechtner, B.; Huck, C.W.; Bonn, G.; Glossmann, H. Histidine77, glutamic acid81, glutamic acid123, threonine126, asparagine194, and tryptophan197 of the human emopamil binding protein are required for in vivo sterol delta 8-delta 7 isomerization. Biochemistry 1999, 38, 1119–1127. [Google Scholar] [CrossRef]
- Herman, G.E.; Kelley, R.I.; Pureza, V.; Smith, D.; Kopacz, K.; Pitt, J.; Sutphen, R.; Sheffield, L.J.; Metzenberg, A.B. Characterization of mutations in 22 females with X-linked dominant chondrodysplasia punctata (Happle syndrome). Genet. Med. 2002, 4, 434–438. [Google Scholar] [CrossRef]
- Long, T.; Hassan, A.; Thompson, B.M.; McDonald, J.G.; Wang, J.; Li, X. Structural basis for human sterol isomerase in cholesterol biosynthesis and multidrug recognition. Nat. Commun. 2019, 10, 2452. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, C.J.; Carpenter, A.B.; Waitkus, M.S.; Wang, C.Y.; Zhu, H.; Hansen, L.J.; Chen, L.H.; Greer, P.K.; Feng, J.; Wang, Y.; et al. Mutant IDH1 Disrupts the Mouse Subventricular Zone and Alters Brain Tumor Progression. Mol. Cancer Res. 2017, 15, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef]
- Rahme, G.J.; Luikart, B.W.; Cheng, C.; Israel, M.A. A recombinant lentiviral PDGF-driven mouse model of proneural glioblastoma. Neuro-Oncol. 2018, 20, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Tchougounova, E.; Kastemar, M.; Brasater, D.; Holland, E.C.; Westermark, B.; Uhrbom, L. Loss of Arf causes tumor progression of PDGFB-induced oligodendroglioma. Oncogene 2007, 26, 6289–6296. [Google Scholar] [CrossRef]
- Alessandrini, F.; Ceresa, D.; Appolloni, I.; Pagani, F.; Poliani, P.L.; Marubbi, D.; Malatesta, P. Glioblastoma models driven by different mutations converge to the proneural subtype. Cancer Lett. 2020, 469, 447–455. [Google Scholar] [CrossRef]
- Becher, O.J.; Hambardzumyan, D.; Walker, T.R.; Helmy, K.; Nazarian, J.; Albrecht, S.; Hiner, R.L.; Gall, S.; Huse, J.T.; Jabado, N.; et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res. 2010, 70, 2548–2557. [Google Scholar] [CrossRef]
- Lei, L.; Sonabend, A.M.; Guarnieri, P.; Soderquist, C.; Ludwig, T.; Rosenfeld, S.; Bruce, J.N.; Canoll, P. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS ONE 2011, 6, e20041. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, J.D.; Fomchenko, E.I.; Akuffo, A.A.; Schmidt, E.; Helmy, K.Y.; Bazzoli, E.; Brennan, C.W.; Holland, E.C.; Milosevic, A. Candidate Pathways for Promoting Differentiation or Quiescence of Oligodendrocyte Progenitor-like Cells in Glioma. Cancer Res. 2012, 72, 4856–4868. [Google Scholar] [CrossRef] [PubMed]
- Le Joncour, V.; Filppu, P.; Hyvonen, M.; Holopainen, M.; Turunen, S.P.; Sihto, H.; Burghardt, I.; Joensuu, H.; Tynninen, O.; Jaaskelainen, J.; et al. Vulnerability of invasive glioblastoma cells to lysosomal membrane destabilization. EMBO Mol. Med. 2019, 11, e9034. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Platanias, L.C. Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers 2023, 15, 3458. [Google Scholar] [CrossRef] [PubMed]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.-Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef]
- Jernigan, F.; Branstrom, A.; Baird, J.D.; Cao, L.; Dali, M.; Furia, B.; Kim, M.J.; O’Keefe, K.; Kong, R.; Laskin, O.L.; et al. Preclinical and Early Clinical Development of PTC596, a Novel Small-Molecule Tubulin-Binding Agent. Mol. Cancer Ther. 2021, 20, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; Cavallo, G.; Lonardi, S.; Magrini, E.; Tosoni, A.; Grosso, D.; Scopece, L.; Blatt, V.; Urbini, B.; Pession, A.; et al. Gefitinib in patients with progressive high-grade gliomas: A multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br. J. Cancer 2007, 96, 1047–1051. [Google Scholar] [CrossRef]
- Westphal, M.; Heese, O.; Steinbach, J.P.; Schnell, O.; Schackert, G.; Mehdorn, M.; Schulz, D.; Simon, M.; Schlegel, U.; Senft, C.; et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur. J. Cancer 2015, 51, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Yang, R.; Mirzaei, R.; Rawji, K.; Poon, C.; Mishra, M.K.; Zemp, F.J.; Bose, P.; Kelly, J.; Dunn, J.F.; et al. Control of brain tumor growth by reactivating myeloid cells with niacin. Sci. Transl. Med. 2020, 12, eaay9924. [Google Scholar] [CrossRef]
- Gerstmeier, J.; Possmayer, A.L.; Bozkurt, S.; Hoffmann, M.E.; Dikic, I.; Herold-Mende, C.; Burger, M.C.; Münch, C.; Kögel, D.; Linder, B. Calcitriol Promotes Differentiation of Glioma Stem-Like Cells and Increases Their Susceptibility to Temozolomide. Cancers 2021, 13, 3577. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2—and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500.e488. [Google Scholar] [CrossRef]
- Kang, Z.; Li, S.; Kang, X.; Deng, J.; Yang, H.; Chen, F.; Jiang, J.; Zhang, J.; Li, W. Phase I study of chlorogenic acid injection for recurrent high-grade glioma with long-term follow-up. Cancer Biol. Med. 2023, 20, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Marín-Ramos, N.I.; Pérez-Hernández, M.; Tam, A.; Swenson, S.D.; Cho, H.Y.; Thein, T.Z.; Hofman, F.M.; Chen, T.C. Inhibition of motility by NEO100 through the calpain-1/RhoA pathway. J. Neurosurg. 2019, 133, 1020–1031. [Google Scholar] [CrossRef]
- Da Silva, A.B.; Cerqueira Coelho, P.L.; das Neves Oliveira, M.; Oliveira, J.L.; Oliveira Amparo, J.A.; da Silva, K.C.; Soares, J.R.P.; Pitanga, B.P.S.; dos Santos Souza, C.; de Faria Lopes, G.P.; et al. The flavonoid rutin and its aglycone quercetin modulate the microglia inflammatory profile improving antiglioma activity. Brain Behav. Immun. 2020, 85, 170–185. [Google Scholar] [CrossRef]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242.e228. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.E.; Yang, Y.; Smith, R.C.; Thompson, B.M.; Yamasaki, T.; Soto-Feliciano, Y.M.; Funato, K.; Liang, Y.; Garcia-Bermudez, J.; Wang, X.; et al. Target identification reveals lanosterol synthase as a vulnerability in glioma. Proc. Natl. Acad. Sci. USA 2019, 116, 7957–7962. [Google Scholar] [CrossRef]
- Pavlik, E.J.; Nelson, K.; Srinivasan, S.; Powell, D.E.; Kenady, D.E.; DePriest, P.D.; Gallion, H.H.; van Nagell, J.R., Jr. Resistance to tamoxifen with persisting sensitivity to estrogen: Possible mediation by excessive antiestrogen binding site activity. Cancer Res. 1992, 52, 4106–4112. [Google Scholar] [PubMed]
- Leignadier, J.; Dalenc, F.; Poirot, M.; Silvente-Poirot, S. Improving the efficacy of hormone therapy in breast cancer: The role of cholesterol metabolism in SERM-mediated autophagy, cell differentiation and death. Biochem. Pharmacol. 2017, 144, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Chen, L.H.; Hansen, L.J.; Carpenter, A.B.; Moure, C.J.; Liu, H.; Pirozzi, C.J.; Diplas, B.H.; Waitkus, M.S.; Greer, P.K.; et al. Cic loss promotes gliomagenesis via aberrant neural stem cell proliferation and differentiation. Cancer Res. 2017, 77, 6097–6108. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652.e636. [Google Scholar] [CrossRef] [PubMed]
- Korade, Z.; Kim, H.Y.; Tallman, K.A.; Liu, W.; Koczok, K.; Balogh, I.; Xu, L.; Mirnics, K.; Porter, N.A. The Effect of Small Molecules on Sterol Homeostasis: Measuring 7-Dehydrocholesterol in Dhcr7-Deficient Neuro2a Cells and Human Fibroblasts. J. Med. Chem. 2016, 59, 1102–1115. [Google Scholar] [CrossRef]
- Reardon, D.A.; Narita, Y.; Arakawa, Y.; Goldlust, S.A.; Ansstas, G.; Mei, J.; Dow, E.; Seki, M.; Furuta, Y.; Song, G.; et al. DSP-0390, an oral emopamil binding protein (EBP) inhibitor, in patients with recurrent high-grade glioma: A first-in-human, phase 1 study. J. Clin. Oncol. 2022, 40, TPS2077. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, M.A.; Yang, R.; Liu, H.; Wang, W.; Song, X.; Hu, B.; Reynolds, N.; Roso, K.; Chen, L.H.; Greer, P.K.; et al. Repurposing Clemastine to Target Glioblastoma Cell Stemness. Cancers 2023, 15, 4619. https://doi.org/10.3390/cancers15184619
Sun MA, Yang R, Liu H, Wang W, Song X, Hu B, Reynolds N, Roso K, Chen LH, Greer PK, et al. Repurposing Clemastine to Target Glioblastoma Cell Stemness. Cancers. 2023; 15(18):4619. https://doi.org/10.3390/cancers15184619
Chicago/Turabian StyleSun, Michael A., Rui Yang, Heng Liu, Wenzhe Wang, Xiao Song, Bo Hu, Nathan Reynolds, Kristen Roso, Lee H. Chen, Paula K. Greer, and et al. 2023. "Repurposing Clemastine to Target Glioblastoma Cell Stemness" Cancers 15, no. 18: 4619. https://doi.org/10.3390/cancers15184619
APA StyleSun, M. A., Yang, R., Liu, H., Wang, W., Song, X., Hu, B., Reynolds, N., Roso, K., Chen, L. H., Greer, P. K., Keir, S. T., McLendon, R. E., Cheng, S.-Y., Bigner, D. D., Ashley, D. M., Pirozzi, C. J., & He, Y. (2023). Repurposing Clemastine to Target Glioblastoma Cell Stemness. Cancers, 15(18), 4619. https://doi.org/10.3390/cancers15184619