
Advances in Immunotherapeutics in Pancreatic Ductal Adenocarcinoma

 ,
, 
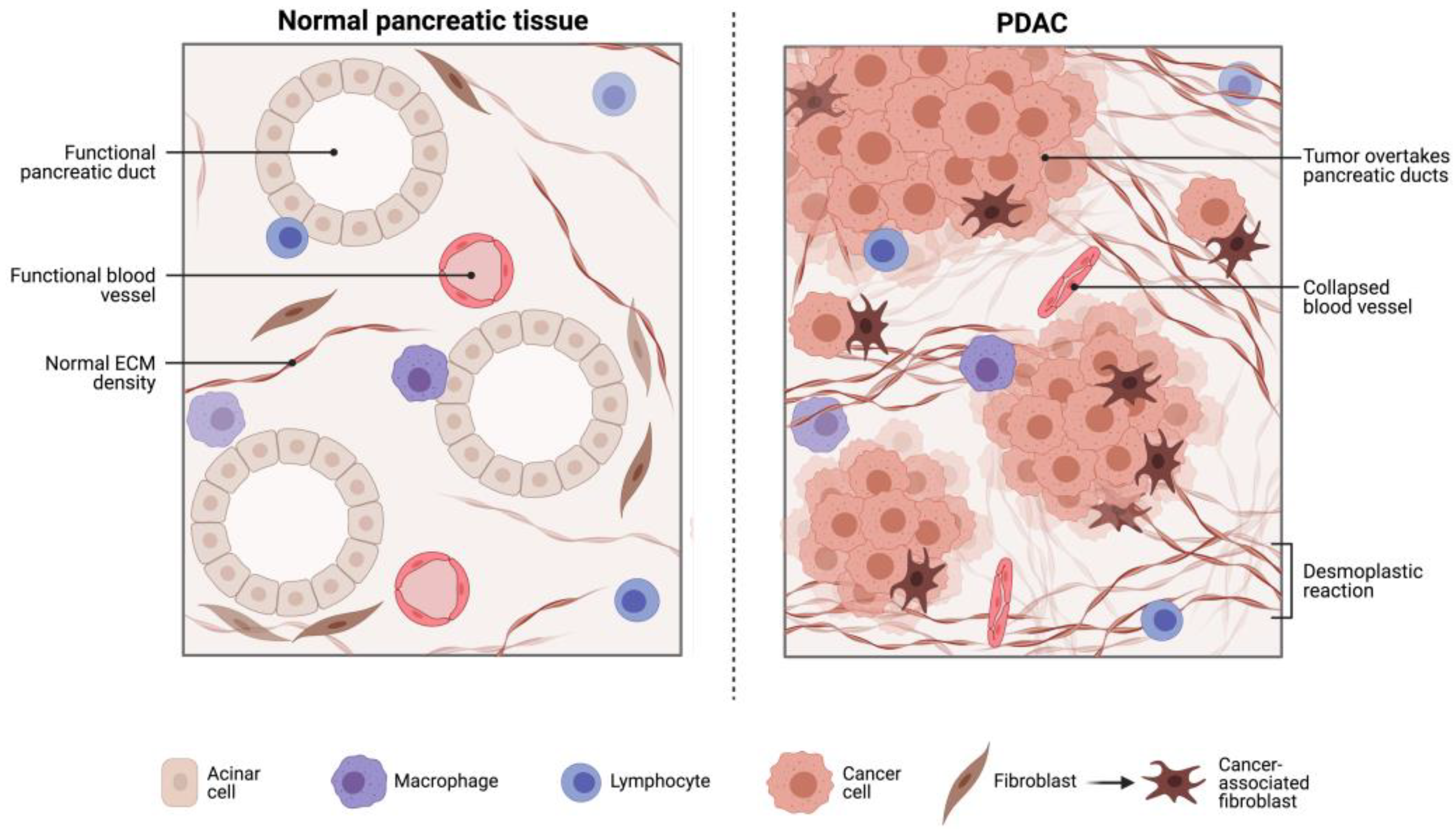{kind=link}
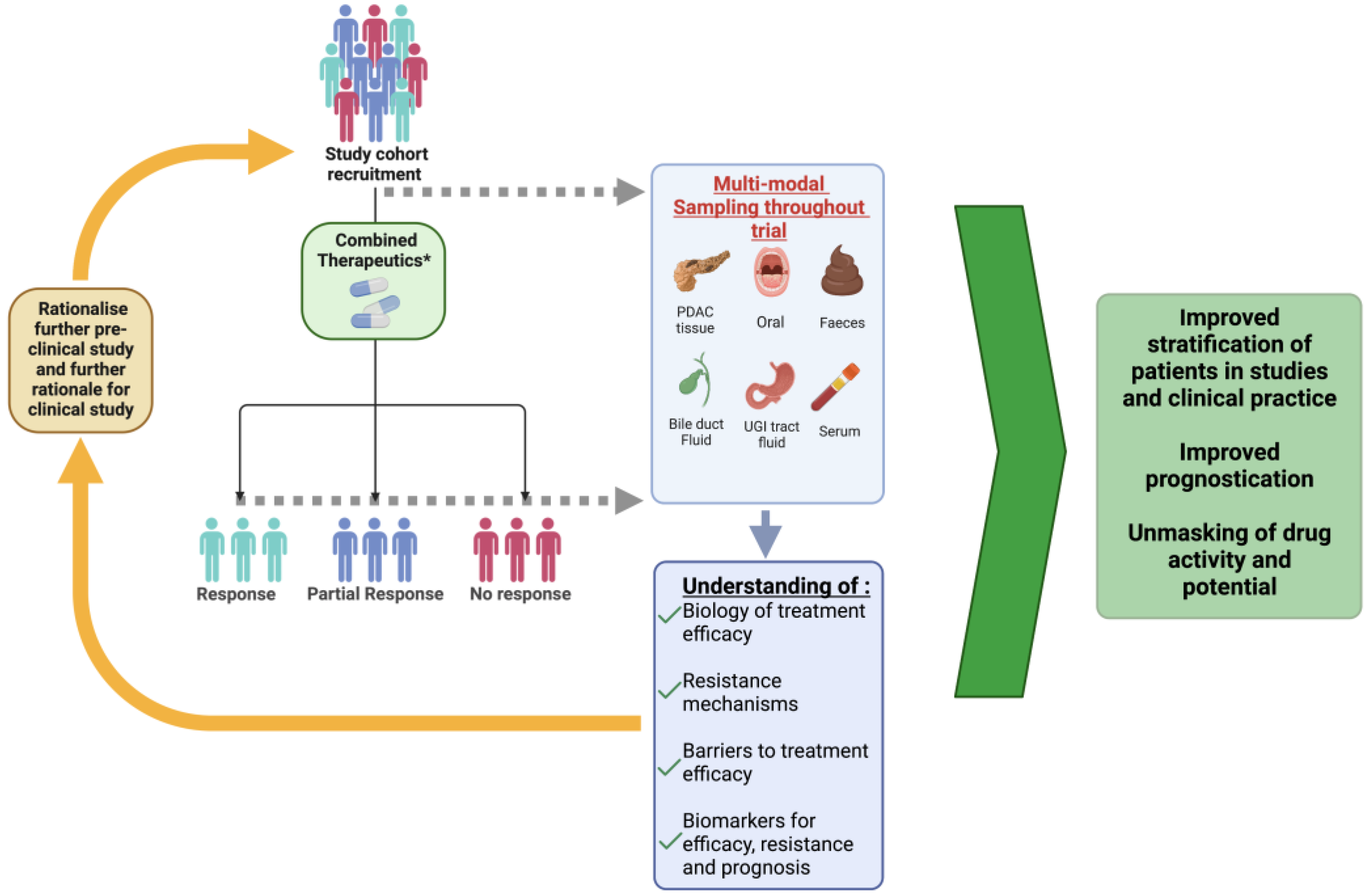{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Immunotherapeutics
2.1. Programmed Death Checkpoint Inhibitors
2.1.1. Combination PD-1 Inhibitor with Anti-CD25
2.1.2. Anti-PD-1 and CTLA-4 Inhibitor Combination
2.1.3. Combination of Chemotherapy and PD-1 Inhibitor Immunotherapy
2.1.4. Combination of Radiotherapy and PD-1 Inhibitor Immunotherapy
2.1.5. Conclusion of Programmed Death Checkpoint Inhibitors
2.2. CTLA-4 Inhibitors
2.3. The Microbiome and Immune Check Point Inhibitors
2.4. CAR T-Cell Therapy
2.5. KRAS Vaccines
2.6. Telomerase Vaccines
2.7. Gastrin Vaccines
2.8. Survivin-Targeting Vaccines
2.9. Heat-Shock Protein (HSP) Peptide Complex-Based Vaccines
2.10. MUC-1 Targeting Vaccines
2.11. Listeria-Based Vaccines
2.12. Dendritic Cell-Based Vaccines
2.13. Oncolytic Viral Therapy
2.14. Immunomodulators of the TME
3. Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694. [Google Scholar] [CrossRef]
- UK, C.R. Pancreatic Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer#heading-Zero (accessed on 14 June 2023).
- Wolfgang, C.L.; Herman, J.M.; Laheru, D.A.; Klein, A.P.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent progress in pancreatic cancer. CA Cancer J. Clin. 2013, 63, 318–348. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Nimura, Y.; Nagino, M. Advances in the treatment of pancreatic cancer: Limitations of surgery and evaluation of new therapeutic strategies. Surg. Today 2009, 39, 466–475. [Google Scholar] [CrossRef]
- Nakamura, M.; Wada, J.; Suzuki, H.; Tanaka, M.; Katano, M.; Morisaki, T. Long-term outcome of immunotherapy for patients with refractory pancreatic cancer. Anticancer Res. 2009, 29, 831–836. [Google Scholar]
- Murphy, J.D.; Chang, D.T.; Abelson, J.; Daly, M.E.; Yeung, H.N.; Nelson, L.M.; Koong, A.C. Cost-effectiveness of modern radiotherapy techniques in locally advanced pancreatic cancer. Cancer 2012, 118, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Sally, Á.; McGowan, R.; Finn, K.; Moran, B.M. Current and Future Therapies for Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 2417. [Google Scholar] [CrossRef]
- Kataja, V. Clinical Oncology-Basic Principles and Practice, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Bailey, P.; Chang, D.K.; Forget, M.A.; Lucas, F.A.; Alvarez, H.A.; Haymaker, C.; Chattopadhyay, C.; Kim, S.H.; Ekmekcioglu, S.; Grimm, E.A.; et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci. Rep. 2016, 6, 35848. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef] [PubMed]
- Brower, V. Checkpoint blockade immunotherapy for cancer comes of age. JNCI J. Natl. Cancer Inst. 2015, 107, djv069. [Google Scholar] [CrossRef]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef]
- Kadono, T. Immune-related adverse events by immune checkpoint inhibitors. Jpn. J. Clin. Immunol. 2017, 40, 83–89. [Google Scholar] [CrossRef]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; Roth, S.; Kong, J.; Guerra, G.; Narasimhan, V.; Pereira, L.; Desai, J.; Heriot, A.; Ramsay, R. An Update on Immunotherapy for Solid Tumors: A Review. Ann. Surg. Oncol. 2018, 25, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Sunami, Y.; Kleeff, J. Immunotherapy of pancreatic cancer. Prog. Mol. Biol. Transl. Sci. 2019, 164, 189–216. [Google Scholar] [CrossRef]
- Young, K.; Hughes, D.J.; Cunningham, D.; Starling, N. Immunotherapy and pancreatic cancer: Unique challenges and potential opportunities. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816281. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Song, X.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017, 407, 57–65. [Google Scholar] [PubMed]
- Ott, P.A.; Bang, Y.-J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.-C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M. T-cell–inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef]
- Frenel, J.S.; Le Tourneau, C.; O’Neil, B.; Ott, P.A.; Piha-Paul, S.A.; Gomez-Roca, C.; van Brummelen, E.M.J.; Rugo, H.S.; Thomas, S.; Saraf, S.; et al. Safety and Efficacy of Pembrolizumab in Advanced, Programmed Death Ligand 1-Positive Cervical Cancer: Results From the Phase Ib KEYNOTE-028 Trial. J. Clin. Oncol. 2017, 35, 4035–4041. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef]
- Brown, M.; Zhang, W.; Yan, D.; Kenath, R.; Le, L.; Wang, H.; Delitto, D.; Ostrov, D.; Robertson, K.; Liu, C.; et al. The role of survivin in the progression of pancreatic ductal adenocarcinoma (PDAC) and a novel survivin-targeted therapeutic for PDAC. PLoS ONE 2020, 15, e0226917. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Mo, S.; Ma, H.; Lu, Z.; Yu, S.; Chen, J. PD-L1 and PD-L2 expression in pancreatic ductal adenocarcinoma and their correlation with immune infiltrates and DNA damage response molecules. J. Pathol. Clin. Res. 2022, 8, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef]
- Merali, N.; Chouari, T.; Kayani, K.; Rayner, C.J.; Jiménez, J.I.; Krell, J.; Giovannetti, E.; Bagwan, I.; Relph, K.; Rockall, T.A.; et al. A Comprehensive Review of the Current and Future Role of the Microbiome in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 1020. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.A.; Balachandran, V.P. Scaling the immune incline in PDAC. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 453–454. [Google Scholar] [CrossRef]
- Zhu, Y.H.; Zheng, J.H.; Jia, Q.Y.; Duan, Z.H.; Yao, H.F.; Yang, J.; Sun, Y.W.; Jiang, S.H.; Liu, D.J.; Huo, Y.M. Immunosuppression, immune escape, and immunotherapy in pancreatic cancer: Focused on the tumor microenvironment. Cell. Oncol. 2023, 46, 17–48. [Google Scholar] [CrossRef]
- Abrantes, R.; Duarte, H.O.; Gomes, C.; Wälchli, S.; Reis, C.A. CAR-Ts: New perspectives in cancer therapy. FEBS Lett. 2022, 596, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Skelton, R.A.; Javed, A.; Zheng, L.; He, J. Overcoming the resistance of pancreatic cancer to immune checkpoint inhibitors. J. Surg. Oncol. 2017, 116, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhou, Y.; Qiao, C.; Ni, T.; Li, Z.; Wang, X.; Guo, Q.; Lu, N.; Wei, L. Correction to: Oroxylin A promotes PTEN-mediated negative regulation of MDM2 transcription via SIRT3-mediated deacetylation to stabilize p53 and inhibit glycolysis in wt-p53 cancer cells. J. Hematol. Oncol. 2019, 12, 143. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Arce Vargas, F.; Furness, A.J.S.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Miranda Rota, E.; Dahan, R.; Georgiou, A.; Sledzinska, A.; et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017, 46, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Pu, N.; Zhao, G.; Yin, H.; Li, J.-a.; Nuerxiati, A.; Wang, D.; Xu, X.; Kuang, T.; Jin, D.; Lou, W. CD25 and TGF-β blockade based on predictive integrated immune ratio inhibits tumor growth in pancreatic cancer. J. Transl. Med. 2018, 16, 294. [Google Scholar]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccines Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Oh, U.; Blevins, G.; Griffith, C.; Richert, N.; Maric, D.; Lee, C.R.; McFarland, H.; Jacobson, S. Regulatory T cells are reduced during anti-CD25 antibody treatment of multiple sclerosis. Arch. Neurol. 2009, 66, 471–479. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Ji, H.H.; Tang, X.W.; Dong, Z.; Song, L.; Jia, Y.T. Adverse Event Profiles of Anti-CTLA-4 and Anti-PD-1 Monoclonal Antibodies Alone or in Combination: Analysis of Spontaneous Reports Submitted to FAERS. Clin. Drug Investig. 2019, 39, 319–330. [Google Scholar] [CrossRef]
- Meserve, J.; Facciorusso, A.; Holmer, A.K.; Annese, V.; Sandborn, W.J.; Singh, S. Systematic review with meta-analysis: Safety and tolerability of immune checkpoint inhibitors in patients with pre-existing inflammatory bowel diseases. Aliment. Pharmacol. Ther. 2021, 53, 374–382. [Google Scholar]
- McDonnell, A.M.; Lesterhuis, W.J.; Khong, A.; Nowak, A.K.; Lake, R.A.; Currie, A.J.; Robinson, B.W. Tumor-infiltrating dendritic cells exhibit defective cross-presentation of tumor antigens, but is reversed by chemotherapy. Eur. J. Immunol. 2015, 45, 49–59. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, R.; Li, C.; Song, Y.; Liu, G.; Huang, Q.; Yu, L.; Zhu, D.; Lu, C.; Lu, A.; et al. Nab-paclitaxel promotes the cancer-immunity cycle as a potential immunomodulator. Am. J. Cancer Res. 2021, 11, 3445–3460. [Google Scholar] [PubMed]
- Ho, T.T.B.; Nasti, A.; Seki, A.; Komura, T.; Inui, H.; Kozaka, T.; Kitamura, Y.; Shiba, K.; Yamashita, T.; Mizukoshi, E.; et al. Combination of gemcitabine and anti-PD-1 antibody enhances the anticancer effect of M1 macrophages and the Th1 response in a murine model of pancreatic cancer liver metastasis. J. Immunother. Cancer 2020, 8, e001367. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Correction to: Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2019, 37, 797. [Google Scholar] [CrossRef] [PubMed]
- Mkrtichyan, M.; Najjar, Y.G.; Raulfs, E.C.; Abdalla, M.Y.; Samara, R.; Rotem-Yehudar, R.; Cook, L.; Khleif, S.N. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur. J. Immunol. 2011, 41, 2977–2986. [Google Scholar] [CrossRef]
- Wu, M.; Huang, Q.; Xie, Y.; Wu, X.; Ma, H.; Zhang, Y.; Xia, Y. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J. Hematol. Oncol. 2022, 15, 24. [Google Scholar] [CrossRef]
- McCarthy, P.M.; Rendo, M.J.; Uy, M.D.; Adams, A.M.; O’Shea, A.E.; Nelson, D.W.; Fenderson, J.L.; Cebe, K.M.; Krell, R.W.; Clifton, G.T.; et al. Near Complete Pathologic Response to PD-1 Inhibitor and Radiotherapy in a Patient with Locally Advanced Pancreatic Ductal Adenocarcinoma. Onco Targets Ther. 2021, 14, 3537–3544. [Google Scholar] [CrossRef]
- Azad, A.; Yin Lim, S.; D’Costa, Z.; Jones, K.; Diana, A.; Sansom, O.J.; Kruger, P.; Liu, S.; McKenna, W.G.; Dushek, O.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2017, 9, 167–180. [Google Scholar] [CrossRef]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS⁺/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef]
- Pu, N.; Lou, W.; Yu, J. PD-1 immunotherapy in pancreatic cancer: Current status. J. Pancreatol. 2019, 2, 6–10. [Google Scholar] [CrossRef]
- Bengsch, F.; Knoblock, D.M.; Liu, A.; McAllister, F.; Beatty, G.L. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immunother. 2017, 66, 1609–1617. [Google Scholar] [CrossRef]
- Mocellin, S.; Nitti, D. CTLA-4 blockade and the renaissance of cancer immunotherapy. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2013, 1836, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.B.; Benson, A.; Yaghmai, V.; Costa, R.L.B.; Zhou, H.; Behdad, A.; Kaplan, J.B.; Sadim, M.; Talamantes, S.; Kalyan, A. An Extremely Rapid Case of Pneumonitis with the Use of Nivolumab for Pancreatic Adenocarcinoma. Case Rep. Oncol. Med. 2018, 2018, 6314392. [Google Scholar] [CrossRef]
- Magee, D.E.; Hird, A.E.; Klaassen, Z.; Sridhar, S.S.; Nam, R.K.; Wallis, C.J.D.; Kulkarni, G.S. Adverse event profile for immunotherapy agents compared with chemotherapy in solid organ tumors: A systematic review and meta-analysis of randomized clinical trials. Ann. Oncol. 2020, 31, 50–60. [Google Scholar] [CrossRef]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3, e122092. [Google Scholar] [CrossRef]
- Huang, Z.-Q.; Buchsbaum, D.J. Monoclonal antibodies in the treatment of pancreatic cancer. Immunotherapy 2009, 1, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A.; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncologist 2020, 25, e808–e815. [Google Scholar] [CrossRef]
- Carmichael, J.; Fink, U.; Russell, R.C.; Spittle, M.F.; Harris, A.L.; Spiessi, G.; Blatter, J. Phase II study of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 1996, 73, 101–105. [Google Scholar] [CrossRef]
- Casper, E.S.; Green, M.R.; Kelsen, D.P.; Heelan, R.T.; Brown, T.D.; Flombaum, C.D.; Trochanowski, B.; Tarassoff, P.G. Phase II trial of gemcitabine (2,2′-difluorodeoxycytidine) in patients with adenocarcinoma of the pancreas. Investig. New Drugs 1994, 12, 29–34. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Sethi, V.; Kurtom, S.; Tarique, M.; Lavania, S.; Malchiodi, Z.; Hellmund, L.; Zhang, L.; Sharma, U.; Giri, B.; Garg, B.; et al. Gut Microbiota Promotes Tumor Growth in Mice by Modulating Immune Response. Gastroenterology 2018, 155, 33–37.e36. [Google Scholar] [CrossRef]
- Thomas, R.M.; Gharaibeh, R.Z.; Gauthier, J.; Beveridge, M.; Pope, J.L.; Guijarro, M.V.; Yu, Q.; He, Z.; Ohland, C.; Newsome, R.; et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis 2018, 39, 1068–1078. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014, 74, 2907–2912. [Google Scholar] [CrossRef]
- Argani, P.; Iacobuzio-Donahue, C.; Ryu, B.; Rosty, C.; Goggins, M.; Wilentz, R.E.; Murugesan, S.R.; Leach, S.D.; Jaffee, E.; Yeo, C.J.; et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: Identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin. Cancer Res. 2001, 7, 3862–3868. [Google Scholar]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Argani, P.; Rosty, C.; Reiter, R.E.; Wilentz, R.E.; Murugesan, S.R.; Leach, S.D.; Ryu, B.; Skinner, H.G.; Goggins, M.; Jaffee, E.M.; et al. Discovery of new markers of cancer through serial analysis of gene expression: Prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001, 61, 4320–4324. [Google Scholar]
- Chmielewski, M.; Hahn, O.; Rappl, G.; Nowak, M.; Schmidt-Wolf, I.H.; Hombach, A.A.; Abken, H. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology 2012, 143, 1095–1107.e1092. [Google Scholar] [CrossRef]
- Abate-Daga, D.; Lagisetty, K.H.; Tran, E.; Zheng, Z.; Gattinoni, L.; Yu, Z.; Burns, W.R.; Miermont, A.M.; Teper, Y.; Rudloff, U.; et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum. Gene Ther. 2014, 25, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Katari, U.L.; Keirnan, J.M.; Worth, A.C.; Hodges, S.E.; Leen, A.M.; Fisher, W.E.; Vera, J.F. Engineered T cells for pancreatic cancer treatment. HPB 2011, 13, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Maliar, A.; Servais, C.; Waks, T.; Chmielewski, M.; Lavy, R.; Altevogt, P.; Abken, H.; Eshhar, Z. Redirected T cells that target pancreatic adenocarcinoma antigens eliminate tumors and metastases in mice. Gastroenterology 2012, 143, 1375–1384.e1375. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Raj, D.; Nikolaidi, M.; Garces, I.; Lorizio, D.; Castro, N.M.; Caiafa, S.G.; Moore, K.; Brown, N.F.; Kocher, H.M.; Duan, X. CEACAM7 Is an Effective Target for CAR T-cell Therapy of Pancreatic Ductal AdenocarcinomaCEACAM7-directed CAR T-cell Therapy of Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1538–1552. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Fucà, G.; Morano, F.; Gloghini, A.; Corso, S.; Aprile, G.; Perrone, F.; De Vita, F.; Tamborini, E.; Tomasello, G.; et al. Biomarkers of Primary Resistance to Trastuzumab in HER2-Positive Metastatic Gastric Cancer Patients: The AMNESIA Case-Control Study. Clin. Cancer Res. 2018, 24, 1082–1089. [Google Scholar] [CrossRef]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. MEK inhibitors in oncology: A patent review (2015-Present). Expert Opin. Ther. Pat. 2017, 27, 887–906. [Google Scholar] [CrossRef]
- Lin, A.; Feller, E.R. Pancreatic carcinoma as a cause of unexplained pancreatitis: Report of ten cases. Ann. Intern. Med. 1990, 113, 166–167. [Google Scholar] [CrossRef]
- Asimgil, H.; Ertetik, U.; Çevik, N.C.; Ekizce, M.; Doğruöz, A.; Gökalp, M.; Arık-Sever, E.; Istvanffy, R.; Friess, H.; Ceyhan, G.O.; et al. Targeting the undruggable oncogenic KRAS: The dawn of hope. JCI Insight 2022, 7, e153688. [Google Scholar] [CrossRef]
- Cowzer, D.; Zameer, M.; Conroy, M.; Kolch, W.; Duffy, A.G. Targeting KRAS in Pancreatic Cancer. J. Pers. Med. 2022, 12, 1870. [Google Scholar] [CrossRef]
- Middleton, G.W.; Valle, J.W.; Wadsley, J.; Propper, D.; Coxon, F.Y.; Ross, P.J.; Madhusudan, S.; Roques, T.; Cunningham, D.; Corrie, P. A phase III randomized trial of chemoimmunotherapy comprising gemcitabine and capecitabine with or without telomerase vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer. J. Clin. Oncol. 2013, 31, 15. [Google Scholar] [CrossRef]
- Monstein, H.-J.; Ohlsson, B.; Axelson, J. Differential expression of gastrin, cholecystokinin-A and cholecystokinin-B receptor mRNA in human pancreatic cancer cell lines. Scand. J. Gastroenterol. 2001, 36, 738–743. [Google Scholar] [CrossRef]
- Gjertsen, M.K.; Buanes, T.; Rosseland, A.R.; Bakka, A.; Gladhaug, I.; Søreide, O.; Eriksen, J.A.; Møller, M.; Baksaas, I.; Lothe, R.A.; et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int. J. Cancer 2001, 92, 441–450. [Google Scholar] [CrossRef]
- Hiyama, E.; Kodama, T.; Shinbara, K.; Iwao, T.; Itoh, M.; Hiyama, K.; Shay, J.W.; Matsuura, Y.; Yokoyama, T. Telomerase activity is detected in pancreatic cancer but not in benign tumors. Cancer Res. 1997, 57, 326–331. [Google Scholar] [PubMed]
- Gilliam, A.D.; Broome, P.; Topuzov, E.G.; Garin, A.M.; Pulay, I.; Humphreys, J.; Whitehead, A.; Takhar, A.; Rowlands, B.J.; Beckingham, I.J. An international multicenter randomized controlled trial of G17DT in patients with pancreatic cancer. Pancreas 2012, 41, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Fonkoua, L.K.; Moody, T.W. The role of gastrin and CCK receptors in pancreatic cancer and other malignancies. Int. J. Biol. Sci. 2016, 12, 283. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, K.M.; Sampson, J.H. Temozolomide treatment outcomes and immunotherapy efficacy in brain tumor. J. Neurooncol. 2021, 151, 55–62. [Google Scholar] [CrossRef]
- Kyte, J.A.; Gaudernack, G.; Dueland, S.; Trachsel, S.; Julsrud, L.; Aamdal, S. Telomerase peptide vaccination combined with temozolomide: A clinical trial in stage IV melanoma patients. Clin. Cancer Res. 2011, 17, 4568–4580. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Staff, C.; Mozaffari, F.; Frödin, J.E.; Mellstedt, H.; Liljefors, M. Telomerase (GV1001) vaccination together with gemcitabine in advanced pancreatic cancer patients. Int. J. Oncol. 2014, 45, 1293–1303. [Google Scholar] [CrossRef]
- Bernhardt, S.L.; Gjertsen, M.K.; Trachsel, S.; Møller, M.; Eriksen, J.A.; Meo, M.; Buanes, T.; Gaudernack, G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: A dose escalating phase I/II study. Br. J. Cancer 2006, 95, 1474–1482. [Google Scholar] [CrossRef]
- Black, J.W. Reflections on some pilot trials of gastrin receptor blockade in pancreatic cancer. Eur. J. Cancer 2009, 45, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Blanc-Brude, O.P.; Mesri, M.; Wall, N.R.; Plescia, J.; Dohi, T.; Altieri, D.C. Therapeutic targeting of the survivin pathway in cancer: Initiation of mitochondrial apoptosis and suppression of tumor-associated angiogenesis. Clin. Cancer Res. 2003, 9, 2683–2692. [Google Scholar]
- Ishizaki, H.; Manuel, E.R.; Song, G.Y.; Srivastava, T.; Sun, S.; Diamond, D.J.; Ellenhorn, J.D. Modified vaccinia Ankara expressing survivin combined with gemcitabine generates specific antitumor effects in a murine pancreatic carcinoma model. Cancer Immunol. Immunother. 2011, 60, 99–109. [Google Scholar] [CrossRef]
- Zhu, K.; Qin, H.; Cha, S.-C.; Neelapu, S.S.; Overwijk, W.; Lizee, G.A.; Abbruzzese, J.L.; Hwu, P.; Radvanyi, L.; Kwak, L.W. Survivin DNA vaccine generated specific antitumor effects in pancreatic carcinoma and lymphoma mouse models. Vaccine 2007, 25, 7955–7961. [Google Scholar] [CrossRef]
- Osborne, N.; Sundseth, R.; Burks, J.; Cao, H.; Liu, X.; Kroemer, A.H.; Sutton, L.; Cato, A.; Smith, J.P. Gastrin vaccine improves response to immune checkpoint antibody in murine pancreatic cancer by altering the tumor microenvironment. Cancer Immunol. Immunother. 2019, 68, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Kameshima, H.; Tsuruma, T.; Kutomi, G.; Shima, H.; Iwayama, Y.; Kimura, Y.; Imamura, M.; Torigoe, T.; Takahashi, A.; Hirohashi, Y. Immunotherapeutic benefit of α-interferon (IFNα) in survivin2 B-derived peptide vaccination for advanced pancreatic cancer patients. Cancer Sci. 2013, 104, 124–129. [Google Scholar] [CrossRef]
- Nogueira-Ferreira, R.; Vitorino, R.; Ferreira-Pinto, M.J.; Ferreira, R.; Henriques-Coelho, T. Exploring the role of post-translational modifications on protein–protein interactions with survivin. Arch. Biochem. Biophys. 2013, 538, 64–70. [Google Scholar] [CrossRef]
- Bastea, L.I.; Hollant, L.; Döppler, H.R.; Reid, E.M.; Storz, P. Sangivamycin and its derivatives inhibit Haspin-Histone H3-survivin signaling and induce pancreatic cancer cell death. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Shima, H.; Tsurita, G.; Wada, S.; Hirohashi, Y.; Yasui, H.; Hayashi, H.; Miyakoshi, T.; Watanabe, K.; Murai, A.; Asanuma, H.; et al. Randomized phase II trial of survivin 2B peptide vaccination for patients with HLA-A24-positive pancreatic adenocarcinoma. Cancer Sci. 2019, 110, 2378–2385. [Google Scholar] [CrossRef]
- Wobser, M.; Keikavoussi, P.; Kunzmann, V.; Weininger, M.; Andersen, M.H.; Becker, J.C. Complete remission of liver metastasis of pancreatic cancer under vaccination with a HLA-A2 restricted peptide derived from the universal tumor antigen survivin. Cancer Immunol. Immunother. 2006, 55, 1294–1298. [Google Scholar] [CrossRef]
- Shevtsov, M.; Multhoff, G. Heat shock protein–peptide and HSP-based immunotherapies for the treatment of cancer. Front. Immunol. 2016, 7, 171. [Google Scholar] [CrossRef]
- Kubo, T.; Tsurita, G.; Hirohashi, Y.; Yasui, H.; Ota, Y.; Watanabe, K.; Murai, A.; Matsuo, K.; Asanuma, H.; Shima, H.; et al. Immunohistological analysis of pancreatic carcinoma after vaccination with survivin 2B peptide: Analysis of an autopsy series. Cancer Sci. 2019, 110, 2386–2395. [Google Scholar] [CrossRef]
- Oki, Y.; Younes, A. Heat shock protein-based cancer vaccines. Expert Rev. Vaccines 2004, 3, 403–411. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Cayado-Gutierrez, N.; Maccioni, M.; Cuello-Carrion, F.D. Heat shock proteins (HSPs) based anti-cancer vaccines. Curr. Mol. Med. 2012, 12, 1183–1197. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef]
- Easton, D.P.; Kaneko, Y.; Subjeck, J.R. The hsp110 and Grp1 70 stress proteins: Newly recognized relatives of the Hsp70s. Cell Stress Chaperones 2000, 5, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.G.; Livingston, P.O.; Lewis, J.J.; Janetzki, S.; Klimstra, D.; Desantis, D.; Srivastava, P.K.; Brennan, M.F. A phase I pilot study of autologous heat shock protein vaccine HSPPC-96 in patients with resected pancreatic adenocarcinoma. Dig. Dis. Sci. 2007, 52, 1964–1972. [Google Scholar] [CrossRef]
- Ciborowski, P.; Finn, O.J. Recombinant epithelial cell mucin (MUC-1) expressed in baculovirus resembles antigenically tumor associated mucin, target for cancer immunotherapy. Biomed. Pept. Proteins Nucleic Acids 1995, 1, 193–198. [Google Scholar]
- Kondo, H.; Hazama, S.; Kawaoka, T.; Yoshino, S.; Yoshida, S.; Tokuno, K.; Takashima, M.; Ueno, T.; Hinoda, Y.; Oka, M. Adoptive immunotherapy for pancreatic cancer using MUC1 peptide-pulsed dendritic cells and activated T lymphocytes. Anticancer Res. 2008, 28, 379–387. [Google Scholar] [PubMed]
- Shindo, Y.; Hazama, S.; Maeda, Y.; Matsui, H.; Iida, M.; Suzuki, N.; Yoshimura, K.; Ueno, T.; Yoshino, S.; Sakai, K.; et al. Adoptive immunotherapy with MUC1-mRNA transfected dendritic cells and cytotoxic lymphocytes plus gemcitabine for unresectable pancreatic cancer. J. Transl. Med. 2014, 12, 175. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kim-Schulze, S.; Manson, K.; DeRaffele, G.; Mitcham, J.; Seo, K.S.; Kim, D.W.; Marshall, J. Poxvirus-based vaccine therapy for patients with advanced pancreatic cancer. J. Transl. Med. 2007, 5, 60. [Google Scholar] [CrossRef]
- Madan, R.A.; Arlen, P.M.; Gulley, J.L. PANVAC-VF: Poxviral-based vaccine therapy targeting CEA and MUC1 in carcinoma. Expert Opin. Biol. Ther. 2007, 7, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Brockstedt, D.G.; Nir-Paz, R.; Hampl, J.; Mathur, S.; Nemunaitis, J.; Sterman, D.H.; Hassan, R.; Lutz, E.; Moyer, B.; et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase I studies of safety and immune induction. Clin. Cancer Res. 2012, 18, 858–868. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Crocenzi, T.; Durham, J.N.; Sugar, E.A.; Wu, A.A.; Onners, B.; Nauroth, J.M.; Anders, R.A.; Fertig, E.J.; Laheru, D.A.; et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with or without Nivolumab in Patients with Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 3578–3588. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse, M.A.; Zeh, H.J.; Weekes, C.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef]
- Kim, V.M.; Blair, A.B.; Lauer, P.; Foley, K.; Che, X.; Soares, K.; Xia, T.; Muth, S.T.; Kleponis, J.; Armstrong, T.D. Anti-pancreatic tumor efficacy of a Listeria-based, Annexin A2-targeting immunotherapy in combination with anti-PD-1 antibodies. J. Immunother. Cancer 2019, 7, 132. [Google Scholar] [CrossRef] [PubMed]
- Selvanesan, B.C.; Chandra, D.; Quispe-Tintaya, W.; Jahangir, A.; Patel, A.; Meena, K.; Alves Da Silva, R.A.; Friedman, M.; Gabor, L.; Khouri, O.; et al. delivers tetanus toxoid protein to pancreatic tumors and induces cancer cell death in mice. Sci. Transl. Med. 2022, 14, eabc1600. [Google Scholar] [CrossRef]
- Okamoto, M.; Kobayashi, M.; Yonemitsu, Y.; Koido, S.; Homma, S. Dendritic cell-based vaccine for pancreatic cancer in Japan. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 133. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yanagimoto, H.; Satoi, S.; Toyokawa, H.; Yamao, J.; Kim, S.; Terakawa, N.; Takahashi, K.; Kwon, A.H. Circulating myeloid dendritic cells as prognostic factors in patients with pancreatic cancer who have undergone surgical resection. J. Surg. Res. 2012, 173, 299–308. [Google Scholar] [CrossRef]
- Tjomsland, V.; Sandström, P.; Spångeus, A.; Messmer, D.; Emilsson, J.; Falkmer, U.; Falkmer, S.; Magnusson, K.E.; Borch, K.; Larsson, M. Pancreatic adenocarcinoma exerts systemic effects on the peripheral blood myeloid and plasmacytoid dendritic cells: An indicator of disease severity? BMC Cancer 2010, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Pei, Q.; Pan, J.; Zhu, H.; Ding, X.; Liu, W.; Lv, Y.; Zou, X.; Luo, H. Gemcitabine-treated pancreatic cancer cell medium induces the specific CTL antitumor activity by stimulating the maturation of dendritic cells. Int. Immunopharmacol. 2014, 19, 10–16. [Google Scholar] [CrossRef]
- Pei, Q.; Pan, J.; Ding, X.; Wang, J.; Zou, X.; Lv, Y. Gemcitabine sensitizes pancreatic cancer cells to the CTLs antitumor response induced by BCG-stimulated dendritic cells via a Fas-dependent pathway. Pancreatology 2015, 15, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Tsukada, J.; Tomoda, T.; Takahashi, H.; Imai, K.; Shimamura, K.; Sunamura, M.; Yonemitsu, Y.; Shimodaira, S.; Koido, S.; et al. Clinical and immunologic evaluation of dendritic cell-based immunotherapy in combination with gemcitabine and/or S-1 in patients with advanced pancreatic carcinoma. Pancreas 2012, 41, 195–205. [Google Scholar] [CrossRef]
- Mayanagi, S.; Kitago, M.; Sakurai, T.; Matsuda, T.; Fujita, T.; Higuchi, H.; Taguchi, J.; Takeuchi, H.; Itano, O.; Aiura, K.; et al. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci. 2015, 106, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Takakura, K.; Mori, M.; Yoshizaki, S.; Tsukinaga, S.; Odahara, S.; Koyama, S.; Imazu, H.; et al. Treatment with chemotherapy and dendritic cells pulsed with multiple Wilms’ tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin. Cancer Res. 2014, 20, 4228–4239. [Google Scholar] [CrossRef]
- Koido, S.; Homma, S.; Okamoto, M.; Takakura, K.; Gong, J.; Sugiyama, H.; Ohkusa, T.; Tajiri, H. Chemoimmunotherapy targeting Wilms’ tumor 1 (WT1)-specific cytotoxic T lymphocyte and helper T cell responses for patients with pancreatic cancer. Oncoimmunology 2014, 3, e958950. [Google Scholar] [CrossRef][Green Version]
- Takakura, K.; Koido, S.; Kan, S.; Yoshida, K.; Mori, M.; Hirano, Y.; Ito, Z.; Kobayashi, H.; Takami, S.; Matsumoto, Y.; et al. Prognostic markers for patient outcome following vaccination with multiple MHC Class I/II-restricted WT1 peptide-pulsed dendritic cells plus chemotherapy for pancreatic cancer. Anticancer Res. 2015, 35, 555–562. [Google Scholar] [PubMed]
- Katsuda, M.; Miyazawa, M.; Ojima, T.; Katanuma, A.; Hakamada, K.; Sudo, K.; Asahara, S.; Endo, I.; Ueno, M.; Hara, K.; et al. A double-blind randomized comparative clinical trial to evaluate the safety and efficacy of dendritic cell vaccine loaded with WT1 peptides (TLP0-001) in combination with S-1 in patients with advanced pancreatic cancer refractory to standard chemotherapy. Trials 2019, 20, 242. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.P.; Klaase, L.; Vink, M.; Dumas, J.; Bezemer, K.; van Krimpen, A.; van der Breggen, R.; Wismans, L.V.; Doukas, M.; de Koning, W.; et al. Autologous dendritic cells pulsed with allogeneic tumour cell lysate induce tumour-reactive T-cell responses in patients with pancreatic cancer: A phase I study. Eur. J. Cancer 2022, 169, 20–31. [Google Scholar] [CrossRef] [PubMed]
- McCormick, K.A.; Coveler, A.L.; Rossi, G.R.; Vahanian, N.N.; Link, C.; Chiorean, E.G. Pancreatic cancer: Update on immunotherapies and algenpantucel-L. Hum. Vaccines Immunother. 2016, 12, 563–575. [Google Scholar] [CrossRef]
- Cintolo, J.A.; Datta, J.; Mathew, S.J.; Czerniecki, B.J. Dendritic cell-based vaccines: Barriers and opportunities. Future Oncol. 2012, 8, 1273–1299. [Google Scholar] [CrossRef] [PubMed]
- Marelli, G.; Dunmall, L.S.C.; Yuan, M.; Di Gioia, C.; Miao, J.; Cheng, Z.; Zhang, Z.; Liu, P.; Ahmed, J.; Gangeswaran, R. A systemically deliverable Vaccinia virus with increased capacity for intertumoral and intratumoral spread effectively treats pancreatic cancer. J. Immunother. Cancer 2021, 9, e001624. [Google Scholar] [PubMed]
- Abd-Aziz, N.; Poh, C.L. Development of oncolytic viruses for cancer therapy. Transl. Res. 2021, 237, 98–123. [Google Scholar] [CrossRef]
- Timmer, F.E.F.; Geboers, B.; Nieuwenhuizen, S.; Dijkstra, M.; Schouten, E.A.C.; Puijk, R.S.; de Vries, J.J.J.; van den Tol, M.P.; Bruynzeel, A.M.E.; Streppel, M.M.; et al. Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers 2021, 13, 4138. [Google Scholar] [CrossRef]
- Mahalingam, D.; Wilkinson, G.A.; Eng, K.H.; Fields, P.; Raber, P.; Moseley, J.L.; Cheetham, K.; Coffey, M.; Nuovo, G.; Kalinski, P.; et al. Pembrolizumab in Combination with the Oncolytic Virus Pelareorep and Chemotherapy in Patients with Advanced Pancreatic Adenocarcinoma: A Phase Ib Study. Clin. Cancer Res. 2020, 26, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Noonan, A.M.; Farren, M.R.; Geyer, S.M.; Huang, Y.; Tahiri, S.; Ahn, D.; Mikhail, S.; Ciombor, K.K.; Pant, S.; Aparo, S.; et al. Randomized Phase 2 Trial of the Oncolytic Virus Pelareorep (Reolysin) in Upfront Treatment of Metastatic Pancreatic Adenocarcinoma. Mol. Ther. 2016, 24, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Hirooka, Y.; Kasuya, H.; Ishikawa, T.; Kawashima, H.; Ohno, E.; Villalobos, I.B.; Naoe, Y.; Ichinose, T.; Koyama, N.; Tanaka, M.; et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer 2018, 18, 596. [Google Scholar] [CrossRef]
- Singh, H.M.; Leber, M.F.; Bossow, S.; Engeland, C.E.; Dessila, J.; Grossardt, C.; Zaoui, K.; Bell, J.C.; Jäger, D.; von Kalle, C. MicroRNA-sensitive oncolytic measles virus for chemovirotherapy of pancreatic cancer. Mol. Ther.-Oncolytics 2021, 21, 340–355. [Google Scholar] [PubMed]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Pasca di Magliano, M. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017, 66, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef]
- Clark, C.E.; Beatty, G.L.; Vonderheide, R.H. Immunosurveillance of pancreatic adenocarcinoma: Insights from genetically engineered mouse models of cancer. Cancer Lett. 2009, 279, 1–7. [Google Scholar] [CrossRef]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Di Caro, G.; Cortese, N.; Castino, G.F.; Grizzi, F.; Gavazzi, F.; Ridolfi, C.; Capretti, G.; Mineri, R.; Todoric, J.; Zerbi, A.; et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut 2016, 65, 1710–1720. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Pfirschke, C.; Zilionis, R.; Engblom, C.; Messemaker, M.; Zou, A.E.; Rickelt, S.; Gort-Freitas, N.A.; Lin, Y.; Bill, R.; Siwicki, M.; et al. Macrophage-Targeted Therapy Unlocks Antitumoral Cross-talk between IFNγ-Secreting Lymphocytes and IL12-Producing Dendritic Cells. Cancer Immunol. Res. 2022, 10, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Almahariq, M.F.; Quinn, T.J.; Kesarwani, P.; Kant, S.; Miller, C.R.; Chinnaiyan, P. Inhibition of Colony-Stimulating Factor-1 Receptor Enhances the Efficacy of Radiotherapy and Reduces Immune Suppression in Glioblastoma. In Vivo 2021, 35, 119–129. [Google Scholar] [CrossRef]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef]
- Lu, J.; Liu, X.; Liao, Y.P.; Salazar, F.; Sun, B.; Jiang, W.; Chang, C.H.; Jiang, J.; Wang, X.; Wu, A.M.; et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat. Commun. 2017, 8, 1811. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Wang, W.; Wang, S.; Yang, T.; Zhang, G.; Wang, D.; Ju, R.; Lu, Y.; Wang, H.; Wang, L. Tumor microenvironment remodeling and tumor therapy based on M2-like tumor associated macrophage-targeting nano-complexes. Theranostics 2021, 11, 2892–2916. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Yang, T.; Yu, J.; Li, F.; Zhao, X. Integrated transcriptional analysis reveals macrophage heterogeneity and macrophage-tumor cell interactions in the progression of pancreatic ductal adenocarcinoma. BMC Cancer 2023, 23, 199. [Google Scholar] [CrossRef]
- Anu RI, S.K.-K.a.K.K. The immunomodulatory role of IDO1- Kynurenine-NAD+ pathway in switching cold tumor microenvironment in PDAC. Front. Oncol. 2023, 13, 1142838. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: An open-label, multicentre, phase 1b study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef]
- Byrne, K.T.; Betts, C.B.; Mick, R.; Sivagnanam, S.; Bajor, D.L.; Laheru, D.A.; Chiorean, E.G.; O’Hara, M.H.; Liudahl, S.M.; Newcomb, C.; et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 4574–4586. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.P.; van Montfoort, N.; Kinderman, P.; Lukkes, M.; Klaase, L.; van Nimwegen, M.; van Gulijk, M.; Dumas, J.; Mustafa, D.A.M.; Lievense, S.L.A.; et al. Dendritic cell vaccination and CD40-agonist combination therapy licenses T cell-dependent antitumor immunity in a pancreatic carcinoma murine model. J. Immunother. Cancer 2020, 8, e000772. [Google Scholar] [CrossRef]
- Strobel, O.; Lorenz, P.; Hinz, U.; Gaida, M.; König, A.K.; Hank, T.; Niesen, W.; Kaiser, J.R.; Al-Saeedi, M.; Bergmann, F.; et al. Actual Five-year Survival After Upfront Resection for Pancreatic Ductal Adenocarcinoma: Who Beats the Odds? Ann. Surg. 2022, 275, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Heatherton, K.R.; O’Connell, K.P.; Alexander, I.S.; Katz, S.C. Assessing the Future of Solid Tumor Immunotherapy. Biomedicines 2022, 10, 655. [Google Scholar] [CrossRef]
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384. [Google Scholar] [CrossRef]
- Rucki, A.A.; Zheng, L. Pancreatic cancer stroma: Understanding biology leads to new therapeutic strategies. World J. Gastroenterol. WJG 2014, 20, 2237. [Google Scholar] [CrossRef]
- Aiello, N.M.; Bajor, D.L.; Norgard, R.J.; Sahmoud, A.; Bhagwat, N.; Pham, M.N.; Cornish, T.C.; Iacobuzio-Donahue, C.A.; Vonderheide, R.H.; Stanger, B.Z. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat. Commun. 2016, 7, 12819. [Google Scholar] [CrossRef]
- Gkolfakis, P.; Crinò, S.F.; Tziatzios, G.; Ramai, D.; Papaefthymiou, A.; Papanikolaou, I.S.; Triantafyllou, K.; Arvanitakis, M.; Lisotti, A.; Fusaroli, P.; et al. Comparative diagnostic performance of end-cutting fine-needle biopsy needles for EUS tissue sampling of solid pancreatic masses: A network meta-analysis. Gastrointest. Endosc. 2022, 95, 1067–1077.e1015. [Google Scholar] [CrossRef]
- Madurantakam Royam, M.; Ramesh, R.; Shanker, R.; Sabarimurugan, S.; Kumarasamy, C.; Ramesh, N.; Gothandam, K.M.; Baxi, S.; Gupta, A.; Krishnan, S.; et al. miRNA Predictors of Pancreatic Cancer Chemotherapeutic Response: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 900. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.N.; Goodwin, R.A.; Vickers, M.M. mutated pancreatic cancer: A change is coming. World J. Gastroenterol. 2021, 27, 1943–1958. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Hechtman, J.F. Tumour response to TRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK gene fusion. Ann. Oncol. 2019, 30, viii36–viii40. [Google Scholar] [CrossRef] [PubMed]
- Di Federico, A.; Mosca, M.; Pagani, R.; Carloni, R.; Frega, G.; De Giglio, A.; Rizzo, A.; Ricci, D.; Tavolari, S.; Di Marco, M.; et al. Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers 2022, 14, 2429. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Odintsov, I.; Espinosa-Cotton, M.; Khodos, I.; Sisso, W.J.; Mattar, M.S.; Lui, A.J.W.; Vojnic, M.; Shameem, S.H.; Chauhan, T.; et al. Zenocutuzumab, a HER2xHER3 Bispecific Antibody, Is Effective Therapy for Tumors Driven by NRG1 Gene Rearrangements. Cancer Discov. 2022, 12, 1233–1247. [Google Scholar] [CrossRef]
- Huff, A.L.; Zaidi, N. Vaccine boosts T cells that target pancreatic tumours. Nature 2023, 618, 37–38. [Google Scholar] [CrossRef]
- Pompella, L.; Tirino, G.; Pappalardo, A.; Caterino, M.; Ventriglia, A.; Nacca, V.; Orditura, M.; Ciardiello, F.; De Vita, F. Pancreatic cancer molecular classifications: From bulk genomics to single cell analysis. Int. J. Mol. Sci. 2020, 21, 2814. [Google Scholar] [CrossRef] [PubMed]
- Cardenes, H.R.; Chiorean, E.G.; DeWitt, J.; Schmidt, M.; Loehrer, P. Locally advanced pancreatic cancer: Current therapeutic approach. Oncologist 2006, 11, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.J.; Morra, D.; Caesar, M.; Carter, M.W.; Abrams, H. Understanding hospital and emergency department congestion: An examination of inpatient admission trends and bed resources. Can. J. Emerg. Med. 2010, 12, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Tetzlaff, J.; Pham, B.; Brehaut, J.; Moher, D. Non-Cochrane vs. Cochrane reviews were twice as likely to have positive conclusion statements: Cross-sectional study. J. Clin. Epidemiol. 2009, 62, 380–386.e381. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chouari, T.; La Costa, F.S.; Merali, N.; Jessel, M.-D.; Sivakumar, S.; Annels, N.; Frampton, A.E. Advances in Immunotherapeutics in Pancreatic Ductal Adenocarcinoma. Cancers 2023, 15, 4265. https://doi.org/10.3390/cancers15174265
Chouari T, La Costa FS, Merali N, Jessel M-D, Sivakumar S, Annels N, Frampton AE. Advances in Immunotherapeutics in Pancreatic Ductal Adenocarcinoma. Cancers. 2023; 15(17):4265. https://doi.org/10.3390/cancers15174265
Chicago/Turabian StyleChouari, Tarak, Francesca Soraya La Costa, Nabeel Merali, Maria-Danae Jessel, Shivan Sivakumar, Nicola Annels, and Adam E. Frampton. 2023. "Advances in Immunotherapeutics in Pancreatic Ductal Adenocarcinoma" Cancers 15, no. 17: 4265. https://doi.org/10.3390/cancers15174265
APA StyleChouari, T., La Costa, F. S., Merali, N., Jessel, M.-D., Sivakumar, S., Annels, N., & Frampton, A. E. (2023). Advances in Immunotherapeutics in Pancreatic Ductal Adenocarcinoma. Cancers, 15(17), 4265. https://doi.org/10.3390/cancers15174265