
Vascular Immune Evasion of Mesenchymal Glioblastoma Is Mediated by Interaction and Regulation of VE-Cadherin on PD-L1

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Database Analyses and RNA-Sequencing
2.3. Generation of 293T Cells Stably Expressing PD-L1 and VE-Cad
2.4. Gene Knockdown
2.5. Cross-Link Assay, Immunoblotting and Immunoprecipitation
2.6. Cytosol and Nuclear Fragment Protein Isolation
2.7. Quantitative Real-Time PCR (qPCR)
2.8. Cell Adhesion and Migration
2.9. Cell Permeability Assay
2.10. Tube Formation Assay
2.11. Cell Viability Assay
2.12. Human Tissue Collection, Immunohistochemistry and Immunofluorescence
2.13. Immunocytochemistry
2.14. Dual Luciferase Reporter Assay
2.15. Intracranial Xenograft Tumor Models
2.16. Statistical Analysis
3. Results
3.1. YKL-40 Expressed by Tumor Cells Is Correlated with Vascular PD-L1 Levels in mGBM
3.2. YKL-40 Over-Expressed by GL261 Cells Induces PD-L1 + Vessel Angiogenesis and Restricts CTL Infiltration In Vivo
3.3. YKL-40 Regulates PD-L1 through Activating the VE-Cad/β-Catenin/LEF Pathway in Endothelial Cells
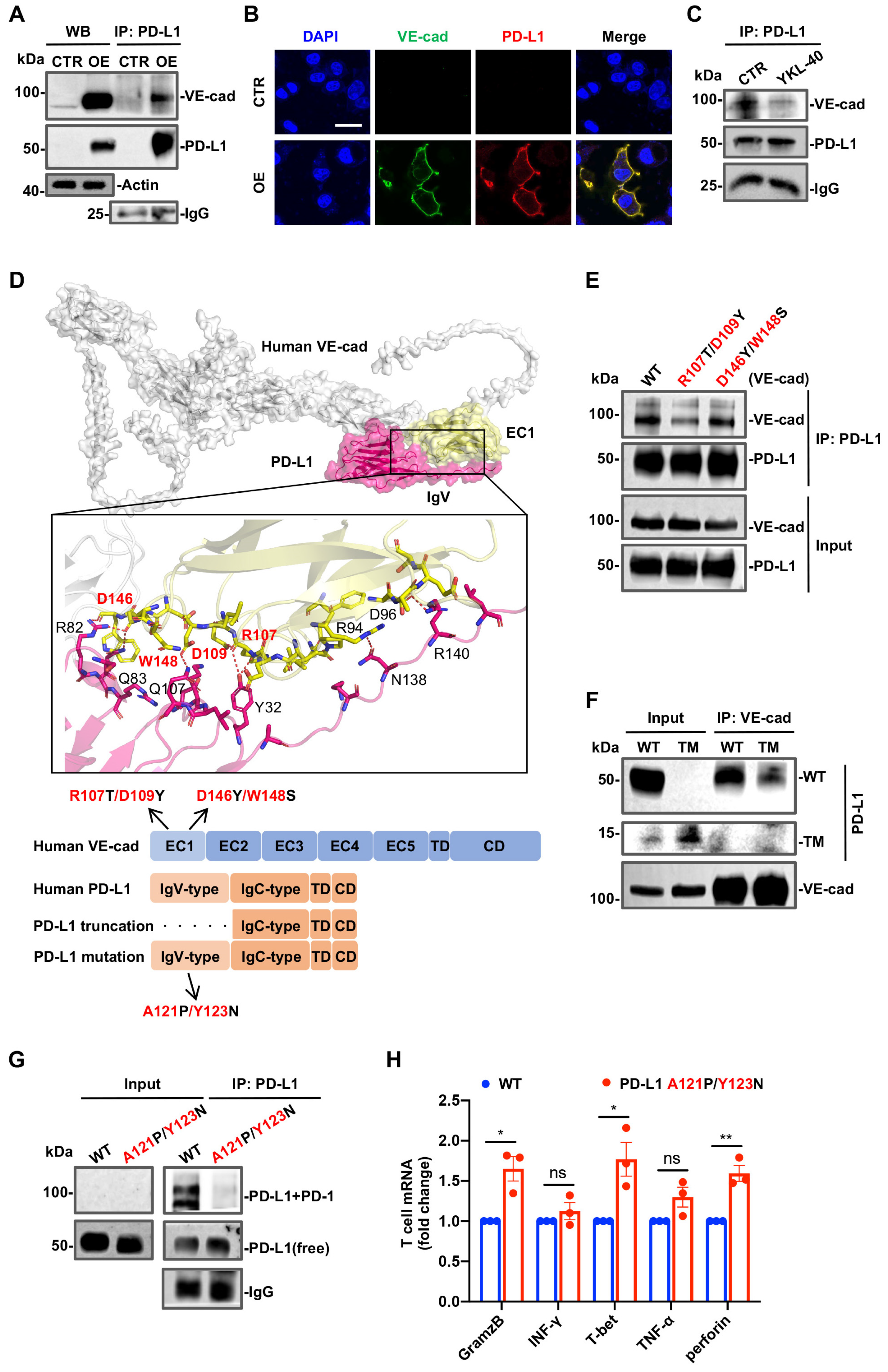
3.4. Physical Interactions between PD-L1 and VE-Cad or PD-1
3.5. YKL-40 Promotes the Interaction between PD-L1 and PD-1, Which Drives Vascular Immune Evasion
3.6. VE-Cad and PD-L1 shRNA in HMVECs Restore Cytotoxicity of CTLs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Van den Bent, M.J.; Bromberg, J.E. Neuro-oncology: The many challenges of treating elderly glioblastoma patients. Nat. Rev. Neurol. 2015, 11, 374–375. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Rimkus, T.K.; Carpenter, R.L.; Sirkisoon, S.; Zhu, D.; Pasche, B.C.; Chan, M.D.; Lesser, G.J.; Tatter, S.B.; Watabe, K.; Debinski, W.; et al. Truncated Glioma-Associated Oncogene Homolog 1 (tGLI1) Mediates Mesenchymal Glioblastoma via Transcriptional Activation of CD44. Cancer Res. 2018, 78, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Jahangiri, A.; Chen, W.; Nguyen, A.T.; Yagnik, G.; Pereira, M.P.; Jain, S.; Garcia, J.H.; Shah, S.S.; Wadhwa, H.; et al. Clonal ZEB1-Driven Mesenchymal Transition Promotes Targetable Oncologic Antiangiogenic Therapy Resistance. Cancer Res. 2020, 80, 1498–1511. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Antiangiogenic therapies for high-grade glioma. Nat. Rev. Neurol. 2009, 5, 610–620. [Google Scholar] [CrossRef]
- Reardon, D.A.; Wen, P.Y. Glioma in 2014: Unravelling tumour heterogeneity-implications for therapy. Nat. Rev. Clin. Oncol. 2015, 12, 69–70. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef]
- Ruscetti, M.; Morris, J.P.T.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.J.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441.e21. [Google Scholar] [CrossRef]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Francescone, R.A.; Scully, S.; Faibish, M.; Taylor, S.L.; Oh, D.; Moral, L.; Yan, W.; Bentley, B.; Shao, R. Role of YKL-40 in the angiogenesis, radioresistance, and progression of glioblastoma. J. Biol. Chem. 2011, 286, 15332–15343. [Google Scholar] [CrossRef]
- Junker, N.; Johansen, J.S.; Hansen, L.T.; Lund, E.L.; Kristjansen, P.E. Regulation of YKL-40 expression during genotoxic or microenvironmental stress in human glioblastoma cells. Cancer Sci. 2005, 96, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Pelloski, C.E.; Mahajan, A.; Maor, M.; Chang, E.L.; Woo, S.; Gilbert, M.; Colman, H.; Yang, H.; Ledoux, A.; Blair, H.; et al. YKL-40 expression is associated with poorer response to radiation and shorter overall survival in glioblastoma. Clin. Cancer Res. 2005, 11, 3326–3334. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Hamel, K.; Petersen, L.; Cao, Q.J.; Arenas, R.B.; Bigelow, C.; Bentley, B.; Yan, W. YKL-40, a secreted glycoprotein, promotes tumor angiogenesis. Oncogene 2009, 28, 4456–4468. [Google Scholar] [CrossRef] [PubMed]
- Shao, R. YKL-40 acts as an angiogenic factor to promote tumor angiogenesis. Front. Physiol. 2013, 4, 122. [Google Scholar] [CrossRef]
- Francescone, R.; Ngernyuang, N.; Yan, W.; Bentley, B.; Shao, R. Tumor-derived mural-like cells coordinate with endothelial cells: Role of YKL-40 in mural cell-mediated angiogenesis. Oncogene 2014, 33, 2110–2122. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kuang, X.; Zhang, Y.; Ye, Y.; Li, J.; Liang, L.; Xie, Z.; Weng, L.; Guo, J.; Li, H.; et al. ADORA1 Inhibition Promotes Tumor Immune Evasion by Regulating the ATF3-PD-L1 Axis. Cancer Cell 2020, 37, 324–339.e328. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.L.; Sparbier, C.E.; Chan, Y.C.; Williamson, J.C.; Woods, K.; Beavis, P.A.; Lam, E.Y.N.; Henderson, M.A.; Bell, C.C.; Stolzenburg, S.; et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 2017, 549, 101–105. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9, eaak9670. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef]
- Shao, R.; Guo, X. Human microvascular endothelial cells immortalized with human telomerase catalytic protein: A model for the study of in vitro angiogenesis. Biochem. Biophys. Res. Commun. 2004, 321, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Scully, S.; Francescone, R.; Faibish, M.; Bentley, B.; Taylor, S.L.; Oh, D.; Schapiro, R.; Moral, L.; Yan, W.; Shao, R. Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J. Neurosci. 2012, 32, 12950–12960. [Google Scholar] [CrossRef] [PubMed]
- DiDomenico, J.; Lamano, J.B.; Oyon, D.; Li, Y.; Veliceasa, D.; Kaur, G.; Ampie, L.; Choy, W.; Lamano, J.B.; Bloch, O. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 2018, 7, e1448329. [Google Scholar] [CrossRef]
- Wang, S.; Yao, F.; Lu, X.; Li, Q.; Su, Z.; Lee, J.H.; Wang, C.; Du, L. Temozolomide promotes immune escape of GBM cells via upregulating PD-L1. Am. J. Cancer Res. 2019, 9, 1161–1171. [Google Scholar]
- Preusser, M.; Berghoff, A.S.; Wick, W.; Weller, M. Clinical Neuropathology mini-review 6-2015: PD-L1: Emerging biomarker in glioblastoma? Clin. Neuropathol. 2015, 34, 313–321. [Google Scholar] [CrossRef]
- Du, L.; Lee, J.H.; Jiang, H.; Wang, C.; Wang, S.; Zheng, Z.; Shao, F.; Xu, D.; Xia, Y.; Li, J.; et al. β-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J. Exp. Med. 2020, 217, e20191115. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Gomez, G.G.; Read, S.B.; Gerschenson, L.E.; Santoli, D.; Zweifach, A.; Kruse, C.A. Interactions of the allogeneic effector leukemic T cell line, TALL-104, with human malignant brain tumors. Neuro Oncol. 2004, 6, 83–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kruse, C.A.; Visonneau, S.; Kleinschmidt-DeMasters, B.K.; Gup, C.J.; Gomez, G.G.; Paul, D.B.; Santoli, D. The human leukemic T-cell line, TALL-104, is cytotoxic to human malignant brain tumors and traffics through brain tissue: Implications for local adoptive immunotherapy. Cancer Res. 2000, 60, 5731–5739. [Google Scholar] [PubMed]
- Cesano, A.; Visonneau, S.; Pasquini, S.; Rovera, G.; Santoli, D. Antitumor efficacy of a human major histocompatibility complex nonrestricted cytotoxic T-cell line (TALL-104) in immunocompetent mice bearing syngeneic leukemia. Cancer Res. 1996, 56, 4444–4452. [Google Scholar]
- Geoerger, B.; Tang, C.B.; Cesano, A.; Visonneau, S.; Marwaha, S.; Judy, K.D.; Sutton, L.N.; Santoli, D.; Phillips, P.C. Antitumor activity of a human cytotoxic T-cell line (TALL-104) in brain tumor xenografts. Neuro Oncol. 2000, 2, 103–113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suffee, N.; Hlawaty, H.; Meddahi-Pelle, A.; Maillard, L.; Louedec, L.; Haddad, O.; Martin, L.; Laguillier, C.; Richard, B.; Oudar, O.; et al. RANTES/CCL5-induced pro-angiogenic effects depend on CCR1, CCR5 and glycosaminoglycans. Angiogenesis 2012, 15, 727–744. [Google Scholar] [CrossRef]
- Wang, S.W.; Liu, S.C.; Sun, H.L.; Huang, T.Y.; Chan, C.H.; Yang, C.Y.; Yeh, H.I.; Huang, Y.L.; Chou, W.Y.; Lin, Y.M.; et al. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis 2015, 36, 104–114. [Google Scholar] [CrossRef]
- Xue, S.; Hu, M.; Li, P.; Ma, J.; Xie, L.; Teng, F.; Zhu, Y.; Fan, B.; Mu, D.; Yu, J. Relationship between expression of PD-L1 and tumor angiogenesis, proliferation, and invasion in glioma. Oncotarget 2017, 8, 49702–49712. [Google Scholar] [CrossRef]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef]
- Liu, Y.; Zugazagoitia, J.; Ahmed, F.S.; Henick, B.S.; Gettinger, S.N.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Immune Cell PD-L1 Colocalizes with Macrophages and Is Associated with Outcome in PD-1 Pathway Blockade Therapy. Clin. Cancer Res. 2020, 26, 970–977. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Ju, X.; Zhang, H.; Zhou, Z.; Wang, Q. Regulation of PD-L1 expression in cancer and clinical implications in immunotherapy. Am. J. Cancer Res. 2020, 10, 1–11. [Google Scholar]
- Carmeliet, P.; Lampugnani, M.G.; Moons, L.; Breviario, F.; Compernolle, V.; Bono, F.; Balconi, G.; Spagnuolo, R.; Oosthuyse, B.; Dewerchin, M.; et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999, 98, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Orlova, V.V.; Economopoulou, M.; Lupu, F.; Santoso, S.; Chavakis, T. Junctional adhesion molecule-C regulates vascular endothelial permeability by modulating VE-cadherin-mediated cell-cell contacts. J. Exp. Med. 2006, 203, 2703–2714. [Google Scholar] [CrossRef]
- Lin, D.Y.; Tanaka, Y.; Iwasaki, M.; Gittis, A.G.; Su, H.P.; Mikami, B.; Okazaki, T.; Honjo, T.; Minato, N.; Garboczi, D.N. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef]
- Jacobelli, J.; Estin Matthews, M.; Chen, S.; Krummel, M.F. Activated T cell trans-endothelial migration relies on myosin-IIA contractility for squeezing the cell nucleus through endothelial cell barriers. PLoS ONE 2013, 8, e75151. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case No. | Age | Sex | Protein Levels a | DFS b (Months) | Tumor Size (mm) | ||||
|---|---|---|---|---|---|---|---|---|---|
| YKL-40 | Vimentin | PD-L1 | VE-Cad | CD8 | |||||
| 1 | 69 | Male | 5 | 4.5 | 0 | 3 | 0 | 5 | 28 × 35 |
| 2 | 53 | Female | 5 | 2.5 | 0 | 0 | 0 | 9 | 40 × 51 × 39 |
| 3 | 72 | Male | 5 | 0 | 5 | 4 | 3 | 2 | 26 × 34 |
| 4 | 59 | Male | 5 | 4.5 | 3 | 5 | 3 | 8 | 62 × 30 |
| 5 | 69 | Female | 3.5 | 4 | 0 | 4 | 0 | 6 | 35 × 39 × 48 |
| 6 | 46 | Female | 5 | 6 | 0 | 3 | 5 | 10 | 50 × 44 |
| 7 | 33 | Male | 3.5 | 3 | 0 | 4 | ND | 41 | 2.5 |
| 8 | 52 | Male | 5 | ND | 4 | 2 | 0 | 11 | 47 × 41 × 34 |
| 9 | 60 | Male | 2.5 | 3 | 0 | 2 | 3 | 9 | 42 × 60 |
| 10 | 53 | Male | 2.5 | 2.5 | 0 | 3 | 2 | 32 | 57.5 × 50 |
| 11 | 47 | Male | 5 | 5 | 5 | 4 | 0 | 10 | 45 × 15 × 21 |
| 12 | 57 | Female | 3 | 3.5 | 0 | 6 | 4 | 7 | 33 × 34 × 34 |
| 13 | 40 | Male | 3.5 | 3.5 | 0 | 4 | 0 | 4 | 29 × 53 |
| 14 | 51 | Male | 4 | 5 | 0 | 2 | 0 | 2 | 38 × 43 × 55 |
| 15 | 35 | Male | 2.5 | 4.5 | 0 | 0 | 0 | >48 | 47 × 45 × 50 |
| 16 | 51 | Male | 5 | 4 | 0 | 2 | 4 | 5 | 44.5 × 40.9 × 54.9 |
| 17 | 52 | Male | 4 | 5.5 | 5 | 5 | 5 | 8 | 65 × 43 |
| 18 | 40 | Female | 2 | 5.5 | 3 | 2 | 4 | >19 | 21 × 25 × 30 |
| 19 | 47 | Male | 4.5 | 5.5 | 0 | 5 | 5 | 8 | 26 × 33 |
| 20 | 44 | Male | ND d | ND | 2 | 3 | 3 | 5 | 40 × 55 |
| 21 | 62 | Female | 3.5 | 6 | 2 | 3 | 5 | 3 | 48 × 48 × 38 |
| 22 | 48 | Female | 5 | 5 | 2 | 4 | 4 | NA e | 29 × 19 |
| 23 | 66 | Male | 3 | 5.5 | 2 | 4 | 4 | NA | 47 × 67 |
| 24 | 43 | Male | 5.5 | 5.5 | 6 | 4 | 3 | NA | 50 × 40 |
| 25 | 71 | Female | 5 | 4.5 | 0 | 5 | 0 | 3.5 | 35 × 50 |
| 26 | 62 | Male | 4.5 | 4.5 | 0 | 6 | 3 | NA | 33 × 30 |
| 27 | 78 | Male | 2 | 3 | 0 | 5 | 5 | 1 | 24 × 19 |
| 28 | 62 | Female | 3.5 | 4 | 5 | 3 | 4 | 3 | 47 × 51 × 46 |
| 29 | 35 | Female | 2 | 3 | 0 | 6 | 5 | NA | 28 × 37 |
| 30 | 56 | Male | 3 | 3 | 4 | 6 | 3 | NA | 34 × 51 × 47 |
| 31 | 65 | Male | 3.5 | 3.5 | 0 | 4 | 5 | NA | 6 × 5 × 4 |
| 32 | 60 | Male | 4.5 | 5.5 | 0 | 4 | 6 | 1 | 47 × 41 × 32 |
| 33 | 62 | Female | 1 | 3.5 | 0 | 3 | 4 | NA | 33 × 41 × 32 |
| 34 | 46 | Male | 4 | 5.5 | 0 | 4 | 3 | 3 | 69 × 51 |
| 35 | 51 | Female | 1 | 3 | 3 | 0 | ND | NA | 38 × 37 × 43 |
| 36 | 76 | Male | 1 | 3 | 0 | 4 | 0 | 5 | 49 × 30 |
| 37 | 53 | Female | 1 | 5 | 0 | 3 | 3 | NA | 25 × 33 × 35 |
| 38 | 51 | Male | 2.5 | 3 | 0 | 3 | 4 | NA | 17.9 |
| Total 38 | Mean 54.6 | F/M c 13/25 | P/N 26/11 | P/N 33/3 | P/N 10/28 | P/N 30/8 | P/N 25/11 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Wang, Z.; Zhang, X.; Yu, H.; Chen, H.; Song, K.; Zhang, Y.; Schwartz, L.M.; Chen, H.; Liu, Y.; et al. Vascular Immune Evasion of Mesenchymal Glioblastoma Is Mediated by Interaction and Regulation of VE-Cadherin on PD-L1. Cancers 2023, 15, 4257. https://doi.org/10.3390/cancers15174257
Luo J, Wang Z, Zhang X, Yu H, Chen H, Song K, Zhang Y, Schwartz LM, Chen H, Liu Y, et al. Vascular Immune Evasion of Mesenchymal Glioblastoma Is Mediated by Interaction and Regulation of VE-Cadherin on PD-L1. Cancers. 2023; 15(17):4257. https://doi.org/10.3390/cancers15174257
Chicago/Turabian StyleLuo, Jing, Ziyi Wang, Xuemei Zhang, Haihui Yu, Hui Chen, Kun Song, Yang Zhang, Lawrence M. Schwartz, Hongzhuan Chen, Yingbin Liu, and et al. 2023. "Vascular Immune Evasion of Mesenchymal Glioblastoma Is Mediated by Interaction and Regulation of VE-Cadherin on PD-L1" Cancers 15, no. 17: 4257. https://doi.org/10.3390/cancers15174257
APA StyleLuo, J., Wang, Z., Zhang, X., Yu, H., Chen, H., Song, K., Zhang, Y., Schwartz, L. M., Chen, H., Liu, Y., & Shao, R. (2023). Vascular Immune Evasion of Mesenchymal Glioblastoma Is Mediated by Interaction and Regulation of VE-Cadherin on PD-L1. Cancers, 15(17), 4257. https://doi.org/10.3390/cancers15174257