
IGF1R Contributes to Cell Proliferation in ALK-Mutated Neuroblastoma with Preference for Activating the PI3K-AKT Signaling Pathway

 , , and
, , and 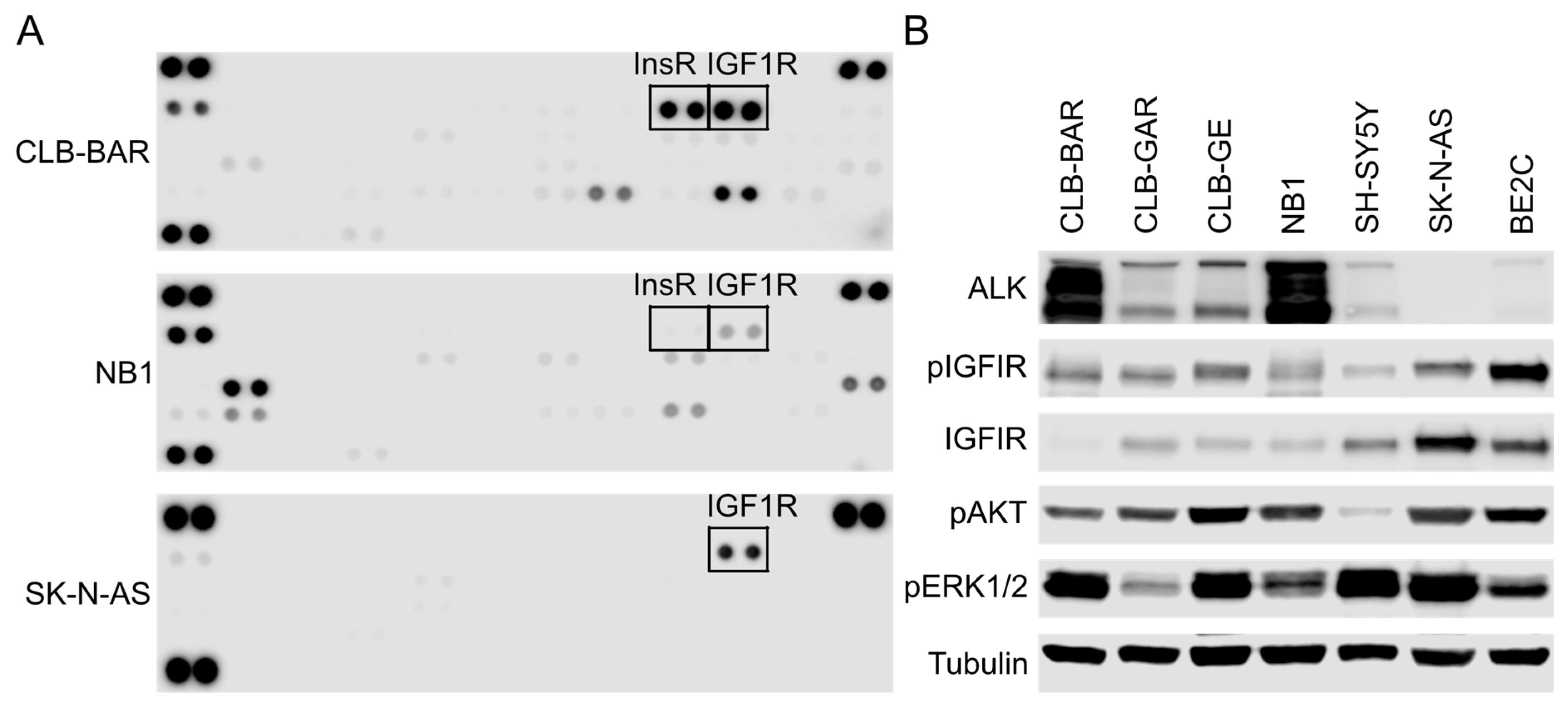{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies, Inhibitors, and Reagents
2.2. Cell Lines and Cell Culture
2.3. Cell Growth and Proliferation Assay
2.4. Colony Formation Assay
2.5. siRNAs and Cell Transfection
2.6. Cell Lysis, Immunoprecipitation, and Immunoblotting Analysis
2.7. Cell Cycle Analysis
3. Results
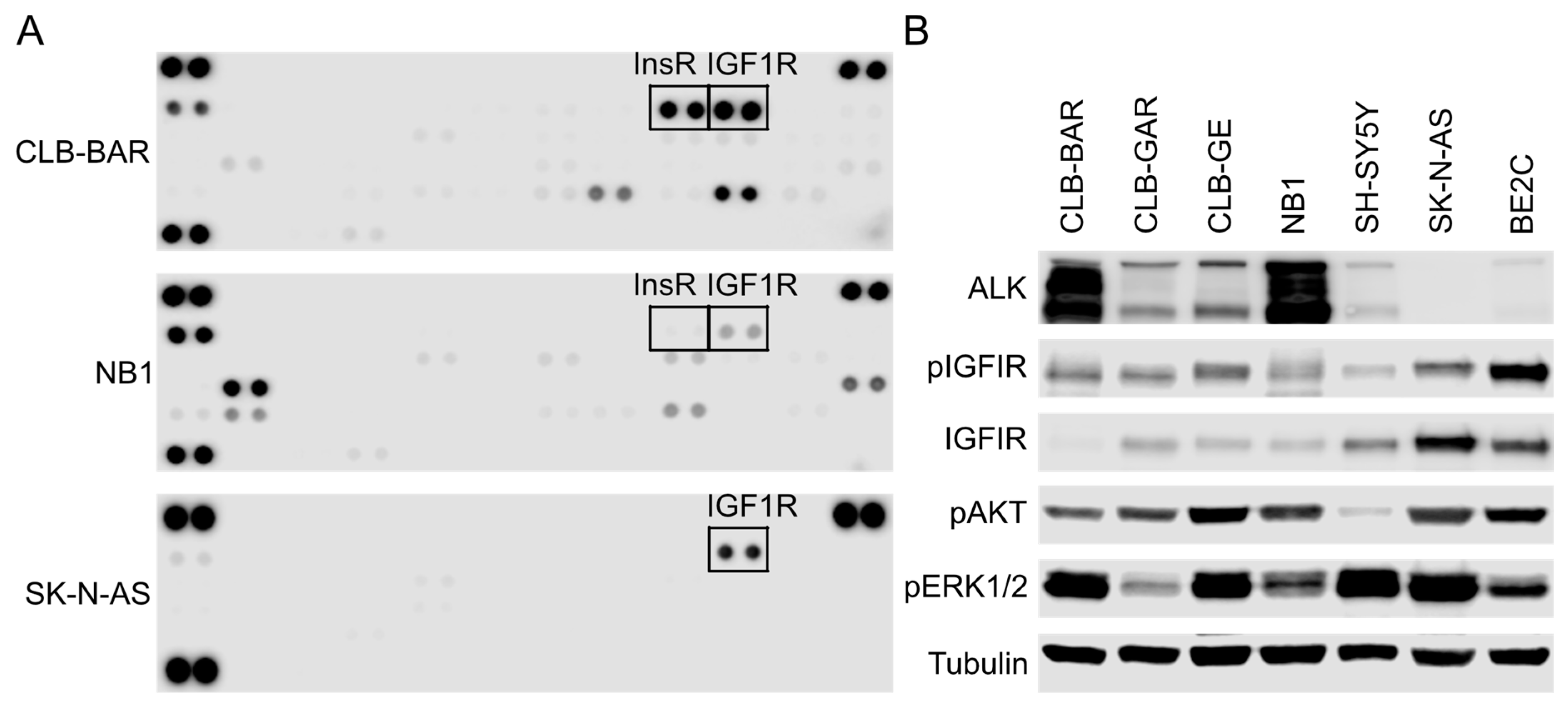
3.1. IGF1R Activity Is Present in ALK-Driven NB Cells
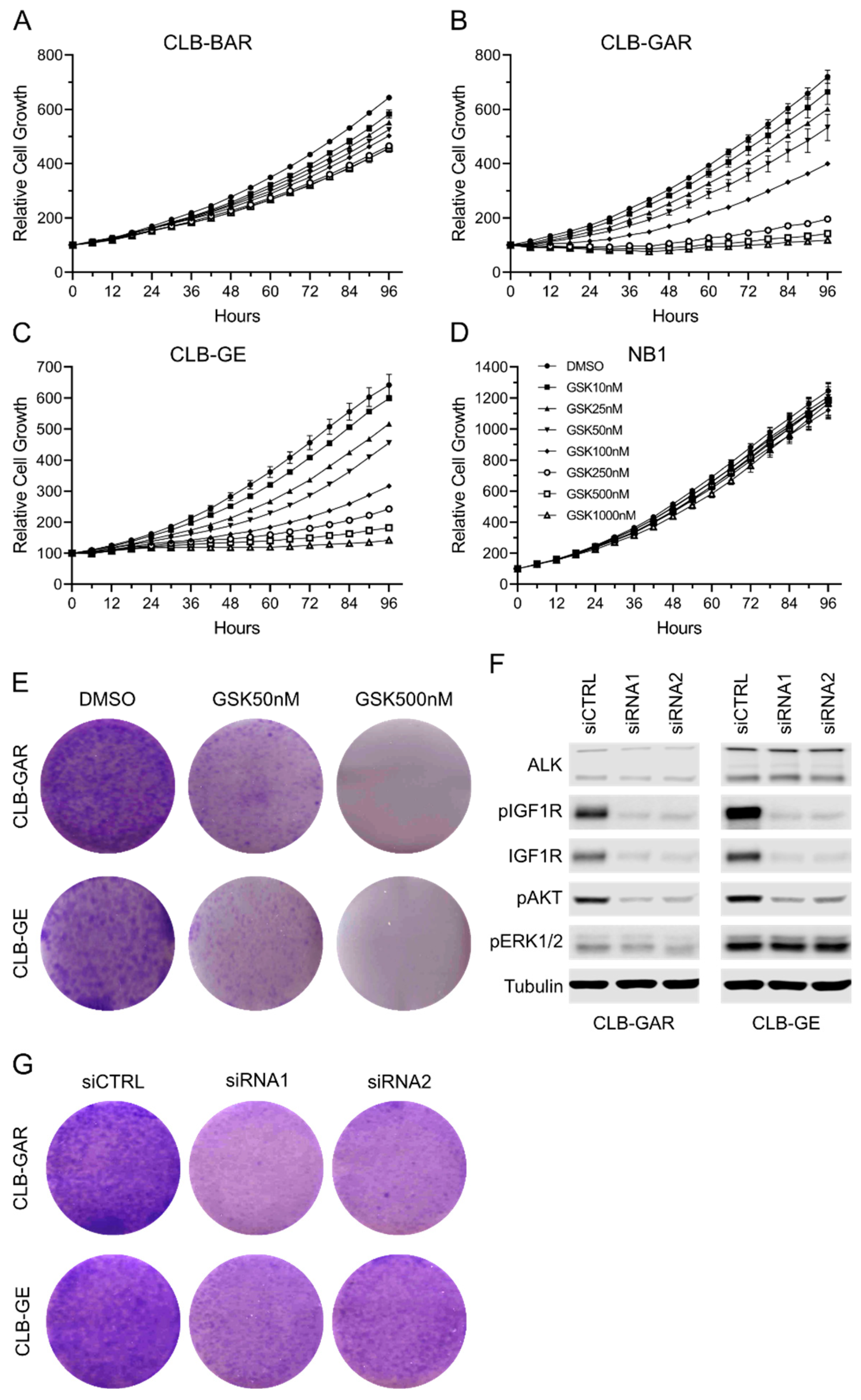
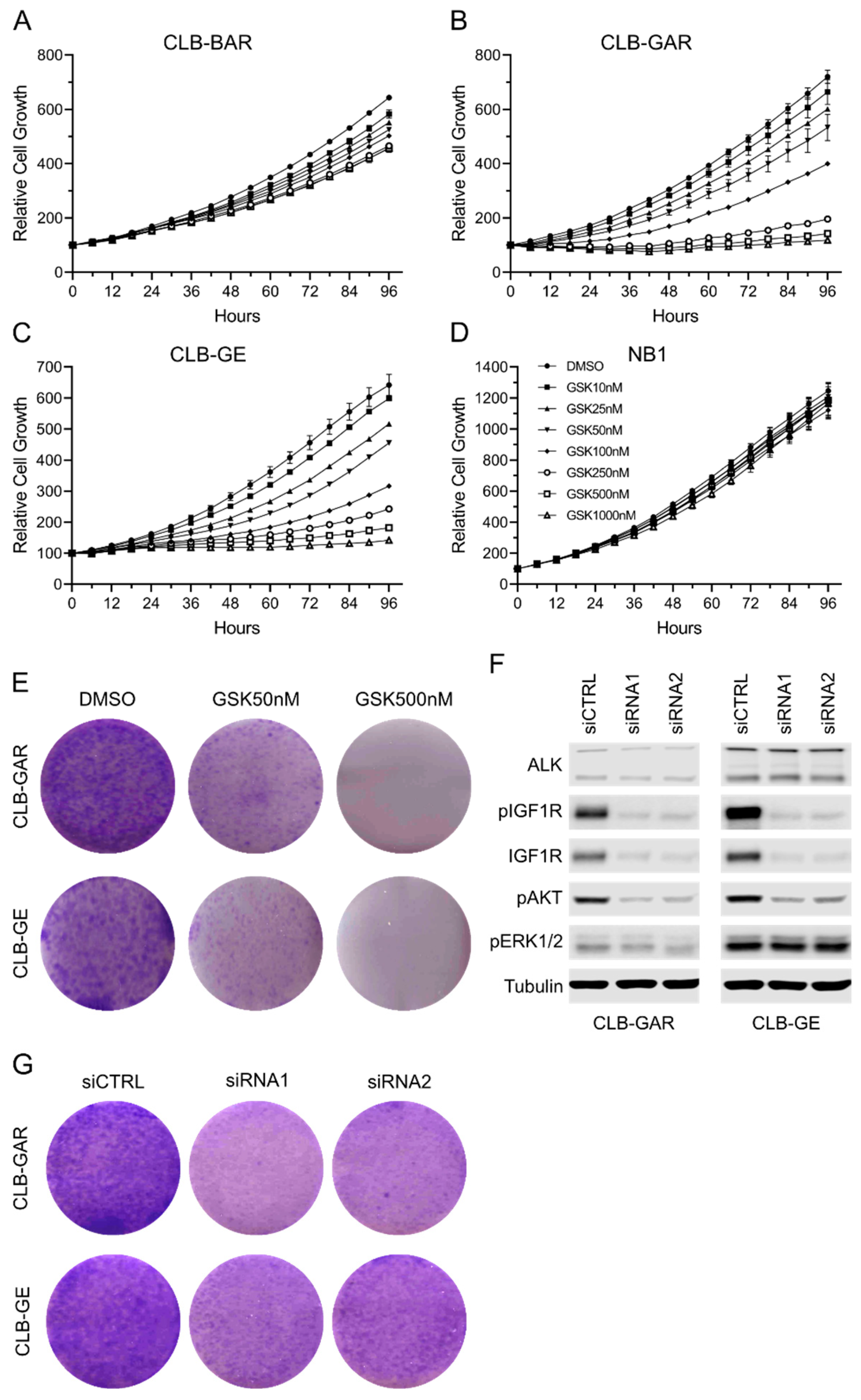
3.2. Differential Sensitivity to IGF1R Inhibition in ALK-Driven NBs
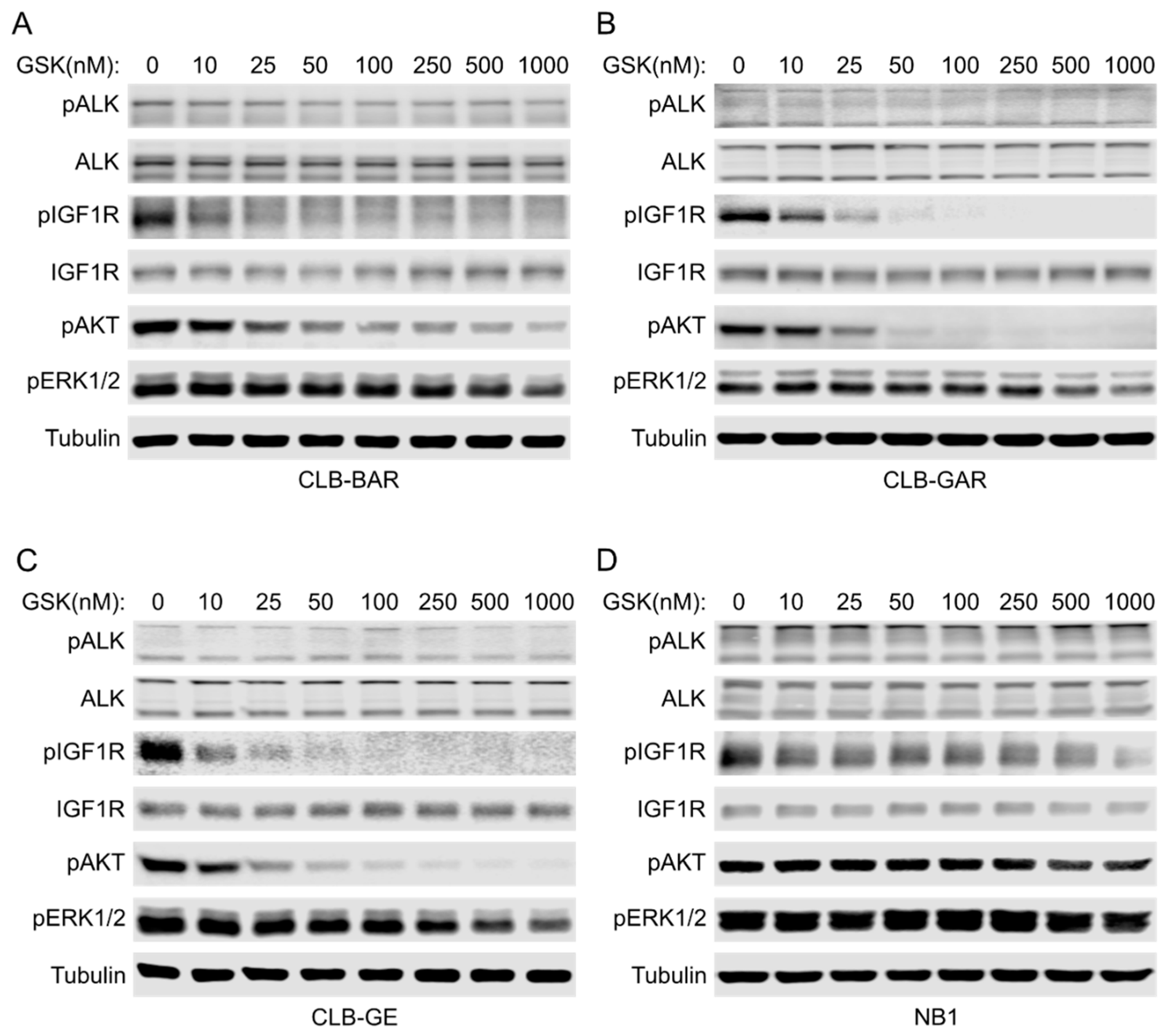
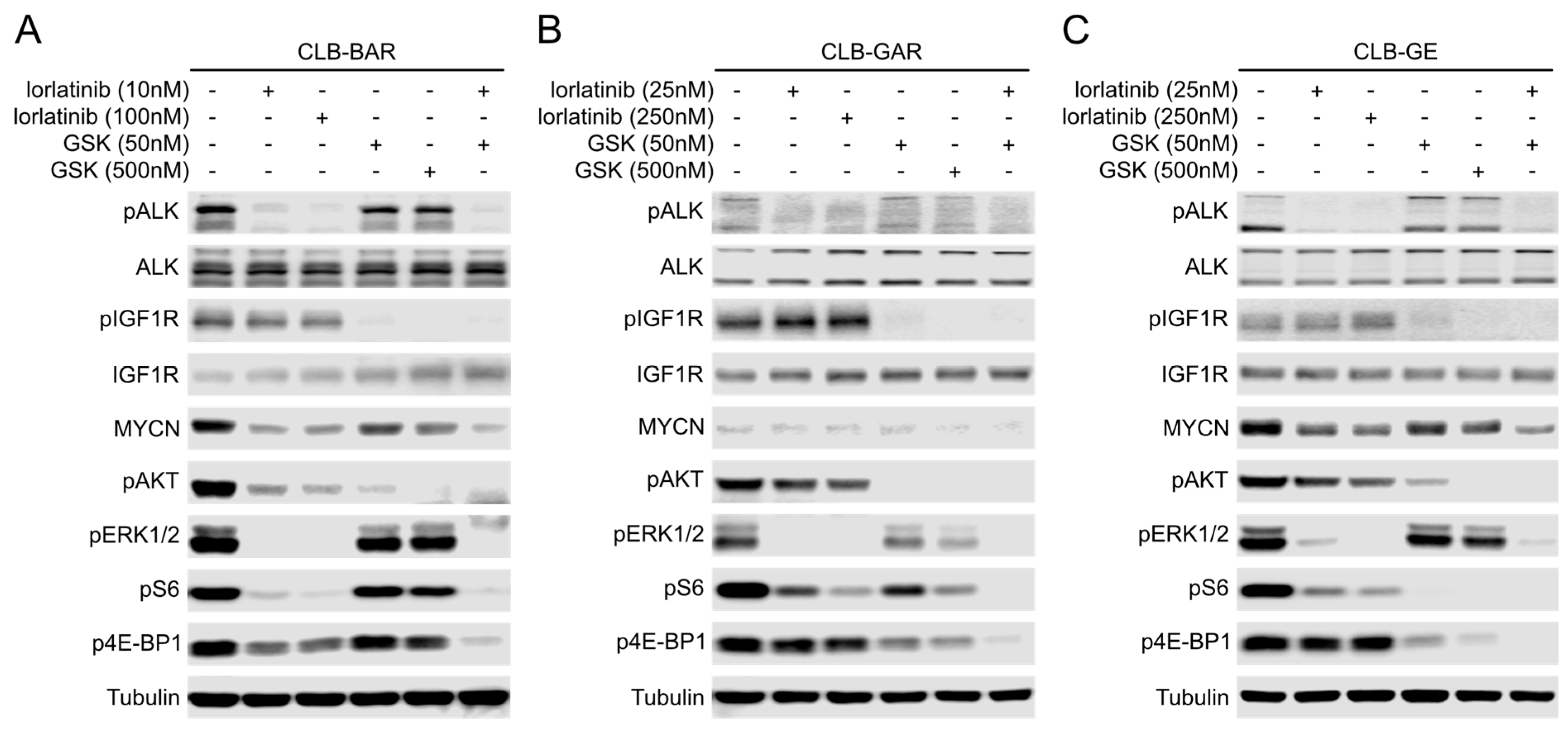
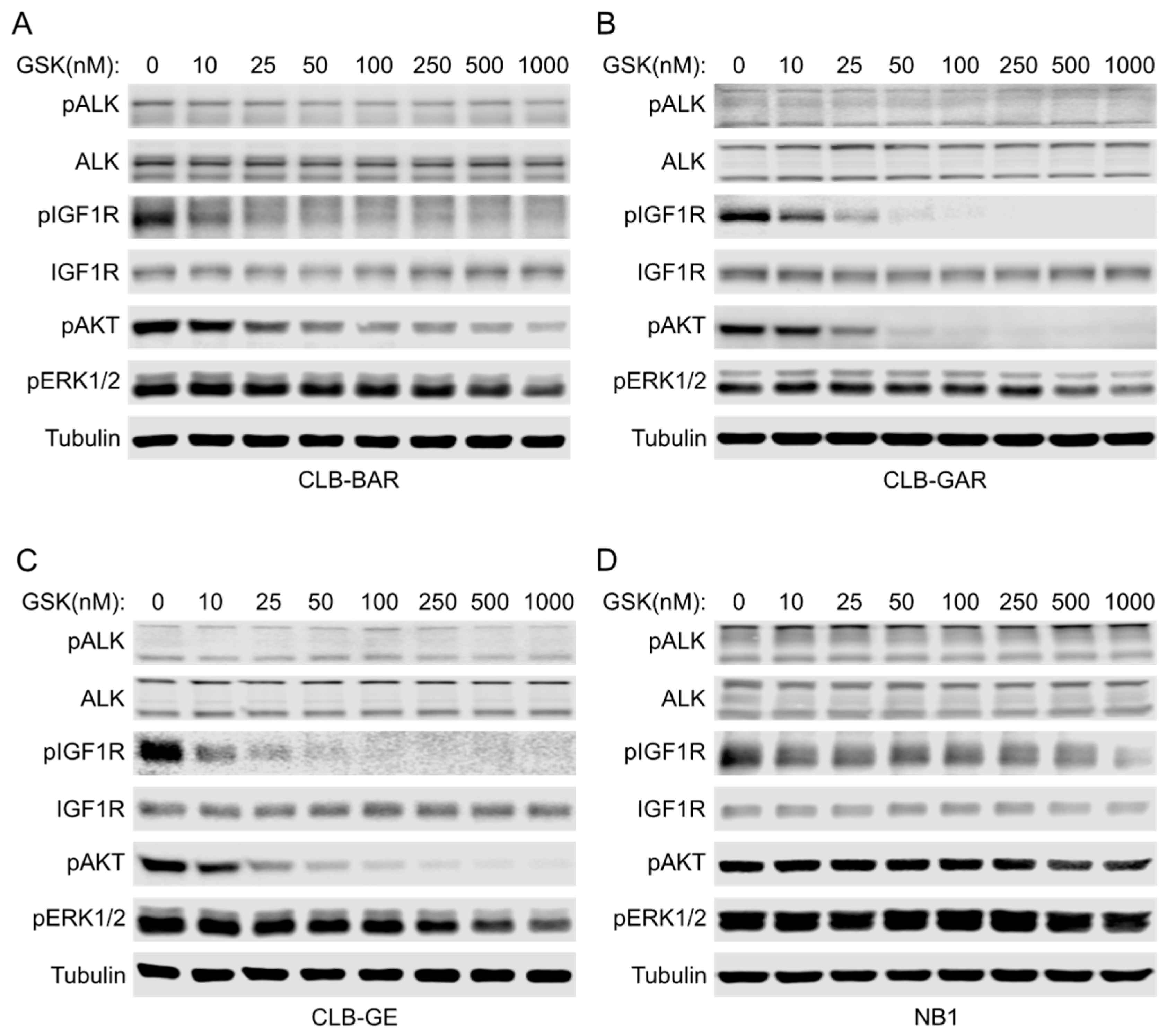
3.3. IGF1R Inhibition Affects Downstream AKT and ERK1/2 Signaling
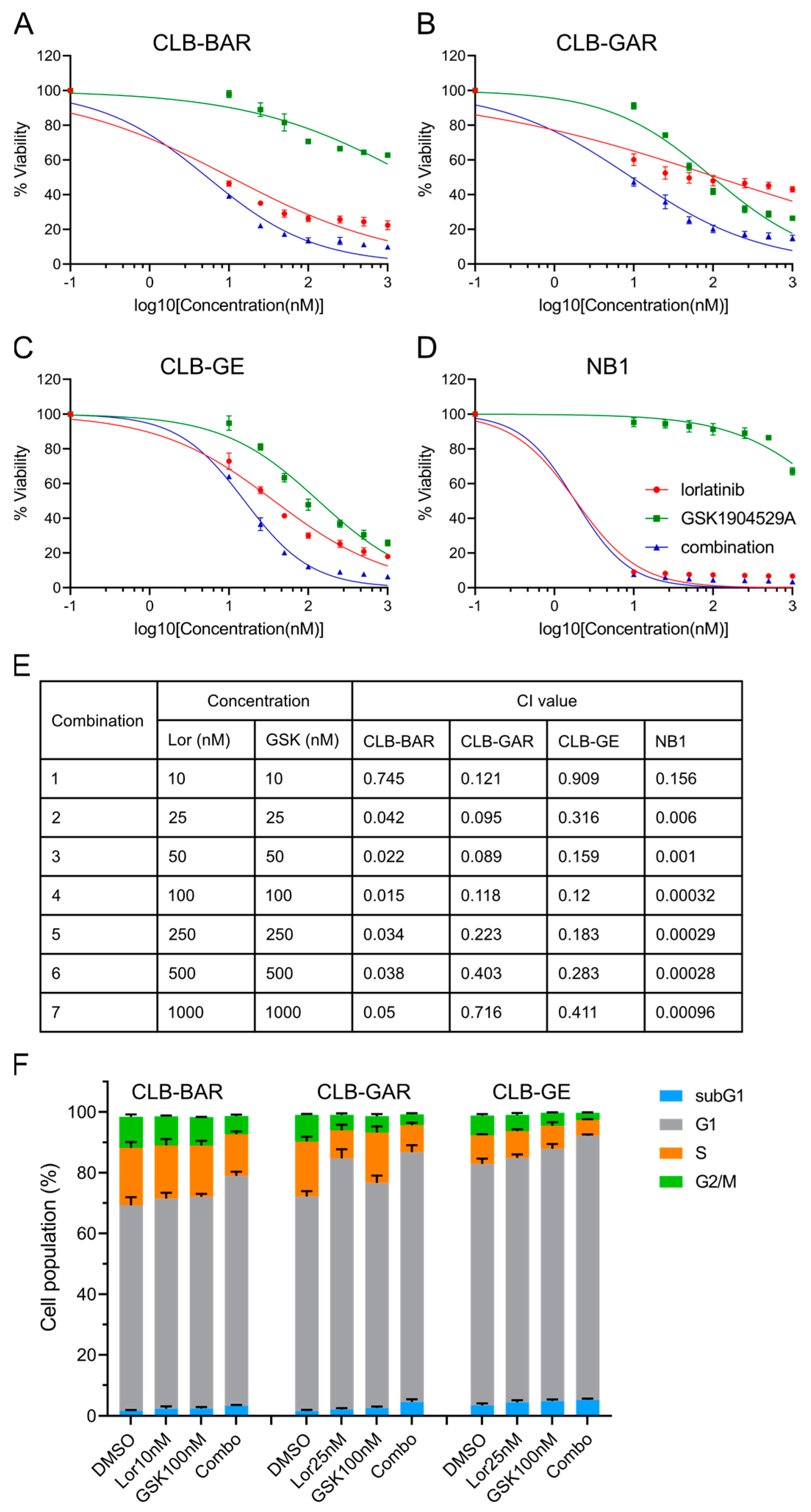
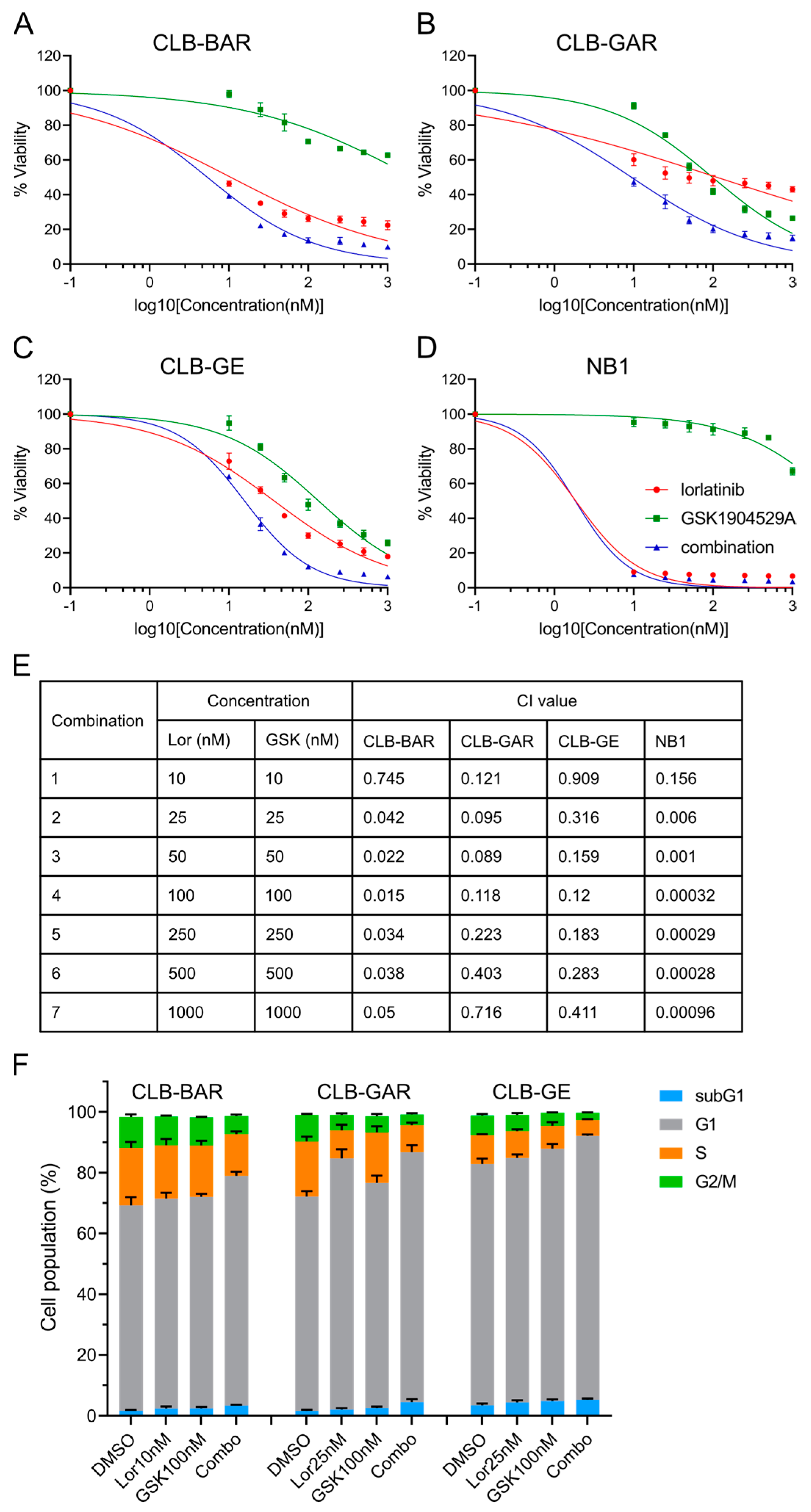
3.4. Combined Inhibition of ALK and IGF1R Synergistically Blocks Cell Proliferation and Promotes G1/S Phase Cell Cycle Arrest
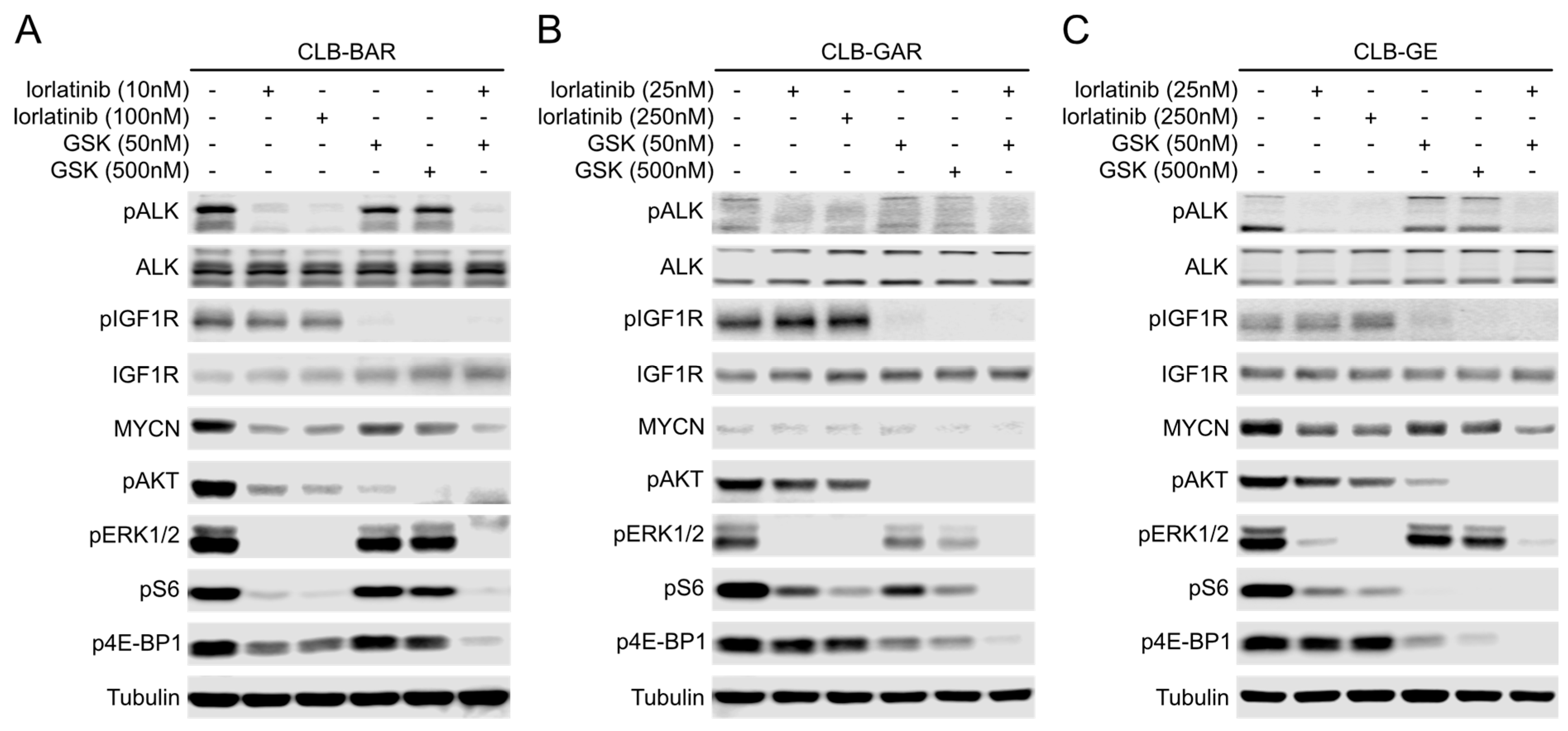
3.5. Both ALK and IGF1R Contribute to Downstream AKT and ERK1/2 Signaling
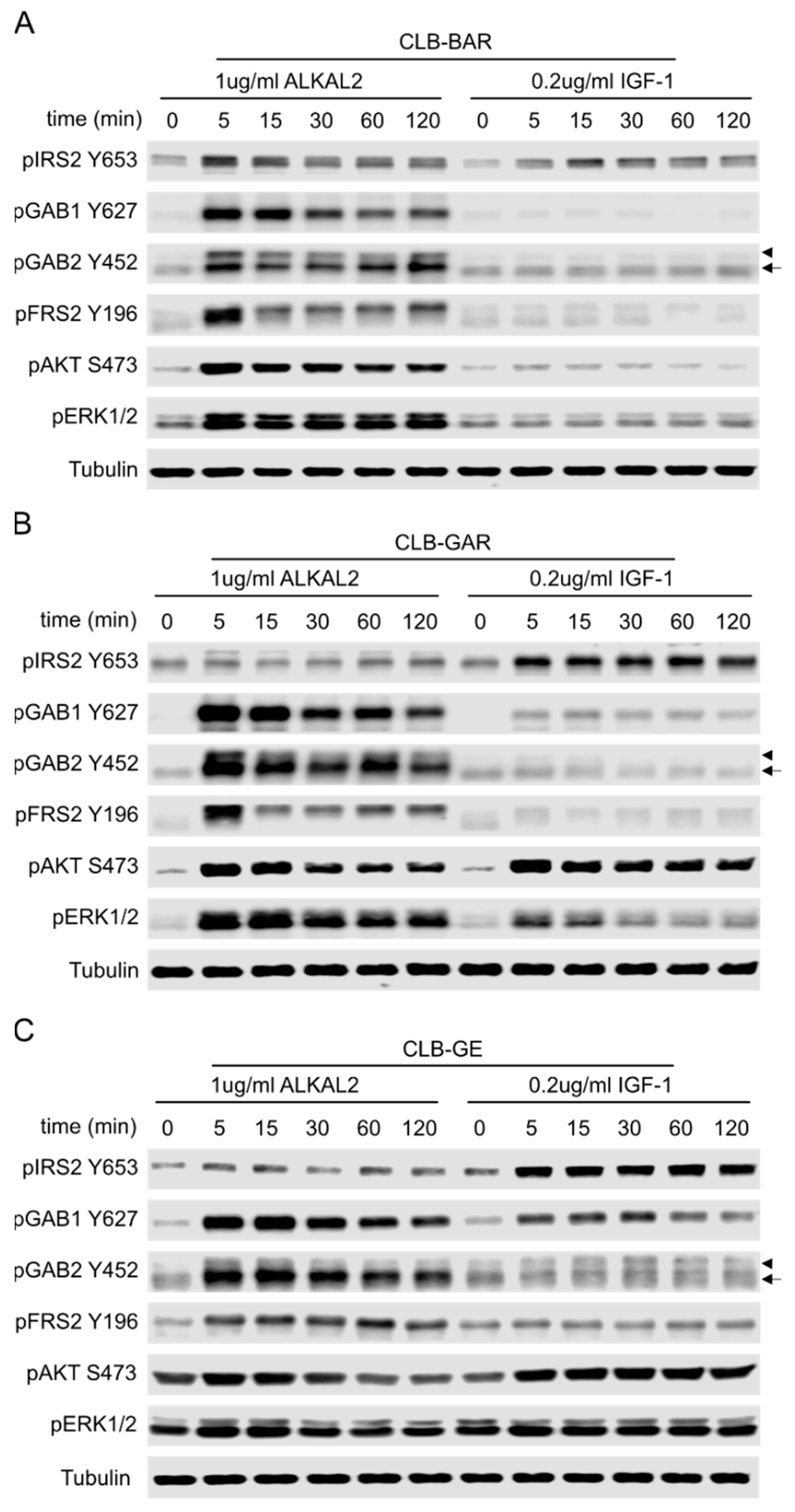
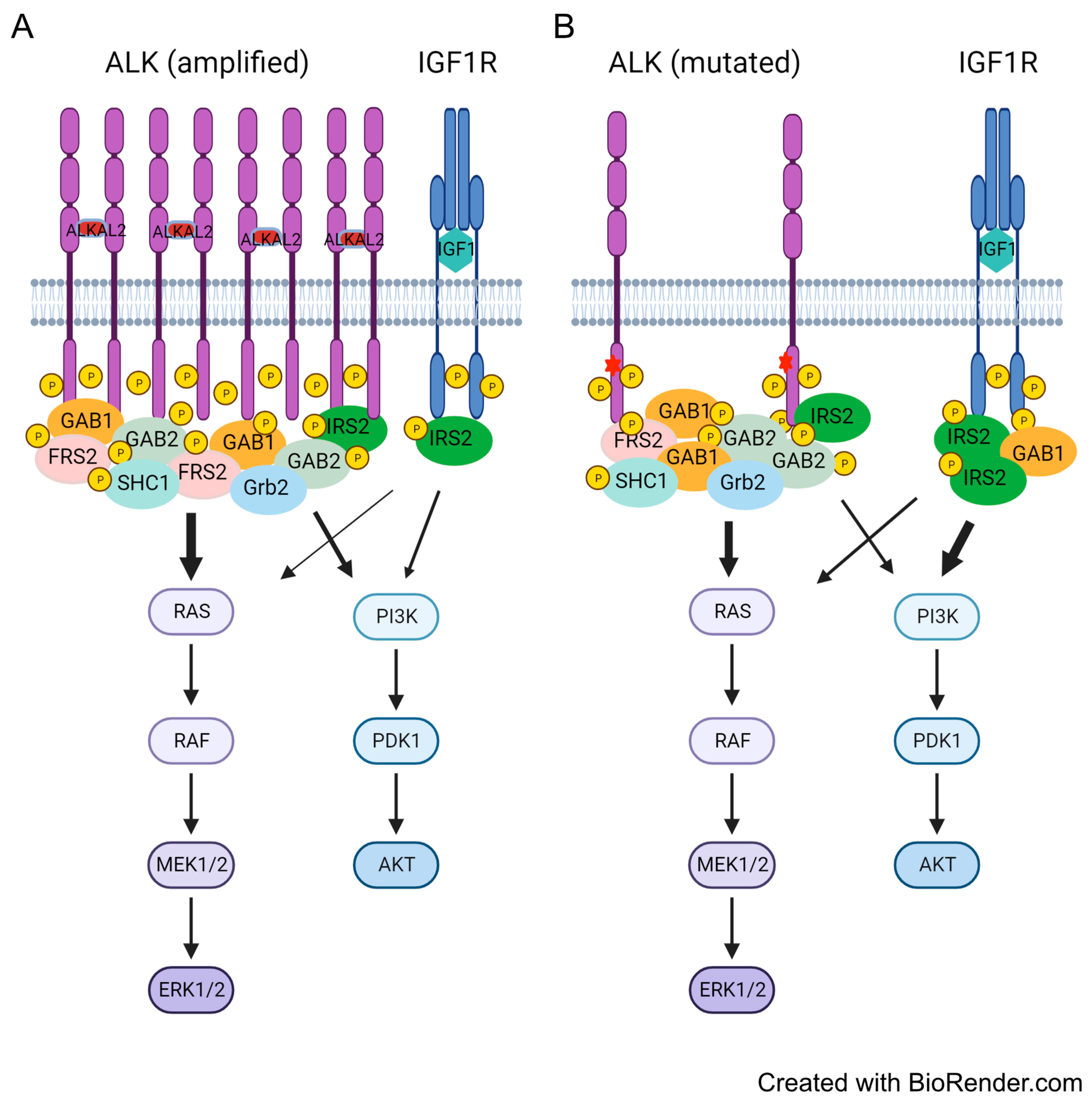
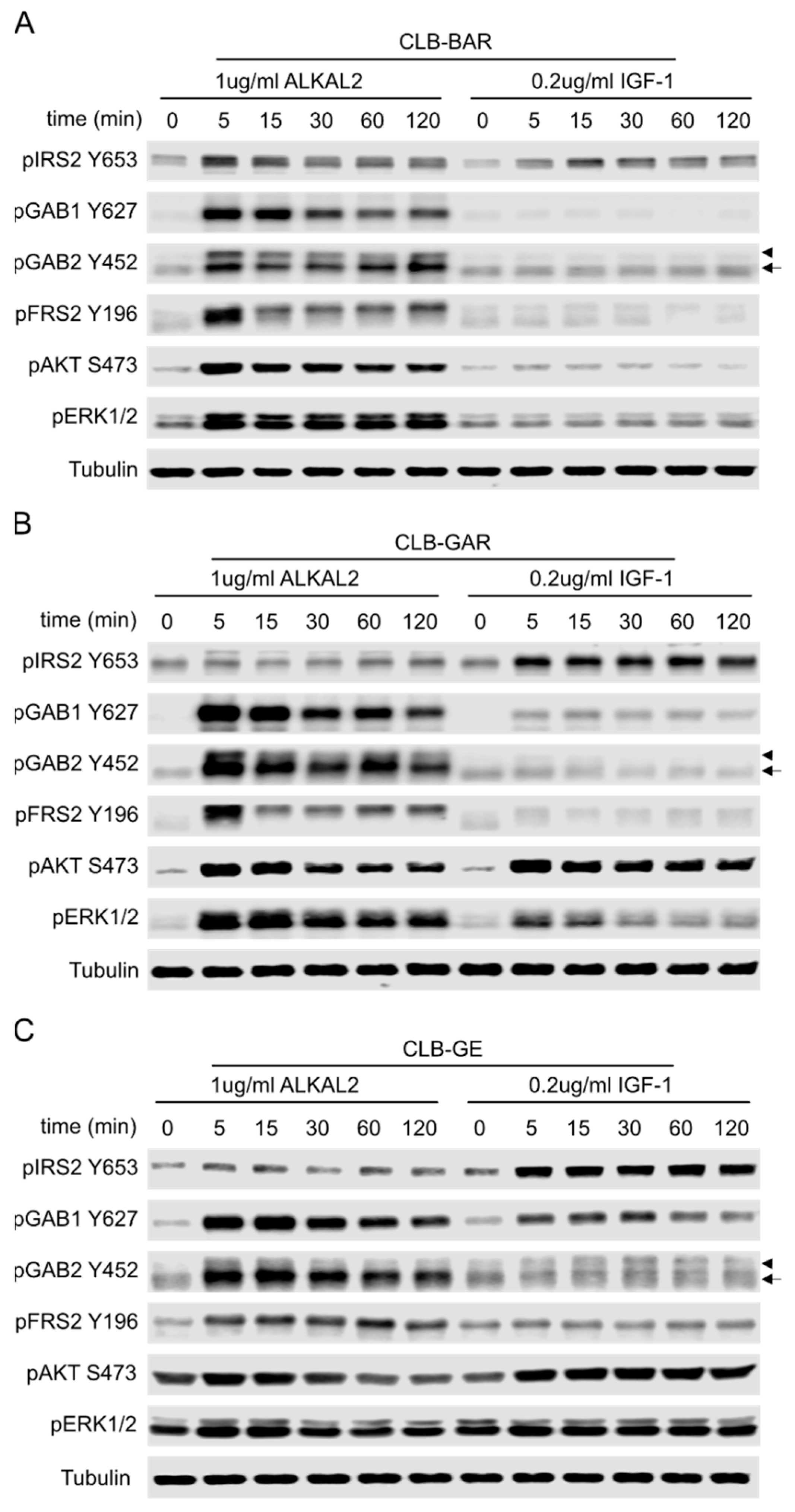
3.6. Differential Preference for Downstream Adaptor Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef]
- Li, J.; Choi, E.; Yu, H.; Bai, X.C. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat. Commun. 2019, 10, 4567. [Google Scholar] [CrossRef] [PubMed]
- Lero, M.W.; Shaw, L.M. Diversity of insulin and IGF signaling in breast cancer: Implications for therapy. Mol. Cell Endocrinol. 2021, 527, 111213. [Google Scholar] [CrossRef]
- Tognon, C.E.; Sorensen, P.H. Targeting the insulin-like growth factor 1 receptor (IGF1R) signaling pathway for cancer therapy. Expert. Opin. Ther. Targets 2012, 16, 33–48. [Google Scholar] [CrossRef]
- Liu, G.; Zhu, M.; Zhang, M.; Pan, F. Emerging Role of IGF-1 in Prostate Cancer: A Promising Biomarker and Therapeutic Target. Cancers 2023, 15, 1287. [Google Scholar] [CrossRef]
- Ochnik, A.M.; Baxter, R.C. Combination therapy approaches to target insulin-like growth factor receptor signaling in breast cancer. Endocr. Relat. Cancer 2016, 23, R513–R536. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Carén, H.; Abel, F.; Kogner, P.; Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 2008, 416, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Jia, Y.; Li, N.; Sun, X.; Ng, K.; Ambing, E.; Gao, M.Y.; Hua, S.; Chen, C.; Kim, S.; et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem. J. 2010, 430, 425–437. [Google Scholar] [CrossRef]
- Favelyukis, S.; Till, J.H.; Hubbard, S.R.; Miller, W.T. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 2001, 8, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Bellini, A.; Pötschger, U.; Bernard, V.; Lapouble, E.; Baulande, S.; Ambros, P.F.; Auger, N.; Beiske, K.; Bernkopf, M.; Betts, D.R.; et al. Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 3377–3390. [Google Scholar] [CrossRef]
- Rosswog, C.; Fassunke, J.; Ernst, A.; Schömig-Markiefka, B.; Merkelbach-Bruse, S.; Bartenhagen, C.; Cartolano, M.; Ackermann, S.; Theissen, J.; Blattner-Johnson, M.; et al. Genomic ALK alterations in primary and relapsed neuroblastoma. Br. J. Cancer 2023, 128, 1559–1571. [Google Scholar] [CrossRef]
- De Brouwer, S.; De Preter, K.; Kumps, C.; Zabrocki, P.; Porcu, M.; Westerhout, E.M.; Lakeman, A.; Vandesompele, J.; Hoebeeck, J.; Van Maerken, T.; et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin. Cancer Res. 2010, 16, 4353–4362. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Javanmardi, N.; Bernard, V.; Leroy, Q.; Cappo, J.; Rio Frio, T.; Pierron, G.; Lapouble, E.; Combaret, V.; Speleman, F.; et al. Emergence of new ALK mutations at relapse of neuroblastoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 2727–2734. [Google Scholar] [CrossRef]
- Bellini, A.; Bernard, V.; Leroy, Q.; Rio Frio, T.; Pierron, G.; Combaret, V.; Lapouble, E.; Clement, N.; Rubie, H.; Thebaud, E.; et al. Deep Sequencing Reveals Occurrence of Subclonal ALK Mutations in Neuroblastoma at Diagnosis. Clin. Cancer Res. 2015, 21, 4913–4921. [Google Scholar] [CrossRef]
- Javanmardi, N.; Fransson, S.; Djos, A.; Sjoberg, R.M.; Nilsson, S.; Truve, K.; Kogner, P.; Martinsson, T. Low Frequency ALK Hotspots Mutations in Neuroblastoma Tumours Detected by Ultra-deep Sequencing: Implications for ALK Inhibitor Treatment. Sci. Rep. 2019, 9, 2199. [Google Scholar] [CrossRef]
- Umapathy, G.; Mendoza-Garcia, P.; Hallberg, B.; Palmer, R.H. Targeting anaplastic lymphoma kinase in neuroblastoma. Acta Pathol. Microbiol. Nutr. Scand. 2019, 127, 288–302. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27 (Suppl. S3), iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef]
- Foster, J.H.; Voss, S.D.; Hall, D.C.; Minard, C.G.; Balis, F.M.; Wilner, K.; Berg, S.L.; Fox, E.; Adamson, P.C.; Blaney, S.M.; et al. Activity of Crizotinib in Patients with ALK-Aberrant Relapsed/Refractory Neuroblastoma: A Children’s Oncology Group Study (ADVL0912). Clin. Cancer Res. 2021, 27, 3543–3548. [Google Scholar] [CrossRef]
- Guan, J.; Tucker, E.R.; Wan, H.; Chand, D.; Danielson, L.S.; Ruuth, K.; El Wakil, A.; Witek, B.; Jamin, Y.; Umapathy, G.; et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis. Model. Mech. 2016, 9, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Infarinato, N.R.; Park, J.H.; Krytska, K.; Ryles, H.T.; Sano, R.; Szigety, K.M.; Li, Y.; Zou, H.Y.; Lee, N.V.; Smeal, T.; et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016, 6, 96–107. [Google Scholar] [CrossRef]
- Siaw, J.T.; Wan, H.; Pfeifer, K.; Rivera, V.M.; Guan, J.; Palmer, R.H.; Hallberg, B. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells, Drosophila and mice. Oncotarget 2016, 7, 29011–29022. [Google Scholar] [CrossRef]
- Lu, J.; Guan, S.; Zhao, Y.; Yu, Y.; Woodfield, S.E.; Zhang, H.; Yang, K.L.; Bieerkehazhi, S.; Qi, L.; Li, X.; et al. The second-generation ALK inhibitor alectinib effectively induces apoptosis in human neuroblastoma cells and inhibits tumor growth in a TH-MYCN transgenic neuroblastoma mouse model. Cancer Lett. 2017, 400, 61–68. [Google Scholar] [CrossRef]
- Heath, J.A.; Campbell, M.A.; Thomas, A.; Solomon, B. Good clinical response to alectinib, a second generation ALK inhibitor, in refractory neuroblastoma. Pediatr. Blood Cancer 2018, 65, e27055. [Google Scholar] [CrossRef]
- Alam, M.W.; Borenas, M.; Lind, D.E.; Cervantes-Madrid, D.; Umapathy, G.; Palmer, R.H.; Hallberg, B. Alectinib, an Anaplastic Lymphoma Kinase Inhibitor, Abolishes ALK Activity and Growth in ALK-Positive Neuroblastoma Cells. Front. Oncol. 2019, 9, 579. [Google Scholar] [CrossRef]
- Fischer, M.; Moreno, L.; Ziegler, D.S.; Marshall, L.V.; Zwaan, C.M.; Irwin, M.S.; Casanova, M.; Sabado, C.; Wulff, B.; Stegert, M.; et al. Ceritinib in paediatric patients with anaplastic lymphoma kinase-positive malignancies: An open-label, multicentre, phase 1, dose-escalation and dose-expansion study. Lancet Oncol. 2021, 22, 1764–1776. [Google Scholar] [CrossRef]
- Goldsmith, K.C.; Park, J.R.; Kayser, K.; Malvar, J.; Chi, Y.Y.; Groshen, S.G.; Villablanca, J.G.; Krytska, K.; Lai, L.M.; Acharya, P.T.; et al. Lorlatinib with or without chemotherapy in ALK-driven refractory/relapsed neuroblastoma: Phase 1 trial results. Nat. Med. 2023, 29, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Fransson, S.; Siaw, J.T.; Treis, D.; Van den Eynden, J.; Chand, D.; Umapathy, G.; Ruuth, K.; Svenberg, P.; Wessman, S.; et al. Clinical response of the novel activating ALK-I1171T mutation in neuroblastoma to the ALK inhibitor ceritinib. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002550. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Giudice, A.M.; Lane, M.V.; McIntyre, B.; Schurch, P.M.; Pascual-Pasto, G.; Buongervino, S.N.; Suresh, S.; Fitzsimmons, A.; Hyman, A.; et al. Serial Profiling of Circulating Tumor DNA Identifies Dynamic Evolution of Clinically Actionable Genomic Alterations in High-Risk Neuroblastoma. Cancer Discov. 2022, 12, 2800–2819. [Google Scholar] [CrossRef] [PubMed]
- Berlak, M.; Tucker, E.; Dorel, M.; Winkler, A.; McGearey, A.; Rodriguez-Fos, E.; da Costa, B.M.; Barker, K.; Fyle, E.; Calton, E.; et al. Mutations in ALK signaling pathways conferring resistance to ALK inhibitor treatment lead to collateral vulnerabilities in neuroblastoma cells. Mol. Cancer 2022, 21, 126. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Dries, R.; Sengupta, S.; Seruggia, D.; Gao, Y.; Sharma, B.; Huang, H.; Moreau, L.; McLane, M.; Day, D.S.; et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature 2019, 572, 676–680. [Google Scholar] [CrossRef]
- Liu, T.; Merguerian, M.D.; Rowe, S.P.; Pratilas, C.A.; Chen, A.R.; Ladle, B.H. Exceptional response to the ALK and ROS1 inhibitor lorlatinib and subsequent mechanism of resistance in relapsed ALK F1174L-mutated neuroblastoma. Cold Spring Harb. Mol. Case Stud. 2021, 7, a006064. [Google Scholar] [CrossRef]
- Marshall, G.M.; Peaston, A.E.; Hocker, J.E.; Smith, S.A.; Hansford, L.M.; Tobias, V.; Norris, M.D.; Haber, M.; Smith, D.P.; Lorenzo, M.J.; et al. Expression of multiple endocrine neoplasia 2B RET in neuroblastoma cells alters cell adhesion in vitro, enhances metastatic behavior in vivo, and activates Jun kinase. Cancer Res. 1997, 57, 5399–5405. [Google Scholar]
- Brodeur, G.M.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Iyer, R.; Varela, C.R.; Light, J.E.; Kolla, V.; Evans, A.E. Trk receptor expression and inhibition in neuroblastomas. Clin. Cancer Res. 2009, 15, 3244–3250. [Google Scholar] [CrossRef]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef]
- Fadeev, A.; Mendoza-Garcia, P.; Irion, U.; Guan, J.; Pfeifer, K.; Wiessner, S.; Serluca, F.; Singh, A.P.; Nüsslein-Volhard, C.; Palmer, R.H. ALKALs are in vivo ligands for ALK family receptor tyrosine kinases in the neural crest and derived cells. Proc. Natl. Acad. Sci. USA 2018, 115, E630–E638. [Google Scholar] [CrossRef] [PubMed]
- Siaw, J.T.; Javanmardi, N.; Van den Eynden, J.; Lind, D.E.; Fransson, S.; Martinez-Monleon, A.; Djos, A.; Sjoberg, R.M.; Ostensson, M.; Caren, H.; et al. 11q Deletion or ALK Activity Curbs DLG2 Expression to Maintain an Undifferentiated State in Neuroblastoma. Cell Rep. 2020, 32, 108171. [Google Scholar] [CrossRef] [PubMed]
- Siaw, J.T.; Gabre, J.L.; Uckun, E.; Vigny, M.; Zhang, W.C.; Van den Eynden, J.; Hallberg, B.; Palmer, R.H.; Guan, J.K. Loss of RET Promotes Mesenchymal Identity in Neuroblastoma Cells. Cancers 2021, 13, 1909. [Google Scholar] [CrossRef]
- Treis, D.; Umapathy, G.; Fransson, S.; Guan, J.; Mendoza-Garcia, P.; Siaw, J.T.; Wessman, S.; Gordon Murkes, L.; Stenman, J.J.E.; Djos, A.; et al. Sustained Response to Entrectinib in an Infant with a Germline ALKAL2 Variant and Refractory Metastatic Neuroblastoma with Chromosomal 2p Gain and Anaplastic Lymphoma Kinase and Tropomyosin Receptor Kinase Activation. JCO Precis. Oncol. 2022, 6, e2100271. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Delisle, L.; Pierre-Eugene, C.; Louis-Brennetot, C.; Surdez, D.; Raynal, V.; Baulande, S.; Boeva, V.; Grossetete-Lalami, S.; Combaret, V.; Peuchmaur, M.; et al. Activated ALK signals through the ERK-ETV5-RET pathway to drive neuroblastoma oncogenesis. Oncogene 2018, 37, 1417–1429. [Google Scholar] [CrossRef]
- DeNardo, B.D.; Holloway, M.P.; Ji, Q.; Nguyen, K.T.; Cheng, Y.; Valentine, M.B.; Salomon, A.; Altura, R.A. Quantitative phosphoproteomic analysis identifies activation of the RET and IGF-1R/IR signaling pathways in neuroblastoma. PLoS ONE 2013, 8, e82513. [Google Scholar] [CrossRef]
- Van den Eynden, J.; Umapathy, G.; Ashouri, A.; Cervantes-Madrid, D.; Szydzik, J.; Ruuth, K.; Koster, J.; Larsson, E.; Guan, J.; Palmer, R.H.; et al. Phosphoproteome and gene expression profiling of ALK inhibition in neuroblastoma cell lines reveals conserved oncogenic pathways. Sci. Signal 2018, 11, eaar5680. [Google Scholar] [CrossRef]
- Tucker, E.R.; Jiménez, I.; Chen, L.; Bellini, A.; Gorrini, C.; Calton, E.; Gao, Q.; Che, H.; Poon, E.; Jamin, Y.; et al. Combination Therapies Targeting ALK-aberrant Neuroblastoma in Preclinical Models. Clin. Cancer Res. 2023, 29, 1317–1331. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Calero, R.; Morchon, E.; Johnsen, J.I.; Serrano, R. Sunitinib suppress neuroblastoma growth through degradation of MYCN and inhibition of angiogenesis. PLoS ONE 2014, 9, e95628. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.; Rowand, J.L.; Groy, A.; Korenchuk, S.; Liu, Q.; Atkins, C.; Dumble, M.; Yang, J.; Anderson, K.; Wilson, B.J.; et al. Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase. Clin. Cancer Res. 2009, 15, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Moore, N.F.; Azarova, A.M.; Bhatnagar, N.; Ross, K.N.; Drake, L.E.; Frumm, S.; Liu, Q.S.; Christie, A.L.; Sanda, T.; Chesler, L.; et al. Molecular rationale for the use of PI3K/AKT/mTOR pathway inhibitors in combination with crizotinib in ALK-mutated neuroblastoma. Oncotarget 2014, 5, 8737–8749. [Google Scholar] [CrossRef]
- Umapathy, G.; El Wakil, A.; Witek, B.; Chesler, L.; Danielson, L.; Deng, X.; Gray, N.S.; Johansson, M.; Kvarnbrink, S.; Ruuth, K.; et al. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci. Signal 2014, 7, ra102. [Google Scholar] [CrossRef]
- Schönherr, C.; Ruuth, K.; Kamaraj, S.; Wang, C.L.; Yang, H.L.; Combaret, V.; Djos, A.; Martinsson, T.; Christensen, J.G.; Palmer, R.H.; et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene 2012, 31, 5193–5200. [Google Scholar] [CrossRef] [PubMed]
- Cage, T.A.; Chanthery, Y.; Chesler, L.; Grimmer, M.; Knight, Z.; Shokat, K.; Weiss, W.A.; Gustafson, W.C. Downregulation of MYCN through PI3K Inhibition in Mouse Models of Pediatric Neural Cancer. Front. Oncol. 2015, 5, 111. [Google Scholar] [CrossRef]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A.; et al. The ALK(F1174L) Mutation Potentiates the Oncogenic Activity of MYCN in Neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef]
- Chesler, L.; Schlieve, C.; Goldenberg, D.D.; Kenney, A.; Kim, G.; McMillan, A.; Matthay, K.K.; Rowitch, D.; Weiss, W.A. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006, 66, 8139–8146. [Google Scholar] [CrossRef]
- Flynn, D.C. Adaptor proteins. Oncogene 2001, 20, 6270–6272. [Google Scholar] [CrossRef]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef]
- Guan, J.K.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.B.; Hsu, A.W.; et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 2015, 4, e09811. [Google Scholar] [CrossRef]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef]
- Emdal, K.B.; Pedersen, A.K.; Bekker-Jensen, D.B.; Lundby, A.; Claeys, S.; De Preter, K.; Speleman, F.; Francavilla, C.; Olsen, J.V. Integrated proximal proteomics reveals IRS2 as a determinant of cell survival in ALK-driven neuroblastoma. Sci. Signal 2018, 11, eaap9752. [Google Scholar] [CrossRef]
- Uckun, E.; Siaw, J.T.; Guan, J.K.; Anthonydhason, V.; Fuchs, J.; Wolfstetter, G.; Hallberg, B.; Palmer, R.H. BioID-Screening Identifies PEAK1 and SHP2 as Components of the ALK Proximitome in Neuroblastoma Cells. J. Mol. Biol. 2021, 433, 167158. [Google Scholar] [CrossRef]
- Borenas, M.; Umapathy, G.; Lai, W.Y.; Lind, D.E.; Witek, B.; Guan, J.; Mendoza-Garcia, P.; Masudi, T.; Claeys, A.; Chuang, T.P.; et al. ALK ligand ALKAL2 potentiates MYCN-driven neuroblastoma in the absence of ALK mutation. EMBO J. 2021, 40, e105784. [Google Scholar] [CrossRef]
- Wojtalla, A.; Salm, F.; Christiansen, D.G.; Cremona, T.; Cwiek, P.; Shalaby, T.; Gross, N.; Grotzer, M.A.; Arcaro, A. Novel agents targeting the IGF-1R/PI3K pathway impair cell proliferation and survival in subsets of medulloblastoma and neuroblastoma. PLoS ONE 2012, 7, e47109. [Google Scholar] [CrossRef]
- Szydzik, J.; Lind, D.E.; Arefin, B.; Kurhe, Y.; Umapathy, G.; Siaw, J.T.; Claeys, A.; Gabre, J.L.; Van den Eynden, J.; Hallberg, B.; et al. ATR inhibition enables complete tumour regression in ALK-driven NB mouse models. Nat. Commun. 2021, 12, 6813. [Google Scholar] [CrossRef]
- Wood, A.C.; Krytska, K.; Ryles, H.T.; Infarinato, N.R.; Sano, R.; Hansel, T.D.; Hart, L.S.; King, F.J.; Smith, T.R.; Ainscow, E.; et al. Dual ALK and CDK4/6 Inhibition Demonstrates Synergy against Neuroblastoma. Clin. Cancer Res. 2017, 23, 2856–2868. [Google Scholar] [CrossRef]
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mossé, Y.P. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef]
- von Mehren, M.; George, S.; Heinrich, M.C.; Schuetze, S.M.; Yap, J.T.; Yu, J.Q.; Abbott, A.; Litwin, S.; Crowley, J.; Belinsky, M.; et al. Linsitinib (OSI-906) for the Treatment of Adult and Pediatric Wild-Type Gastrointestinal Stromal Tumors, a SARC Phase II Study. Clin. Cancer Res. 2020, 26, 1837–1845. [Google Scholar] [CrossRef]
- Khan, S.; LeBlanc, R.; Gyger, M.; White, D.; Kaufman, J.; Jazubowiak, A.; Gul, E.; Paul, H.; Le, L.W.; Lau, A.; et al. A phase-1 trial of linsitinib (OSI-906) in combination with bortezomib and dexamethasone for the treatment of relapsed/refractory multiple myeloma. Leuk. Lymphoma 2021, 62, 1721–1729. [Google Scholar] [CrossRef]
- Puzanov, I.; Lindsay, C.R.; Goff, L.; Sosman, J.; Gilbert, J.; Berlin, J.; Poondru, S.; Simantov, R.; Gedrich, R.; Stephens, A.; et al. A phase I study of continuous oral dosing of OSI-906, a dual inhibitor of insulin-like growth factor-1 and insulin receptors, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 701–711. [Google Scholar] [CrossRef]
- Fassnacht, M.; Berruti, A.; Baudin, E.; Demeure, M.J.; Gilbert, J.; Haak, H.; Kroiss, M.; Quinn, D.I.; Hesseltine, E.; Ronchi, C.L.; et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: A double-blind, randomised, phase 3 study. Lancet Oncol. 2015, 16, 426–435. [Google Scholar] [CrossRef]
- Macaulay, V.M.; Middleton, M.R.; Eckhardt, S.G.; Rudin, C.M.; Juergens, R.A.; Gedrich, R.; Gogov, S.; McCarthy, S.; Poondru, S.; Stephens, A.W.; et al. Phase I Dose-Escalation Study of Linsitinib (OSI-906) and Erlotinib in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 2897–2907. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, J.; Borenäs, M.; Xiong, J.; Lai, W.-Y.; Palmer, R.H.; Hallberg, B. IGF1R Contributes to Cell Proliferation in ALK-Mutated Neuroblastoma with Preference for Activating the PI3K-AKT Signaling Pathway. Cancers 2023, 15, 4252. https://doi.org/10.3390/cancers15174252
Guan J, Borenäs M, Xiong J, Lai W-Y, Palmer RH, Hallberg B. IGF1R Contributes to Cell Proliferation in ALK-Mutated Neuroblastoma with Preference for Activating the PI3K-AKT Signaling Pathway. Cancers. 2023; 15(17):4252. https://doi.org/10.3390/cancers15174252
Chicago/Turabian StyleGuan, Jikui, Marcus Borenäs, Junfeng Xiong, Wei-Yun Lai, Ruth H. Palmer, and Bengt Hallberg. 2023. "IGF1R Contributes to Cell Proliferation in ALK-Mutated Neuroblastoma with Preference for Activating the PI3K-AKT Signaling Pathway" Cancers 15, no. 17: 4252. https://doi.org/10.3390/cancers15174252
APA StyleGuan, J., Borenäs, M., Xiong, J., Lai, W.-Y., Palmer, R. H., & Hallberg, B. (2023). IGF1R Contributes to Cell Proliferation in ALK-Mutated Neuroblastoma with Preference for Activating the PI3K-AKT Signaling Pathway. Cancers, 15(17), 4252. https://doi.org/10.3390/cancers15174252