
A Case-Only Genome-Wide Interaction Study of Smoking and Bladder Cancer Risk: Results from the COBLAnCE Cohort
 , ,
, ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
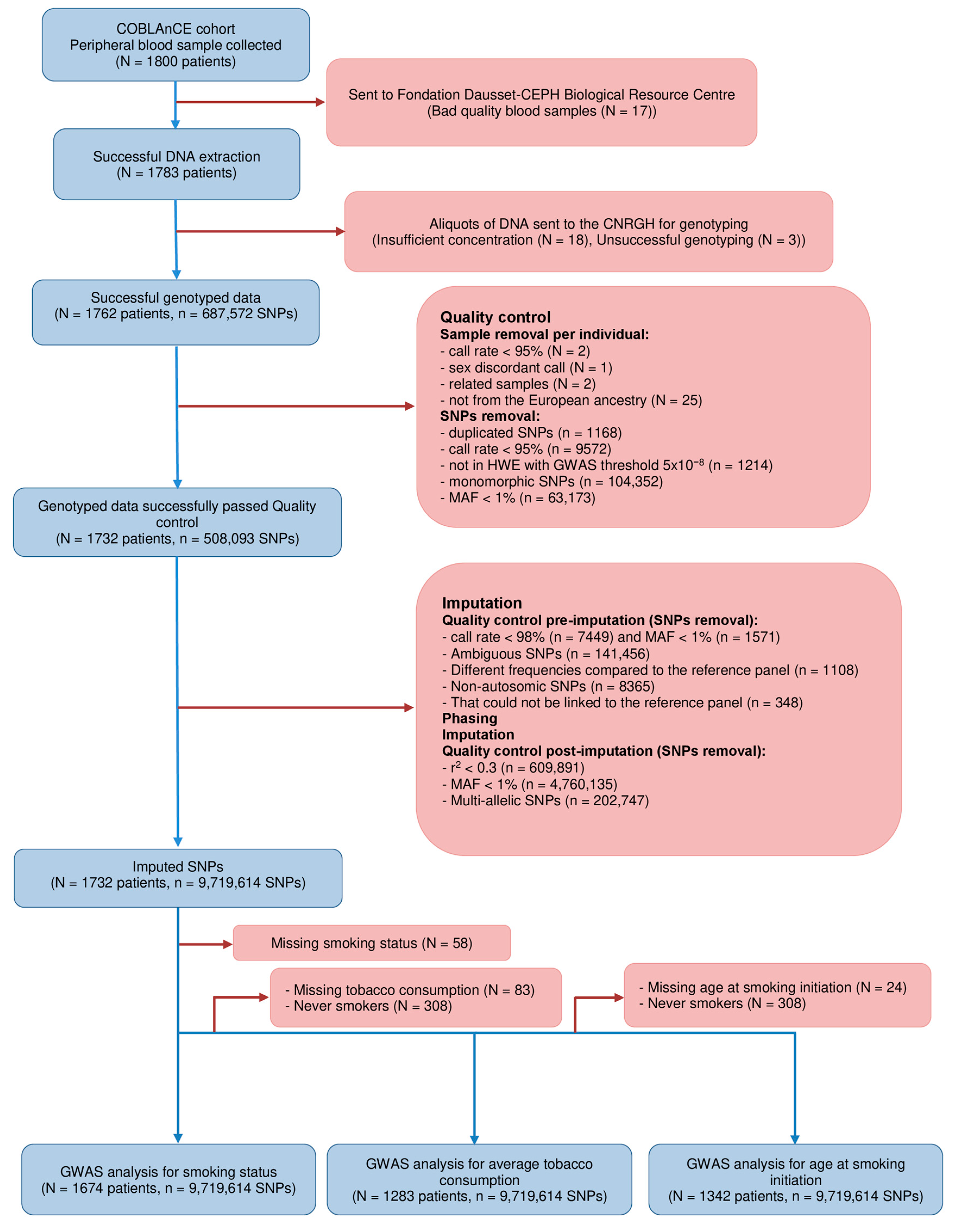
2.1. Study Population
2.2. Smoking Phenotypes
2.3. Genotype Data, Quality Control, and Imputation
2.4. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.C.; Skipper, P.L.; Tannenbaum, S.R.; Chan, K.K.; Ross, R.K. Arylamine exposures and bladder cancer risk. Mutat. Res. Mol. Mech. Mutagen. 2002, 506-507, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-I.; Liou, S.-H.; Loh, C.-H.; Uang, S.-N.; Yu, Y.-C.; Shih, T.-S. Bladder cancer screening and monitoring of 4,4′-Methylenebis(2-chloroaniline) exposure among workers in Taiwan. Urology 2005, 66, 305–310. [Google Scholar] [CrossRef] [PubMed]
- García-Pérez, J.; Pollán, M.; Boldo, E.; Pérez-Gómez, B.; Aragonés, N.; Lope, V.; Ramis, R.; Vidal, E.; López-Abente, G. Mortality due to lung, laryngeal and bladder cancer in towns lying in the vicinity of combustion installations. Sci. Total. Environ. 2009, 407, 2593–2602. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.I.; López, J.F.; Vivaldi, B.; Coz, F. Long-Term Impact of Arsenic in Drinking Water on Bladder Cancer Health Care and Mortality Rates 20 Years After End of Exposure. J. Urol. 2012, 187, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Freedman, N.D.; Silverman, D.T.; Hollenbeck, A.R.; Schatzkin, A.; Abnet, C.C. Association Between Smoking and Risk of Bladder Cancer Among Men and Women. JAMA 2011, 306, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Lammers, R.J.; Witjes, W.P.; Hendricksen, K.; Caris, C.T.; Janzing-Pastors, M.H.; Witjes, J.A. Smoking Status Is a Risk Factor for Recurrence After Transurethral Resection of Non–Muscle-Invasive Bladder Cancer. Eur. Urol. 2011, 60, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Boström, P.J.; Alkhateeb, S.; Trottier, G.; Athanasopoulos, P.Z.; Mirtti, T.; Kortekangas, H.; Laato, M.; van Rhijn, B.; van der Kwast, T.; Fleshner, N.E.; et al. Sex differences in bladder cancer outcomes among smokers with advanced bladder cancer. BJU Int. 2011, 109, 70–76. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Padala, S.A.; Barsouk, A. Epidemiology of Bladder Cancer. Med Sci. 2020, 8, 15. [Google Scholar] [CrossRef]
- Burger, M.; Catto, J.W.F.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and Risk Factors of Urothelial Bladder Cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef]
- de Maturana, E.L.; Rava, M.; Anumudu, C.; Sáez, O.; Alonso, D.; Malats, N. Bladder Cancer Genetic Susceptibility. A Systematic Review. Bl. Cancer 2018, 4, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Guey, L.T.; García-Closas, M.; Murta-Nascimento, C.; Lloreta, J.; Palencia, L.; Kogevinas, M.; Rothman, N.; Vellalta, G.; Calle, M.L.; Marenne, G.; et al. Genetic Susceptibility to Distinct Bladder Cancer Subphenotypes. Eur. Urol. 2010, 57, 283–292. [Google Scholar] [CrossRef] [PubMed]
- A Kiemeney, L.; Sulem, P.; Besenbacher, S.; Vermeulen, S.H.; Sigurdsson, A.; Thorleifsson, G.; Gudbjartsson, D.F.; Stacey, S.N.; Gudmundsson, J.; Zanon, C.; et al. A sequence variant at 4p16.3 confers susceptibility to urinary bladder cancer. Nat. Genet. 2010, 42, 415–419. [Google Scholar] [CrossRef]
- A Kiemeney, L.; Thorlacius, S.; Sulem, P.; Geller, F.; Aben, K.K.H.; Stacey, S.N.; Gudmundsson, J.; Jakobsdottir, M.; Bergthorsson, J.T.; Sigurdsson, A.; et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat. Genet. 2008, 40, 1307–1312. [Google Scholar] [CrossRef]
- Figueroa, J.D.; Ye, Y.; Siddiq, A.; Garcia-Closas, M.; Chatterjee, N.; Prokunina-Olsson, L.; Cortessis, V.K.; Kooperberg, C.; Cussenot, O.; Benhamou, S.; et al. Genome-wide association study identifies multiple loci associated with bladder cancer risk. Hum. Mol. Genet. 2013, 23, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Rothman, N.; Garcia-Closas, M.; Chatterjee, N.; Malats, N.; Wu, X.F.; Figueroa, J.D.; Real, F.X.; Van den Berg, D.; Matullo, G.; Baris, D.; et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat. Genet. 2010, 42, 978–984. [Google Scholar] [CrossRef]
- Hein, D.W. Molecular genetics and function of NAT1 and NAT2: Role in aromatic amine metabolism and carcinogenesis. Mutat. Res. Mol. Mech. Mutagen. 2002, 506-507, 65–77. [Google Scholar] [CrossRef]
- Figueroa, J.D.; Han, S.S.; Garcia-Closas, M.; Baris, D.; Jacobs, E.J.; Kogevinas, M.; Schwenn, M.; Malats, N.; Johnson, A.; Purdue, M.P.; et al. Genome-wide interaction study of smoking and bladder cancer risk. Carcinog. 2014, 35, 1737–1744. [Google Scholar] [CrossRef]
- Koutros, S.; Kiemeney, L.A.; Choudhury, P.P.; Milne, R.L.; de Maturana, E.L.; Ye, Y.; Joseph, V.; Florez-Vargas, O.; Dyrskjøt, L.; Figueroa, J.; et al. Genome-wide Association Study of Bladder Cancer Reveals New Biological and Translational Insights. Eur. Urol. 2023, 84, 127–137. [Google Scholar] [CrossRef]
- Smith, P.G.; E Day, N. The Design of Case-Control Studies: The Influence of Confounding and Interaction Effects. Leuk. Res. 1984, 13, 356–365. [Google Scholar] [CrossRef]
- Piegorsch, W.W.; Weinberg, C.R.; Taylor, J.A. Non-hierarchical logistic models and case-only designs for assessing susceptibility in population-based case-control studies. Stat. Med. 1994, 13, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Gauderman, W.J. Sample Size Requirements for Association Studies of Gene-Gene Interaction. Am. J. Epidemiology 2002, 155, 478–484. [Google Scholar] [CrossRef] [PubMed]
- García-Closas, M.; Malats, N.; Silverman, D.; Dosemeci, M.; Kogevinas, M.; Hein, D.W.; Tardón, A.; Serra, C.; Carrato, A.; García-Closas, R.; et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: Results from the Spanish Bladder Cancer Study and meta-analyses. Lancet 2005, 366, 649–659. [Google Scholar] [CrossRef]
- Garcia-Closas, M.; Rothman, N.; Figueroa, J.D.; Prokunina-Olsson, L.; Han, S.S.; Baris, D.; Jacobs, E.J.; Malats, N.; De Vivo, I.; Albanes, D.; et al. Common Genetic Polymorphisms Modify the Effect of Smoking on Absolute Risk of Bladder Cancer. Cancer Res 2013, 73, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Lubin, J.H.; Kogevinas, M.; Silverman, D.; Malats, N.; Garcia-Closas, M.; Tardón, A.; Hein, D.W.; Garcia-Closas, R.; Serra, C.; Dosemeci, M.; et al. Evidence for an intensity-dependent interaction of NAT2 acetylation genotype and cigarette smoking in the Spanish Bladder Cancer Study. Leuk. Res. 2007, 36, 236–241. [Google Scholar] [CrossRef][Green Version]
- Lebret, T.; Bonastre, J.; Fraslin, A.; Neuzillet, Y.; Droupy, S.; Rebillard, X.; De la Taille, A.; Guy, L.; Villers, A.; Schneider, M.; et al. COBLAnCE: A Prospective Cohort to Study Prognostic and Predictive Factors in Bladder Cancer and to Generate Re-al-World Data on Treatment Patterns, Resource Use and Quality of Life. BMJ Open 2023, in press. [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco Smoke and Involuntary Smoking; IARC: Lyon, France, 2004. [Google Scholar]
- Malhotra, J.; Borron, C.; Freedman, N.D.; Abnet, C.C.; Brandt, P.A.v.D.; White, E.; Milne, R.L.; Giles, G.G.; Boffetta, P. Association between Cigar or Pipe Smoking and Cancer Risk in Men: A Pooled Analysis of Five Cohort Studies. Cancer Prev. Res. 2017, 10, 704–709. [Google Scholar] [CrossRef]
- Purcell, S.; Chang, C. PLINK, [v1.9]. Available online: www.cog-genomics.org/plink/1.9/ (accessed on 6 July 2023).
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Delaneau, O.; McVean, G.A.; Donnelly, P.; Lunter, G.; Marchini, J.L.; Myers, S.; Gupta-Hinch, A.; Iqbal, Z.; Mathieson, I.; Rimmer, A.; et al. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef]
- Howie, B.N.; Donnelly, P.; Marchini, J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PLOS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef] [PubMed]
- Howie, B.; Marchini, J.; Stephens, M. Genotype Imputation with Thousands of Genomes. G3 Genes|Genomes|Genetics 2011, 1, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Murcray, C.E.; Lewinger, J.P.; Gauderman, W.J. Gene-Environment Interaction in Genome-Wide Association Studies. Am. J. Epidemiology 2008, 169, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, Y.; Wedow, R.; Li, Y.; Brazel, D.M.; Chen, F.; Datta, G.; Davila-Velderrain, J.; McGuire, D.; Tian, C.; et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 2019, 51, 237–244. [Google Scholar] [CrossRef]
- Sakornsakolpat, P.; Morrow, J.D.; Castaldi, P.J.; Hersh, C.P.; Bossé, Y.; Silverman, E.K.; Manichaikul, A.; Cho, M.H. Integrative genomics identifies new genes associated with severe COPD and emphysema. Respir. Res. 2018, 19, 46. [Google Scholar] [CrossRef]
- Cheron, J.; D’exaerde, A.d.K. Drug addiction: From bench to bedside. Transl. Psychiatry 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Lin, J.; Peng, J.; Liu, G.; Deng, L. Overexpression of MECP2 attenuates cigarette smoke extracts induced lung epithelial cell injury by promoting CYP1B1 methylation. J. Toxicol. Sci. 2020, 45, 177–186. [Google Scholar] [CrossRef]
- Korytina, G.F.; Akhmadishina, L.Z.; Kochetova, O.V.; Aznabaeva, Y.G.; Zagidullin, S.Z.; Victorova, T.V. Polymorphic variants of glutamate receptor (GRIK5, GRIN2B) and serotonin receptor (HTR2A) genes are associated with chronic. Mol. Biol. 2017, 51, 533–542. [Google Scholar] [CrossRef]
- Vink, J.M.; Smit, A.B.; de Geus, E.J.; Sullivan, P.; Willemsen, G.; Hottenga, J.-J.; Smit, J.H.; Hoogendijk, W.J.; Zitman, F.G.; Peltonen, L.; et al. Genome-wide Association Study of Smoking Initiation and Current Smoking. Am. J. Hum. Genet. 2009, 84, 367–379. [Google Scholar] [CrossRef]
- Pros, E.; Saigi, M.; Alameda, D.; Gomez-Mariano, G.; Martinez-Delgado, B.; Alburquerque-Bejar, J.; Carretero, J.; Tonda, R.; Esteve-Codina, A.; Catala, I.; et al. Genome-wide profiling of non-smoking-related lung cancer cells reveals common RB1 rearrangements associated with histopathologic transformation in EGFR-mutant tumors. Ann. Oncol. 2020, 31, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, M.D. Common and Unique Biological Pathways Associated with Smoking Initiation/Progression, Nicotine Dependence, and Smoking Cessation. Neuropsychopharmacology 2009, 35, 702–719. [Google Scholar] [CrossRef] [PubMed]
- Uhl, G.R.; Liu, Q.-R.; Drgon, T.; Johnson, C.; Walther, D.; Rose, J.E.; David, S.P.; Niaura, R.; Lerman, C. Molecular Genetics of Successful Smoking Cessation. Arch. Gen. Psychiatry 2008, 65, 683–693. [Google Scholar] [CrossRef]
- Zhong, X.; Drgonova, J.; Li, C.-Y.; Uhl, G.R. Human cell adhesion molecules: Annotated functional subtypes and overrepresentation of addiction-associated genes. Ann. New York Acad. Sci. 2015, 1349, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, T.E.; Gudbjartsson, D.F.; Surakka, I.; Vink, J.M.; Amin, N.; Geller, F.; Sulem, P.; Rafnar, T.; Esko, T.; Walter, S.; et al. Sequence variants at CHRNB3–CHRNA6 and CYP2A6 affect smoking behavior. Nat. Genet. 2010, 42, 448–453. [Google Scholar] [CrossRef]
- Fernàndez-Castillo, N.; Gan, G.; van Donkelaar, M.M.; Vaht, M.; Weber, H.; Retz, W.; Meyer-Lindenberg, A.; Franke, B.; Harro, J.; Reif, A.; et al. RBFOX1, encoding a splicing regulator, is a candidate gene for aggressive behavior. Eur. Neuropsychopharmacol. 2017, 30, 44–55. [Google Scholar] [CrossRef]
- O’leary, A.; Fernàndez-Castillo, N.; Gan, G.; Yang, Y.; Yotova, A.Y.; Kranz, T.M.; Grünewald, L.; Freudenberg, F.; Antón-Galindo, E.; Cabana-Domínguez, J.; et al. Behavioural and functional evidence revealing the role of RBFOX1 variation in multiple psychiatric disorders and traits. Mol. Psychiatry 2022, 27, 4464–4473. [Google Scholar] [CrossRef]
- Bavarva, J.H.; Tae, H.; Settlage, R.E.; Garner, H.R. Characterizing the Genetic Basis for Nicotine Induced Cancer Development: A Transcriptome Sequencing Study. PLoS ONE 2013, 8, e67252. [Google Scholar] [CrossRef]
- Samanic, C.; Kogevinas, M.; Dosemeci, M.; Malats, N.; Real, F.X.; Garcia-Closas, M.; Serra, C.; Carrato, A.; García-Closas, R.; Sala, M.; et al. Smoking and Bladder Cancer in Spain: Effects of Tobacco Type, Timing, Environmental Tobacco Smoke, and Gender. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1348–1354. [Google Scholar] [CrossRef]
- De Goeij, L.; Westhoff, E.; Witjes, J.A.; Aben, K.K.; Kampman, E.; Kiemeney, L.A.; Vrieling, A. The UroLife study: Protocol for a Dutch prospective cohort on lifestyle habits in relation to non-muscle-invasive bladder cancer prognosis and health-related quality of life. BMJ Open 2019, 9, e030396. [Google Scholar] [CrossRef]
- Zeegers, M.P.; Bryan, R.T.; Langford, C.; Billingham, L.; Murray, P.; Deshmukh, N.S.; Hussain, S.; James, N.; Wallace, D.M.A.; Cheng, K. The West Midlands Bladder Cancer Prognosis Programme: Rationale and design. BJU Int. 2010, 105, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Kwan, M.L.; Kushi, L.H.; Danforth, K.N.; Roh, J.M.; Ergas, I.J.; Lee, V.S.; Cannavale, K.L.; Harrison, T.N.; Contreras, R.; Loo, R.K.; et al. The Be-Well Study: A prospective cohort study of lifestyle and genetic factors to reduce the risk of recurrence and progression of non-muscle-invasive bladder cancer. Cancer Causes Control. 2019, 30, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, E.; Wu, X.; Kiemeney, L.A.; Lerner, S.P.; Ye, Y.; Huang, M.; Dinney, C.P.; Vrieling, A.; Tu, H. Dietary patterns and risk of recurrence and progression in non-muscle-invasive bladder cancer. Int. J. Cancer 2017, 142, 1797–1804. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall (N = 1732) | Women (N = 303) | Men (N = 1429) | |
|---|---|---|---|
| Age (Years) | |||
| Mean (SD) | 68.55 (10.79) | 68.05 (12.46) | 68.65 (10.41) |
| Median (Q1, Q3) | 68.70 (61.74, 76.34) | 68.25 (60.27, 77.31) | 68.73 (62.07, 76.10) |
| Min; Max | 22.05; 95.31 | 22.2; 93.34 | 22.05; 95.31 |
| Missing | 1 | 0 | 1 |
| Smoking status | |||
| Never smoker | 308 (17.78%) | 115 (37.95%) | 193 (13.51%) |
| Ever smoker | 1366 (78.87%) | 176 (58.09%) | 1190 (83.28%) |
| Missing | 58 (3.35%) | 12 (3.96%) | 46 (3.22%) |
| Duration of smoking (Years) (N = 1366) | |||
| Mean (SD) | 34.10 (14.54) | 33.09 (14.17) | 34.25 (14.59) |
| Median (Q1, Q3) | 35.00 (24.00, 45.00) | 35.00 (22.00, 44.00) | 35.00 (24.00, 45.00) |
| Min; Max | 1; 76 | 1; 62 | 1; 76 |
| Missing | 83 | 17 | 66 |
| Average tobacco consumption in grams (N = 1366) | |||
| Mean (SD) | 19.82 (12.65) | 16.00 (11.45) | 20.36 (12.72) |
| Median (Q1, Q3) | 18.61 (11.43, 23.51) | 14.41 (9.23, 20.00) | 19.11 (12.16, 24.67) |
| Min; Max | 0.5; 115 | 1; 62.06 | 0.5; 115 |
| Missing | 83 | 17 | 66 |
| Age at smoking initiation (Years) (N = 1366) | |||
| Mean (SD) | 18.40 (5.83) | 20.79 (7.46) | 18.06 (5.47) |
| Median (Q1, Q3) | 18.00 (15.00, 20.00) | 18 (17.00, 22.00) | 17 (15.00, 20.00) |
| Min; Max | 7; 73 | 10; 56 | 7; 73 |
| Missing | 24 | 5 | 19 |
| Phenotype | rsID | Chr | Position | Locus | REF | ALT | MAF | OR (95% CI) | β (SE) | p-value | Annotation (Distance) | Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
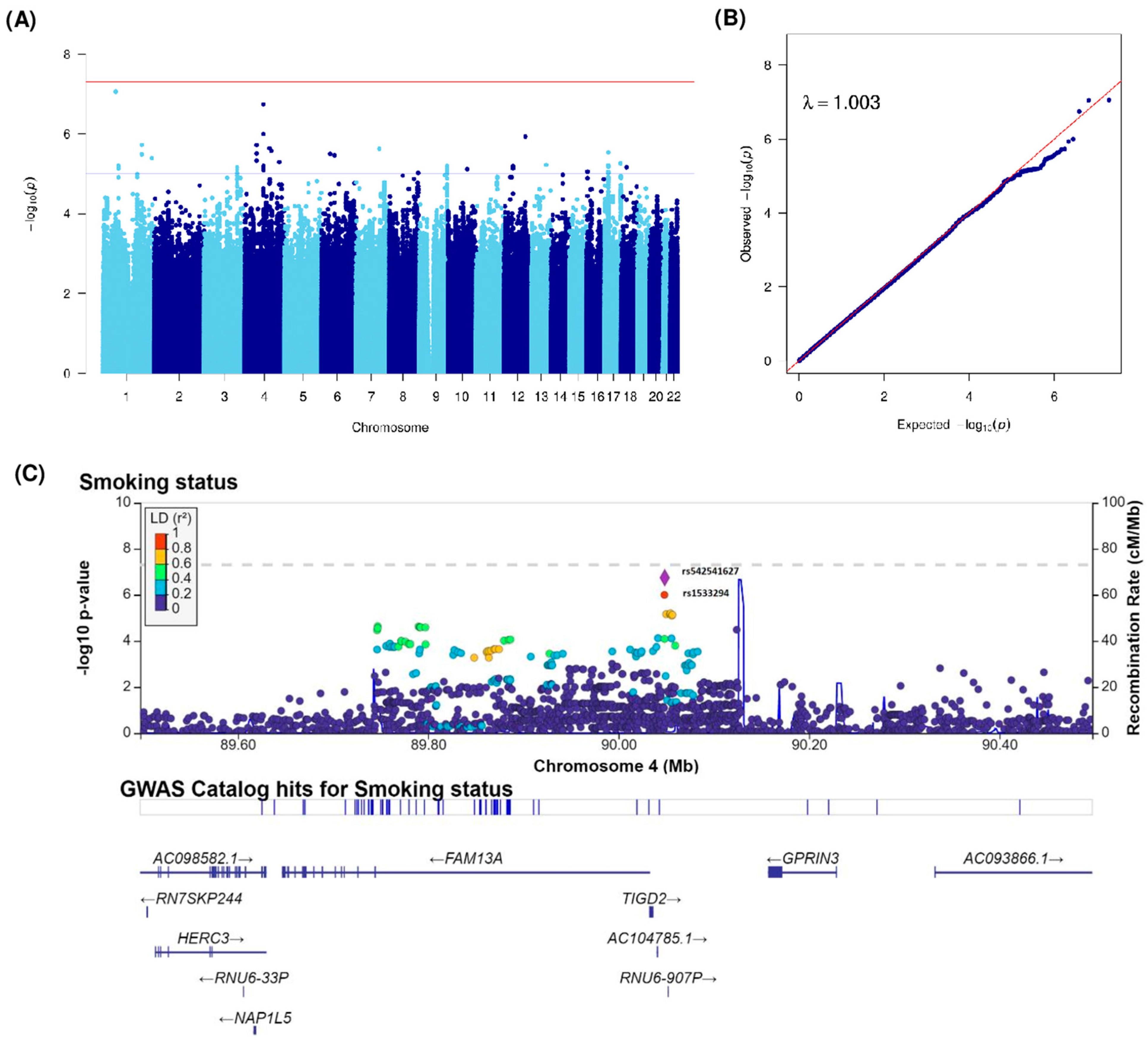
| Smoking status (Ever vs. Never) | rs114073636 | 1 | 62066340 | 1p31.3 | G | A | 0.04 | 0.31 (0.20–0.47) | 8.68 × 10−8 | NFIA (137880), MGC34796 (53574) | intergenic | |
| rs116571608 | 1 | 62062694 | 1p31.3 | G | A | 0.04 | 0.31 (0.20–0.47) | 8.87 × 10−8 | NFIA (134234), MGC34796 (57220) | intergenic | ||
| rs2110040 | 1 | 187842047 | 1q31.1 | G | C | 0.01 | 0.17 (0.08–0.36) | 1.88 × 10−6 | LINC01037 (395693) | intergenic | ||
| rs542541627 | 4 | 90048466 | 4q22.1 | A | AAAAACAAACAAAC | 0.44 | 1.69 (1.39–2.06) | 1.81 × 10−7 | TIGD2 (12414), GPRIN3 (109068) | intergenic | ||
| rs1533294 | 4 | 90048122 | 4q22.1 | C | T | 0.42 | 1.63 (1.34–1.98) | 1.01 × 10−6 | TIGD2 (12070), GPRIN3 (109412) | intergenic | ||
| rs80281369 | 4 | 55856707 | 4q12 | T | C | 0.04 | 0.36 (0.24–0.55) | 1.91 × 10−6 | KIT (249826), KDR (87941) | intergenic | ||
| rs11098419 | 4 | 118939953 | 4q26 | T | G | 0.30 | 1.62 (1.33–1.98) | 2.30 × 10−6 | LINC02264 (148850), NDST3 (15688) | intergenic | ||
| rs115317515 | 4 | 129223251 | 4q28.2 | C | T | 0.02 | 0.29 (0.17–0.49) | 2.64 × 10−6 | PGRMC2 (14283), LINC02615 (125920) | intergenic | ||
| rs76261406 | 7 | 110492301 | 7q31.1 | T | G | 0.03 | 0.23 (0.13–0.42) | 2.39 × 10−6 | IMMP2L | intronic | ||
| rs11112182 | 12 | 105139116 | 12q23.3 | A | G | 0.19 | 0.57 (0.45–0.71) | 1.16 × 10−6 | CHST11 | intronic | ||
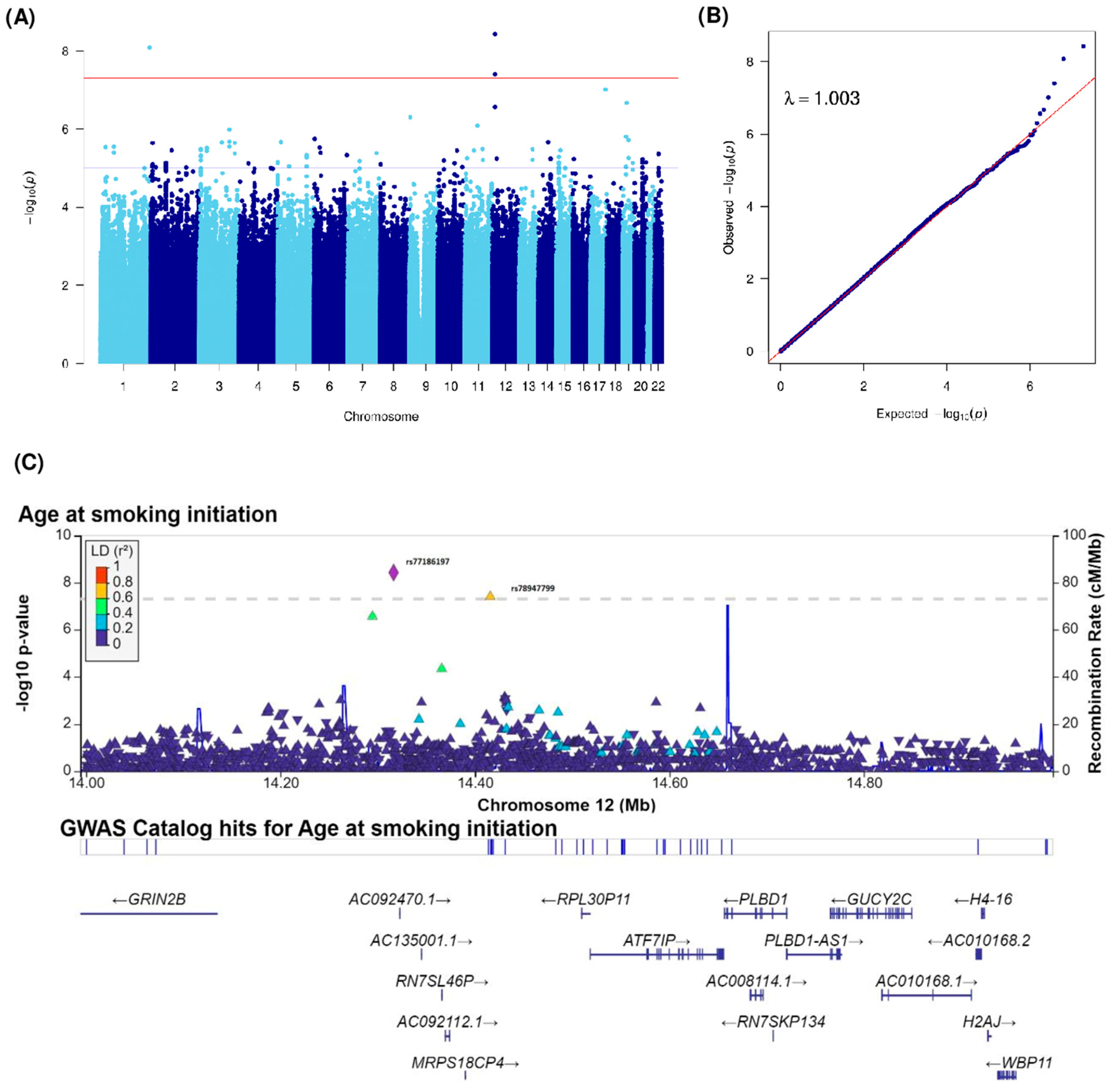
| Age at smoking initiation (Years) | rs531756449 | 1 | 244460577 | 1q44 | C | G | 0.02 | 0.3 (0.05) | 8.26 × 10−9 | ZBTB18 (239797), C1orf100 (55360) | intergenic | |
| rs140538571 | 3 | 149654460 | 3q25.1 | C | A | 0.01 | −0.24 (0.05) | 1.04 × 10−6 | RNF13 | intronic | ||
| rs115421081 | 3 | 149600807 | 3q25.1 | C | G | 0.01 | −0.24 (0.05) | 1.05 × 10−6 | RNF13 | intronic | ||
| rs148961658 | 9 | 4111766 | 9p24.2 | G | C | 0.01 | 0.26 (0.05) | 5.03 × 10−7 | GLIS3 | intronic | ||
| rs117818261 | 11 | 62644214 | 11q12.3 | C | T | 0.01 | 0.21 (0.04) | 8.13 × 10−7 | SLC3A2 | intronic | ||
| rs77186197 | 12 | 14316383 | 12p13.1 | C | T | 0.01 | 0.29 (0.05) | 3.74 × 10−9 | GRIN2B (183093), ATF7IP(202183) | intergenic | ||
| rs78947799 | 12 | 14415811 | 12p13.1 | A | C | 0.01 | 0.31 (0.06) | 3.97 × 10−8 | GRIN2B (282521), ATF7IP (102755) | intergenic | ||
| rs79782126 | 12 | 14294621 | 12p13.1 | T | C | 0.02 | 0.18 (0.03) | 2.76 × 10−7 | GRIN2B (161331), ATF7IP (223945) | intergenic | ||
| rs149790626 | 17 | 72716530 | 17q25.1 | C | T | 0.01 | 0.35 (0.06) | 9.76 × 10−8 | RAB37 | intronic | ||
| rs34177209 | 19 | 18474978 | 19p13.11 | T | A | 0.26 | 0.06 (0.01) | 1.57 × 10−6 | PGPEP1 | UTR3 | ||
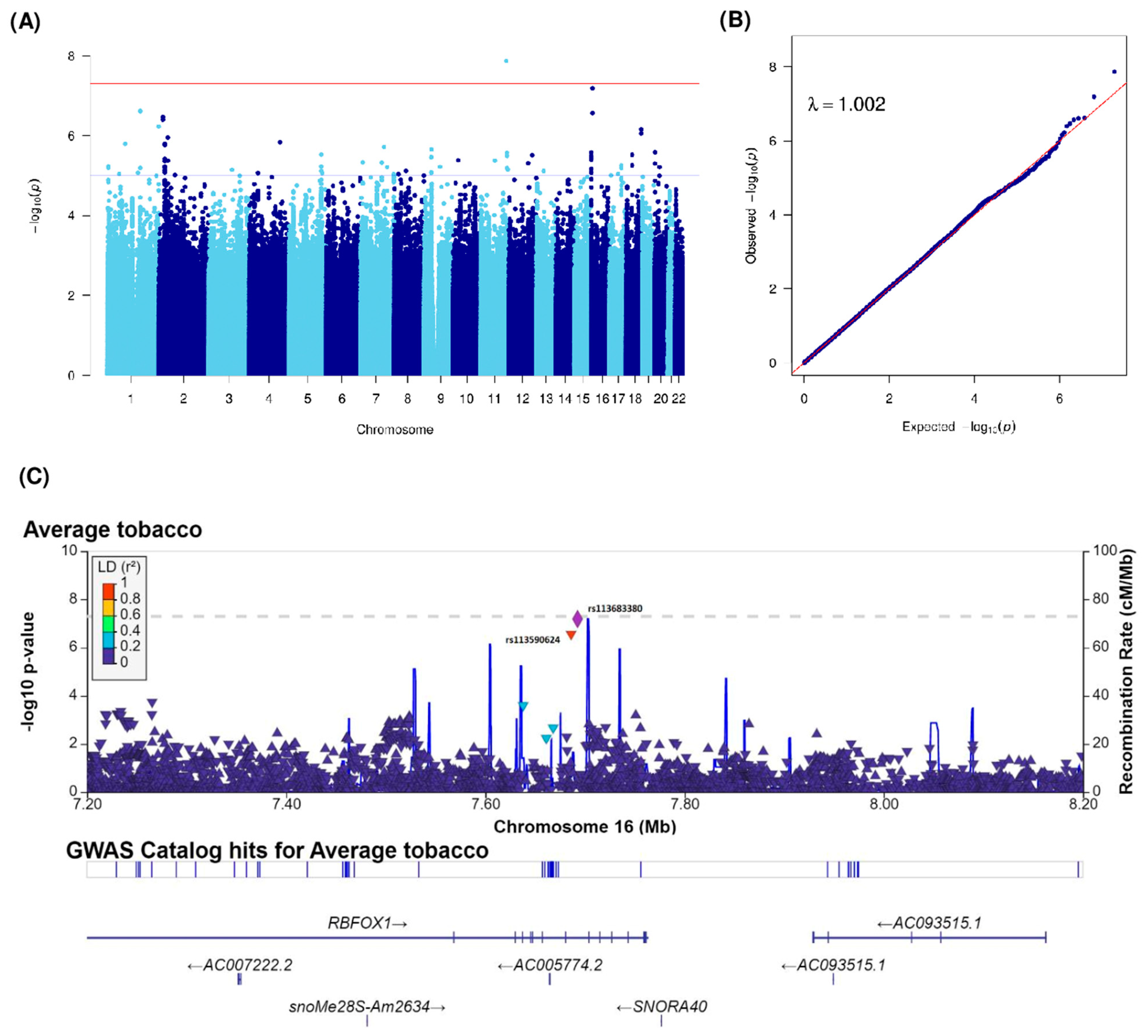
| Average tobacco consumption (grams) | rs114681930 | 1 | 160046712 | 1q23.2 | G | T | 0.01 | −0.7 (0.14) | 2.39 × 10−7 | KCNJ10 (6762), KCNJ9 (4616) | intergenic | |
| rs79752468 | 1 | 160050867 | 1q23.2 | T | A | 0.01 | −0.7 (0.14) | 2.44 × 10−7 | KCNJ9 (461) | upstream | ||
| rs142728151 | 1 | 248166173 | 1q44 | G | A | 0.02 | −0.58 (0.12) | 5.94 × 10−7 | OR2L13 | intronic | ||
| rs114634507 | 2 | 19247230 | 2p24.1 | G | A | 0.02 | −0.65 (0.13) | 3.44 × 10−7 | LINC01376 (20504), MIR4757 (300960) | intergenic | ||
| rs149246142 | 2 | 19292223 | 2p24.1 | G | A | 0.02 | −0.67 (0.13) | 4.01 × 10−7 | LINC01376 (65497), MIR4757 (255967) | intergenic | ||
| rs2714069 | 11 | 123389614 | 11q24.1 | A | G | 0.03 | 0.63 (0.11) | 1.35 × 10−8 | GRAMD1B | intronic | ||
| rs113683380 | 16 | 7692534 | 16p13.3 | G | A | 0.02 | −0.66 (0.12) | 6.44 × 10−8 | RBFOX1 | intronic | ||
| rs113590624 | 16 | 7686089 | 16p13.3 | G | C | 0.02 | −0.64 (0.12) | 2.74 × 10−7 | RBFOX1 | intronic | ||
| rs146671367 | 18 | 72146124 | 18q22.3 | C | T | 0.01 | −0.92 (0.18) | 6.89 × 10−7 | DIPK1C (21621), CNDP2 (17474) | intergenic | ||
| rs151292117 | 18 | 72145473 | 18q22.3 | G | A | 0.01 | −0.94 (0.19) | 8.84 × 10−7 | DIPK1C (20970), CNDP2 (18125) | intergenic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karimi, M.; Mendez-Pineda, S.; Blanché, H.; Boland, A.; Besse, C.; Deleuze, J.-F.; Meng, X.-Y.; Sirab, N.; Groussard, K.; Lebret, T.; et al. A Case-Only Genome-Wide Interaction Study of Smoking and Bladder Cancer Risk: Results from the COBLAnCE Cohort. Cancers 2023, 15, 4218. https://doi.org/10.3390/cancers15174218
Karimi M, Mendez-Pineda S, Blanché H, Boland A, Besse C, Deleuze J-F, Meng X-Y, Sirab N, Groussard K, Lebret T, et al. A Case-Only Genome-Wide Interaction Study of Smoking and Bladder Cancer Risk: Results from the COBLAnCE Cohort. Cancers. 2023; 15(17):4218. https://doi.org/10.3390/cancers15174218
Chicago/Turabian StyleKarimi, Maryam, Sebastian Mendez-Pineda, Hélène Blanché, Anne Boland, Céline Besse, Jean-François Deleuze, Xiang-Yu Meng, Nanor Sirab, Karine Groussard, Thierry Lebret, and et al. 2023. "A Case-Only Genome-Wide Interaction Study of Smoking and Bladder Cancer Risk: Results from the COBLAnCE Cohort" Cancers 15, no. 17: 4218. https://doi.org/10.3390/cancers15174218
APA StyleKarimi, M., Mendez-Pineda, S., Blanché, H., Boland, A., Besse, C., Deleuze, J.-F., Meng, X.-Y., Sirab, N., Groussard, K., Lebret, T., Bonastre, J., Allory, Y., Radvanyi, F., Benhamou, S., & Michiels, S. (2023). A Case-Only Genome-Wide Interaction Study of Smoking and Bladder Cancer Risk: Results from the COBLAnCE Cohort. Cancers, 15(17), 4218. https://doi.org/10.3390/cancers15174218