
A Novel Mouse Model of Combined Hepatocellular-Cholangiocarcinoma Induced by Diethylnitrosamine and Loss of Ppp2r5d

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Mouse Models
2.2. Histological Evaluation of Samples
2.3. Protein Extraction and Immunoblot Analysis
2.4. Statistical Analysis
3. Results
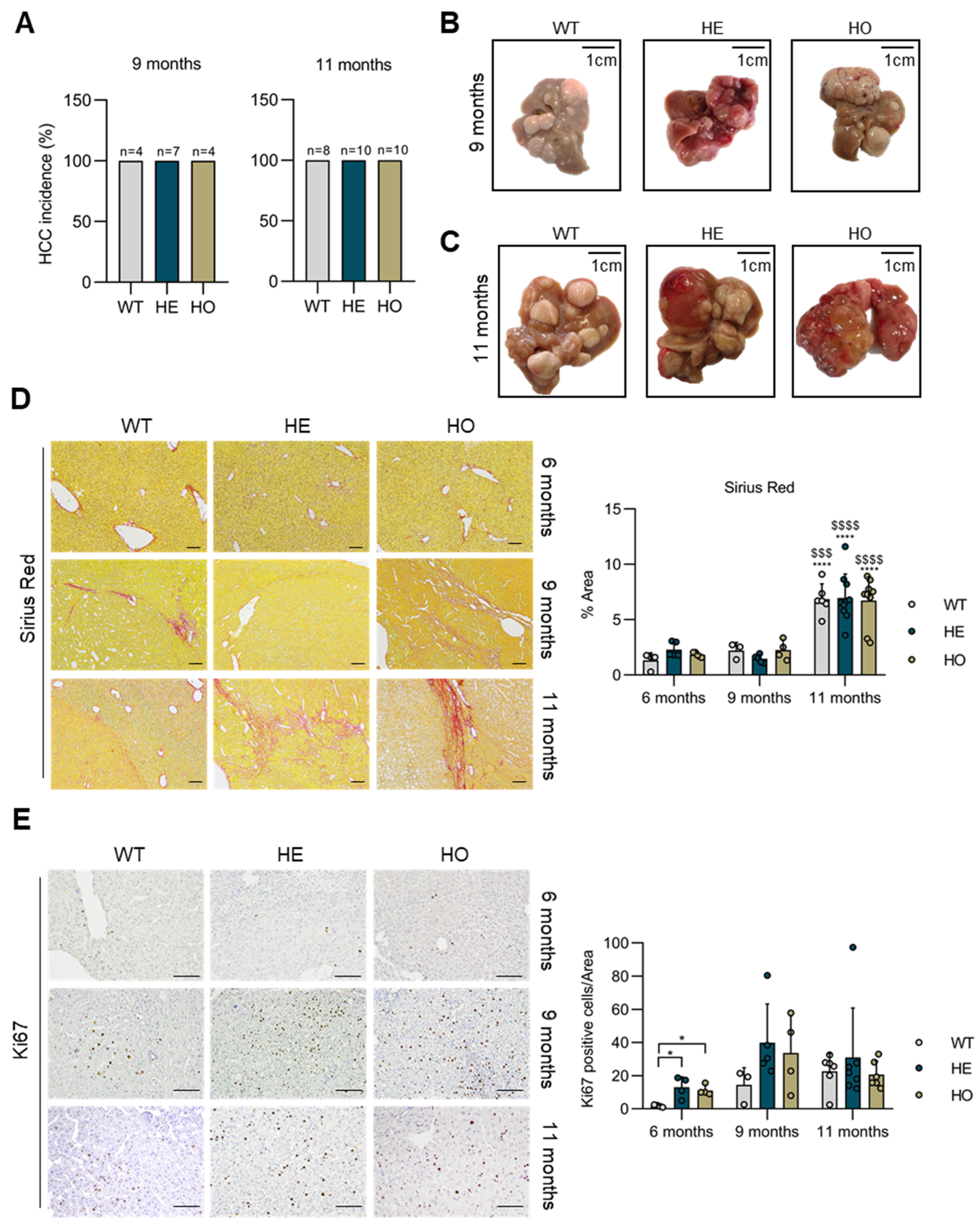
3.1. Analysis of Livers at 6 Months Post-DEN Injection Shows That Either Homozygous or Heterozygous Ppp2r5d Deletion Accelerates HCC Development
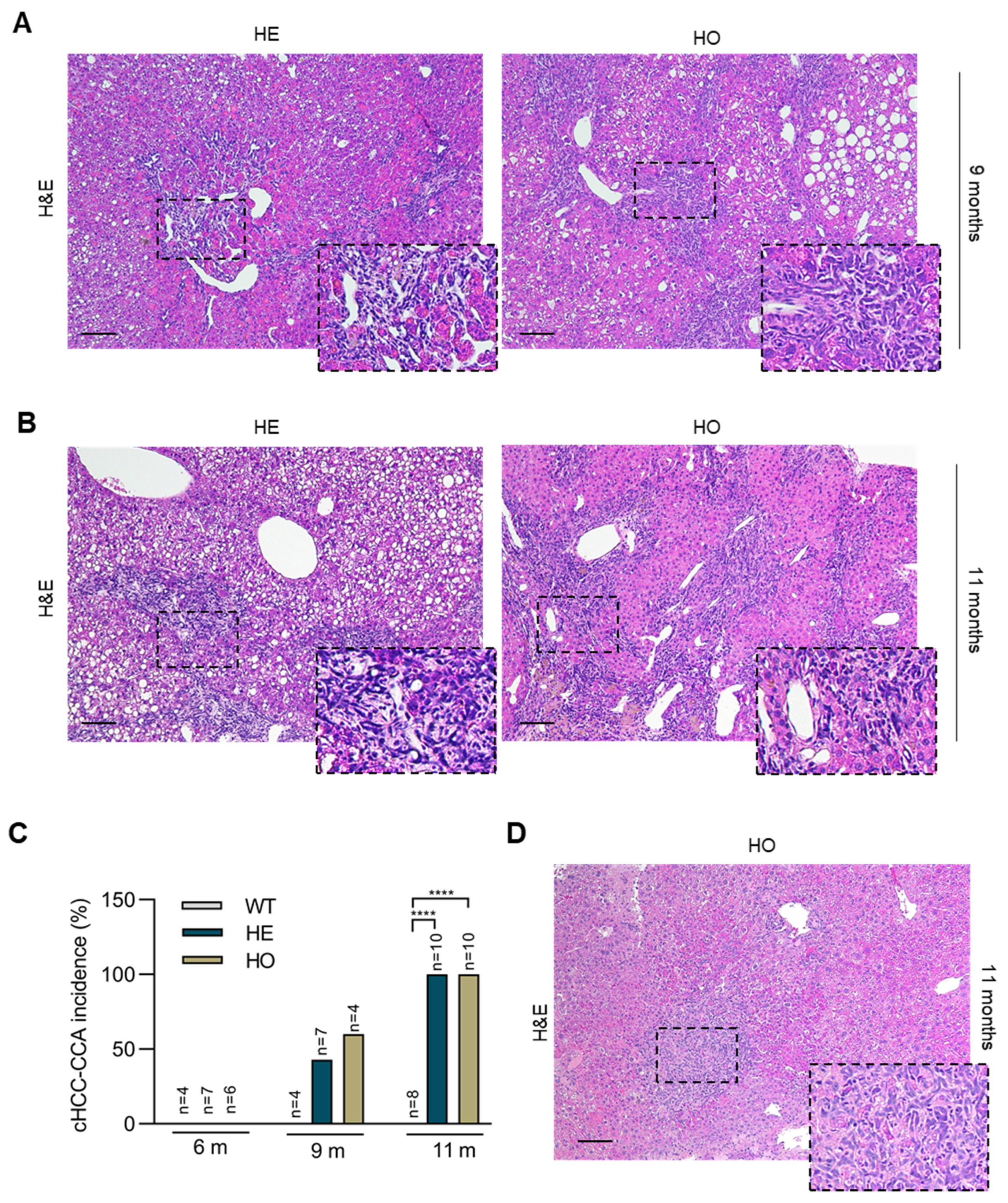
3.2. Analysis of Livers at 9 and 11 Months Post-DEN Injection Highlights the Onset of a Combined HCC-CCA Phenotype Specifically in Homozygous and Heterozygous Ppp2r5d KO Mice
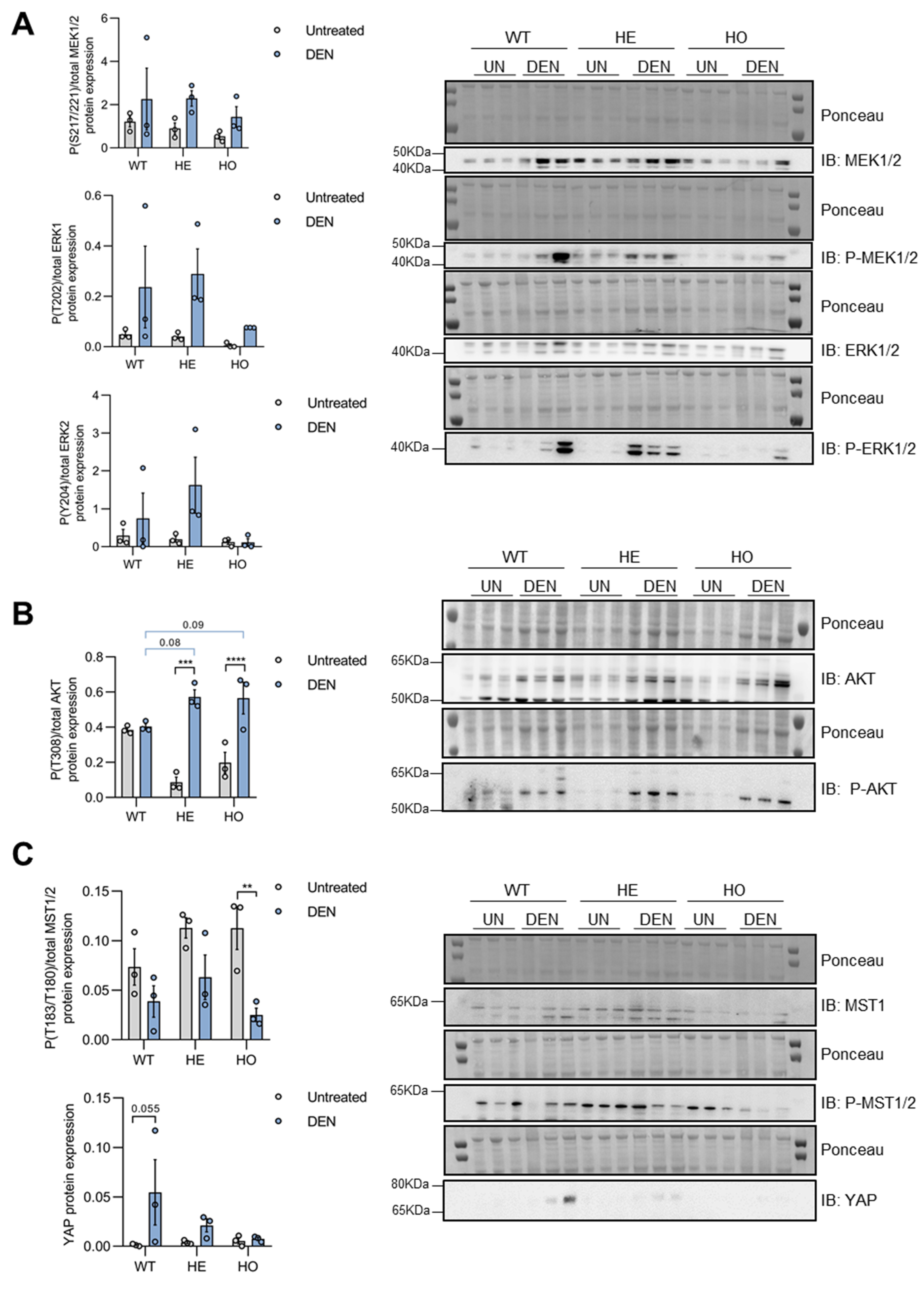
3.3. DEN Treatment Induces Activation of MAPK, AKT, and YAP Pathways in Pre-Malignant Liver Tissues of WT, HE, and HO Mice, with AKT Activation Being Affected by (Partial) Loss of Ppp2r5d
3.4. Only YAP, but Not MAPK or AKT Signaling, Is further Activated in Tumor versus Non-Tumor Tissue of DEN-Treated WT, HE, and HO Mice, with AKT and YAP Phosphorylation Being Affected by Ppp2r5d
3.5. Increased c-MYC Stabilization Contributes to the Liver Tumor Phenotype of HE and HO Mice
3.6. Ppp2r5d Expression Is Upregulated in DEN-Induced Liver Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome Sequencing of Hepatocellular Carcinomas Identifies New Mutational Signatures and Potential Therapeutic Targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-Ancestry Mutational Landscape of Hepatocellular Carcinoma Genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular Carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef]
- Burkhart, R.A.; Ronnekleiv-Kelly, S.M.; Pawlik, T.M. Personalized Therapy in Hepatocellular Carcinoma: Molecular Markers of Prognosis and Therapeutic Response. Surg. Oncol. 2017, 26, 138–145. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next Horizon in Mechanisms and Management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Mu, X.; Español-Suñer, R.; Mederacke, I.; Affò, S.; Manco, R.; Sempoux, C.; Lemaigre, F.P.; Adili, A.; Yuan, D.; Weber, A.; et al. Hepatocellular Carcinoma Originates from Hepatocytes and Not from the Progenitor/Biliary Compartment. J. Clin. Investig. 2015, 125, 3891–3903. [Google Scholar] [CrossRef]
- Tummala, K.S.; Brandt, M.; Teijeiro, A.; Graña, O.; Schwabe, R.F.; Perna, C.; Djouder, N. Hepatocellular Carcinomas Originate Predominantly from Hepatocytes and Benign Lesions from Hepatic Progenitor Cells. Cell Rep. 2017, 19, 584–600. [Google Scholar] [CrossRef]
- Nam, S.W.; Park, J.Y.; Ramasamy, A.; Shevade, S.; Islam, A.; Long, P.M.; Park, C.K.; Park, S.E.; Kim, S.Y.; Lee, S.H.; et al. Molecular Changes from Dysplastic Nodule to Hepatocellular Carcinoma through Gene Expression Profiling. Hepatology 2005, 42, 809–818. [Google Scholar] [CrossRef]
- Zucman-rossi, J.; Villanueva, A.; Nault, J.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. YGAST 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas Can Originate from Hepatocytes in Mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, S.; Suzuki, A. Intrahepatic Cholangiocarcinoma Can Arise from Notch-Mediated Conversion of Hepatocytes. J. Clin. Investig. 2012, 122, 3914–3918. [Google Scholar] [CrossRef]
- Andrews, T.S.; Atif, J.; Liu, J.C.; Perciani, C.T.; Ma, X.Z.; Thoeni, C.; Slyper, M.; Eraslan, G.; Segerstolpe, A.; Manuel, J.; et al. Single-Cell, Single-Nucleus, and Spatial RNA Sequencing of the Human Liver Identifies Cholangiocyte and Mesenchymal Heterogeneity. Hepatol. Commun. 2022, 6, 821–840. [Google Scholar] [CrossRef]
- Jeon, J.; Maeng, L.S.; Bae, Y.J.; Lee, E.J.; Yoon, Y.C.; Yoon, N. Comparing Clonality between Components of Combined Hepatocellular Carcinoma and Cholangiocarcinoma by Targeted Sequencing. Cancer Genom. Proteom 2018, 15, 291–298. [Google Scholar] [CrossRef]
- Malvi, D.; de Biase, D.; Fittipaldi, S.; Grillini, M.; Visani, M.; Pession, A.; D’Errico, A.; Vasuri, F. Immunomorphology and Molecular Biology of Mixed Primary Liver Cancers: Is Nestin a Marker of Intermediate-Cell Carcinoma? Histopathology 2020, 76, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Cholangiocarcinoma: Current Knowledge and Future Perspectives Consensus Statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.; Aishima, S.; Clavien, P.-A.; Fowler, K.; Goodman, Z.; Gores, G.; Gouw, A.; Kagen, A.; Klimstra, D.; Komuta, M.; et al. CHCC-CCA: Consensus Terminology for Primary Liver Carcinomas with Both Hepatocytic and Cholangiocytic Differentation. Hepatology 2018, 68, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J.; Hoof, C. Van PP2A: The Expected Tumor Suppressor. Curr. Opin. Genet. Dev. 2005, 15, 34–41. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J. Protein Phosphatase 2A: A Highly Regulated Family of Serine/Threonine Phosphatases Implicated in Cell Growth and Signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Eichhorn, P.J.A.; Creyghton, M.P.; Bernards, R. Protein Phosphatase 2A Regulatory Subunits and Cancer. Biochim. Biophys. Acta 2009, 1795, 1–15. [Google Scholar] [CrossRef]
- Perrotti, D.; Neviani, P. Protein Phosphatase 2A: A Target for Anticancer Therapy. Lancet Oncol. 2013, 14, 229–238. [Google Scholar] [CrossRef]
- Meeusen, B.; Janssens, V. Tumor Suppressive Protein Phosphatases in Human Cancer: Emerging Targets for Therapeutic Intervention and Tumor Stratification. Int. J. Biochem. Cell Biol. 2018, 96, 98–134. [Google Scholar] [CrossRef] [PubMed]
- Kauko, O.; Westermarck, J. Non-Genomic Mechanisms of Protein Phosphatase 2A (PP2A) Regulation in Cancer. Int. J. Biochem. Cell Biol. 2018, 96, 157–164. [Google Scholar] [CrossRef]
- Pavic, K.; Gupta, N.; Omella, J.D.; Derua, R.; Aakula, A.; Huhtaniemi, R.; Määttä, J.A.; Höfflin, N.; Okkeri, J.; Wang, Z.; et al. Structural Mechanism for Inhibition of PP2A-B56α and Oncogenicity by CIP2A. Nat. Commun. 2023, 14, 1143. [Google Scholar] [CrossRef]
- Narla, G.; Sangodkar, J.; Ryder, C.B. The Impact of Phosphatases on Proliferative and Survival Signaling in Cancer. Cell. Mol. Life Sci. 2018, 75, 2695–2718. [Google Scholar] [CrossRef] [PubMed]
- Aakula, A.; Sharma, M.; Tabaro, F.; Nätkin, R.; Kamila, J.; Honkanen, H.; Schapira, M.; Arrowsmith, C.; Nykter, M.; Westermarck, J. RAS and PP2A Activities Converge on Epigenetic Gene Regulation. Life Sci. Alliance 2023, 6, e202301928. [Google Scholar] [CrossRef] [PubMed]
- Sablina, A.A.; Hector, M.; Colpaert, N.; Hahn, W.C. Identification of PP2A Complexes and Pathways Involved in Cell Transformation. Cancer Res. 2010, 70, 10474–10484. [Google Scholar] [CrossRef]
- Meeusen, B.; Cortesi, E.E.; Omella, J.D.; Sablina, A.; Ventura, J.-J.; Janssens, V. PPP2R4 Dysfunction Promotes KRAS-Mutant Lung Adenocarcinoma Development and Mediates Opposite Responses to MEK and MTOR Inhibition. Cancer Lett. 2021, 520, 57–67. [Google Scholar] [CrossRef]
- Mumby, M. PP2A: Unveiling a Reluctant Tumor Suppressor. Cell 2007, 130, 21–24. [Google Scholar] [CrossRef]
- He, H.; Wu, G.; Li, W.; Cao, Y.; Liu, Y. CIP2A Is Highly Expressed in Hepatocellular Carcinoma and Predicts Poor Prognosis. Diagn. Mol. Pathol. 2012, 21, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.H.; Chen, Y.L.; Chu, P.Y.; Shih, C.T.; Yu, H.C.; Tai, W.T.; Shiau, C.W.; Chen, K.F. Upregulation of the Oncoprotein SET Determines Poor Clinical Outcomes in Hepatocellular Carcinoma and Shows Therapeutic Potential. Oncogene 2016, 35, 4891–4902. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shi, W.Y.; He, W.; Yan, Z.W.; Liu, M.H.; Chen, J.; Yang, Y.S.; Wang, Y.Q.; Chen, G.Q.; Huang, Y. FAM122A Supports the Growth of Hepatocellular Carcinoma Cells and Its Deletion Enhances Doxorubicin-Induced Cytotoxicity. Exp. Cell Res. 2020, 387, 111714. [Google Scholar] [CrossRef]
- Cristóbal, I.; Lamarca, A. Role of the PP2A Pathway in Cholangiocarcinoma: State of the Art and Future Perspectives. Cancers 2022, 14, 5422. [Google Scholar] [CrossRef]
- Xian, L.; Hou, S.; Huang, Z.; Tang, A.; Shi, P.; Wang, Q.; Song, A.; Jiang, S.; Lin, Z.; Guo, S.; et al. Liver-Specific Deletion of Ppp2cα Enhances Glucose Metabolism and Insulin Sensitivity. Aging 2015, 7, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.S.; Seibert, O.; Klöting, N.; Dietrich, A.; Straßburger, K.; Fernández-Veledo, S.; Vendrell, J.J.; Zorzano, A.; Blüher, M.; Herzig, S.; et al. PPP2R5C Couples Hepatic Glucose and Lipid Homeostasis. PLoS Genet. 2015, 11, e1005561. [Google Scholar] [CrossRef]
- Lu, N.; Liu, Y.; Tang, A.; Chen, L.; Miao, D.; Yuan, X. Hepatocyte-Specific Ablation of PP2A Catalytic Subunit α Attenuates Liver Fibrosis Progression via TGF-β 1/Smad Signaling. BioMed Res. Int. 2015, 2015, 794862. [Google Scholar] [CrossRef]
- Lambrecht, C.; Libbrecht, L.; Sagaert, X.; Pauwels, P.; Hoorne, Y.; Crowther, J.; Louis, J.V.; Sents, W.; Sablina, A.; Janssens, V. Loss of Protein Phosphatase 2A Regulatory Subunit B56 δ Promotes Spontaneous Tumorigenesis in Vivo. Oncogene 2018, 37, 544–552. [Google Scholar] [CrossRef]
- Lambrecht, C.; Ferreira, G.; Domenech, J.; Libbrecht, L.; De vos, R.; Derua, R.; Mathieu, C.; Overbergh, L.; Waelkens, E.; Janssens, V. Differential Proteomic Analysis of Hepatocellular Carcinomas from Ppp2r5d Knockout Mice and Normal (Knockout) Livers. Cancer Genom. Proteom. 2020, 17, 669–685. [Google Scholar] [CrossRef]
- Heindryckx, F.; Colle, I.; Van Vlierberghe, H. Experimental Mouse Models for Hepatocellular Carcinoma Research. Int. J. Exp. Pathol. 2009, 90, 367–386. [Google Scholar] [CrossRef]
- Friemel, J.; Frick, L.; Unger, K.; Egger, M.; Parrotta, R.; Böge, Y.T.; Adili, A.; Karin, M.; Luedde, T.; Heikenwalder, M.; et al. Characterization of HCC Mouse Models: Towards an Etiology-Oriented Subtyping Approach. Mol. Cancer Res. 2019, 17, 1493–1502. [Google Scholar] [CrossRef]
- Connor, F.; Rayner, T.F.; Aitken, S.J.; Feig, C.; Lukk, M.; Santoyo-Lopez, J.; Odom, D.T. Mutational Landscape of a Chemically-Induced Mouse Model of Liver Cancer. J. Hepatol. 2018, 69, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Dow, M.; Pyke, R.M.; Tsui, B.Y.; Alexandrov, L.B.; Nakagawa, H.; Taniguchi, K.; Seki, E.; Harismendy, O.; Shalapour, S.; Karin, M.; et al. Integrative Genomic Analysis of Mouse and Human Hepatocellular Carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E9879–E9888. [Google Scholar] [CrossRef]
- Lee, J.S.; Chu, I.S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of Comparative Functional Genomics to Identify Best-Fit Mouse Models to Study Human Cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef]
- Schulien, I.; Hasselblatt, P. Diethylnitrosamine-Induced Liver Tumorigenesis in Mice. Methods Cell Biol. 2021, 163, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Delire, B.; Stärkel, P. The Ras/MAPK Pathway and Hepatocarcinoma: Pathogenesis and Therapeutic Implications. Eur. J. Clin. Invest. 2015, 45, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Louis, J.V.; Martens, E.; Borghgraef, P.; Lambrecht, C.; Sents, W.; Longin, S.; Zwaenepoel, K.; Pijnenborg, R.; Landrieu, I.; Lippens, G.; et al. Mice Lacking Phosphatase PP2A Subunit PR61/B′δ (Ppp2r5d) Develop Spatially Restricted Tauopathy by Deregulation of CDK5 and GSK3β. Proc. Natl. Acad. Sci. USA 2011, 108, 6957–6962. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Mieulet, V.; Burgess, D.; Findlay, G.M.; Sully, K.; Procter, J.; Goris, J.; Janssens, V.; Morrice, N.A.; Lamb, R.F. PP2AT61ε Is an Inhibitor of MAP4K3 in Nutrient Signaling to MTOR. Mol. Cell 2010, 37, 633–642. [Google Scholar] [CrossRef]
- Kurma, K.; Manches, O.; Chuffart, F.; Sturm, N.; Gharzeddine, K.; Zhang, J.; Mercey-ressejac, M.; Rousseaux, S.; Millet, A.; Lerat, H.; et al. DEN-Induced Rat Model Reproduces Key Features of Human Hepatocellular Carcinoma. Cancers 2021, 13, 4981. [Google Scholar] [CrossRef]
- Liu, Y.; Zhuo, S.; Zhou, Y.; Ma, L.; Sun, Z.; Wu, X.; Wang, X.W.; Gao, B.; Yang, Y. Yap-Sox9 Signaling Determines Hepatocyte Plasticity and Lineage-Specific Hepatocarcinogenesis. J. Hepatol. 2022, 76, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, Q.; Liang, N.; Xue, H.; Yang, T.; Chen, X.; Qiu, Z.; Zeng, C.; Sun, T.; Yuan, W.; et al. Oncogenic Driver Genes and Tumor Microenvironment Determine the Type of Liver Cancer. Cell Death Dis. 2020, 11, 313. [Google Scholar] [CrossRef]
- Cheuk-lam Lo, R.; Oi-lin Ng, I. Hepatic Progenitor Cells: Their Role and Functional Significance in the New Classification of Primary Liver Cancers. Liver Cancer 2013, 2, 84–92. [Google Scholar] [CrossRef]
- Morales-Ruiz, M.; Santel, A.; Ribera, J.; Jiménez, W. The Role of Akt in Chronic Liver Disease and Liver Regeneration. Semin. Liver Dis. 2017, 37, 11–16. [Google Scholar] [CrossRef]
- Van Kanegan, M.J.; Adams, D.G.; Wadzinski, B.E.; Strack, S. Distinct Protein Phosphatase 2A Heterotrimers Modulate Growth Factor Signaling to Extracellular Signal-Regulated Kinases and Akt. J. Biol. Chem. 2005, 280, 36029–36036. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Huang, K.Y.; Yang, C.H.; Yang, Y.S.; Lee, W.Y.; Chiang, C.W. Regulation of Phosphorylation of Thr-308 of Akt, Cell Proliferation, and Survival by the B55α Regulatory Subunit Targeting of the Protein Phosphatase 2A Holoenzyme to Akt. J. Biol. Chem. 2008, 283, 1882–1892. [Google Scholar] [CrossRef]
- He, X.; Li, M.; Yu, H.; Liu, G.; Wang, N.; Yin, C.; Tu, Q.; Narla, G.; Tao, Y.; Cheng, S.; et al. Loss of Hepatic Aldolase B Activates Akt and Promotes Hepatocellular Carcinogenesis by Destabilizing the Aldob/Akt/PP2A Protein Complex. PLoS Biol. 2020, 18, e3000803. [Google Scholar] [CrossRef]
- Papke, C.M.; Smolen, K.A.; Swingle, M.R.; Cressey, L.; Heng, R.A.; Toporsian, M.; Deng, L.; Hagen, J.; Shen, Y.; Chung, W.K.; et al. A Disorder-Related Variant (E420K) of a PP2A-Regulatory Subunit (PPP2R5D) Causes Constitutively Active AKT-MTOR Signaling and Uncoordinated Cell Growth. J. Biol. Chem. 2021, 296, 100313. [Google Scholar] [CrossRef]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.S.; et al. Hippo Signaling Is a Potent in Vivo Growth and Tumor Suppressor Pathway in the Mammalian Liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef]
- Perra, A.; Kowalik, M.A.; Ghiso, E.; Ledda-Columbano, G.M.; Di Tommaso, L.; Angioni, M.M.; Raschioni, C.; Testore, E.; Roncalli, M.; Giordano, S.; et al. YAP Activation Is an Early Event and a Potential Therapeutic Target in Liver Cancer Development. J. Hepatol. 2014, 61, 1088–1096. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Kaposi-Novak, P.; Libbrecht, L.; Woo, H.; Lee, Y.; Sears, N.C.; Conner, E.A.; Factor, V.M.; Roskams, T.; Thorgeirsson, S.S. Central Role of C-Myc during Malignant Conversion in Human Hepatocarcinogenesis. Cancer Res. 2009, 69, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Cubero, F.J.; Nevzorova, Y.A. C-MYC-Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease. Genes 2017, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Gerstenlauer, M.; Chan, L.K.; Leithäuser, F.; Yeh, M.M.; Wirth, T.; Maier, H.J. Block of NF-KB Signaling Accelerates MYC-Driven Hepatocellular Carcinogenesis and Modifies the Tumor Phenotype towards Combined Hepatocellular Cholangiocarcinoma. Cancer Lett. 2019, 458, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Hann, S.R. Role of Post-Translational Modifications in Regulating c-Myc Proteolysis, Transcriptional Activity and Biological Function. Semin. Cancer Biol. 2006, 16, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, H.; Li, X.; Vartuli, R.L.; Rowse, M.; Xing, Y.; Rudra, P.; Ghosh, D.; Zhao, R.; Ford, H.L. Eya3 Partners with PP2A to Induce C-Myc Stabilization and Tumor Progression. Nat. Commun. 2018, 9, 1047. [Google Scholar] [CrossRef]
- Arnold, H.K.; Sears, R.C. A Tumor Suppressor Role for PP2A-B56α through Negative Regulation of c-Myc and Other Key Oncoproteins. Cancer Metastasis Rev. 2008, 27, 147–158. [Google Scholar] [CrossRef]
- Liu, L.; Eisenman, R.N. Regulation of C-Myc Protein Abundance by a Protein Phosphatase 2A—Glycogen Synthase Kinase 3 β—Negative Feedback Pathway. Genes Cancer 2012, 3, 23–36. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of Glycogen Synthase Kinase-3 by Insulin Mediated by Protein Kinase, B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P.; Biondi, R.M. A Common Phosphate Binding Site Explains the Unique Substrate Specificity of GSK3 and Its Inactivation by Phosphorylation. Mol. Cell 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Martens, E.; Stevens, I.; Janssens, V.; Vermeesch, J.; Götz, J.; Goris, J.; Van Hoof, C. Genomic Organisation, Chromosomal Localisation Tissue Distribution and Developmental Regulation of the PR61/B′ Regulatory Subunits of Protein Phosphatase 2A in Mice. J. Mol. Biol. 2004, 336, 971–986. [Google Scholar] [CrossRef]
- Maronpot, R.R.; Fox, T.; Malarkey, D.E.; Goldsworthy, T.L. Mutations in the Ras Proto-Oncogene: Clues to Etiology and Molecular Pathogenesis of Mouse Liver Tumors. Toxicology 1995, 101, 125–156. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular Therapies and Precision Medicine for Hepatocellular Carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Steer, C.J.; Cressman, E.N.K. Molecular Mechanisms of Hepatocellular Carcinoma. Hepatology 2008, 48, 2047–2063. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Cressman, E.N.K.; Steer, C.J. Cellular and Molecular Mechanisms of Hepatocellular Carcinoma: An Update. Arch. Toxicol. 2013, 87, 227–247. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.M.; McKillop, I.H.; Cahill, P.A.; Sitzmann, J.V. Increased MAPK Expression and Activity in Primary Human Hepatocellular Carcinoma. Biochem. Biophys. Res. Commun. 1997, 236, 54–58. [Google Scholar] [CrossRef] [PubMed]
- McKillop, I.H.; Schmidt, C.M.; Cahill, P.A.; Sitzmann, J.V. Altered Expression of Mitogen-Activated Protein Kinases in a Rat Model of Experimental Hepatocellular Carcinoma. Hepatology 1997, 26, 1484–1491. [Google Scholar] [CrossRef]
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J.; et al. Activation of Mitogen-Activated Protein Kinases / Extracellular Signal-Regulated Kinases in Human Hepatocellular Carcinoma. Hepatology 1998, 27, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Oshima, H.; Katoh, M.; Tamai, Y.; Oshima, M.; Taketo, M.M. Hepatocarcinogenesis in Mice with β-Catenin and Ha-Ras Gene Mutations. Cancer Res. 2004, 64, 48–54. [Google Scholar] [CrossRef]
- Liu, Y.; Xin, B.; Yamamoto, M.; Goto, M.; Ooshio, T.; Kamikokura, Y.; Tanaka, H.; Meng, L.; Okada, Y.; Mizukami, Y.; et al. Generation of Combined Hepatocellular-Cholangiocarcinoma through Transdifferentiation and Dedifferentiation in P53-Knockout Mice. Cancer Sci. 2021, 112, 3111–3124. [Google Scholar] [CrossRef]
- Arnold, H.K.; Sears, R.C. Protein Phosphatase 2A Regulatory Subunit B56α Associates with C-Myc and Negatively Regulates c-Myc Accumulation. Mol. Cell. Biol. 2006, 26, 2832–2844. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-Dependent Phosphorylation Pathways Regulate Myc Protein Stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Li, L.; Che, L.; Tharp, K.M.; Park, H.M.; Pilo, M.G.; Cao, D.; Cigliano, A.; Latte, G.; Xu, Z.; Ribback, S.; et al. Differential Requirement for de Novo Lipogenesis in Cholangiocarcinoma and Hepatocellular Carcinoma of Mice and Humans. Hepatology 2016, 63, 1900–1913. [Google Scholar] [CrossRef]
- Coral, H.; Chunmei, W.; Mattu, S.; Destefanis, G.; Ladu, S.; Delegu, S.; Ambuster, J.; Fan, L. AKT and N-Ras Co-Activation in the Mouse Liver Promotes Rapid Carcinogenesis via MTORC1, FOXM1/SKP2, and c-Myc Pathways. Hepatology 2012, 55, 833–845. [Google Scholar] [CrossRef]
- Cunningham, C.E.; Li, S.; Vizeacoumar, F.S.; Bhanumathy, K.K.; Lee, J.S.; Parameswaran, S.; Furber, L.; Abuhussein, O.; Paul, J.M.; McDonald, M.; et al. Therapeutic Relevance of the Protein Phosphatase 2A in Cancer. Oncotarget 2016, 7, 61544–61561. [Google Scholar] [CrossRef] [PubMed]
- Baena, E.; Gandarillas, A.; Vallespinos, M.; Zanet, J.; Bachs, O.; Redondo, C.; Fabregat, I.; Martinez-A, C.; de Alboran, I.M.; Vallespinó, M.; et al. C-Myc Regulates Cell Size and Ploidy but Is Not Essential for Postnatal Proliferation in Liver. Proc. Natl. Acad. Sci. USA 2005, 102, 7286–7291. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, S.; Zhang, Y.; Jia, J.; Wang, J.; Liu, X.; Zhang, J.; Song, X.; Ribback, S.; Cigliano, A.; et al. TAZ Is Indispensable for C-MYC-Induced Hepatocarcinogenesis. J. Hepatol. 2022, 76, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Moya, I.M.; Castaldo, S.A.; van den Mooter, L.; Soheily, S.; Sansores-Garcia, L.; Jacobs, J.; Mannaerts, I.; Xie, J.; Verboven, E.; Hillen, H.; et al. Peritumoral Activation of the Hippo Pathway Effectors YAP and TAZ Suppresses Liver Cancer in Mice. Science 2019, 366, 1029–1034. [Google Scholar] [CrossRef]
- Walesky, C.; Edwards, G.; Borude, P.; Gunewardena, S.; O’Neil, M.; Yoo, B.; Apte, U. Hepatocyte Nuclear Factor 4 Alpha Deletion Promotes Diethylnitrosamine-Induced Hepatocellular Carcinoma in Rodents. Hepatology 2013, 57, 2480–2490. [Google Scholar] [CrossRef]
- Wang, W.; Wang, H.; Peters, M.; Ding, N.; Ribback, S.; Utpatel, K.; Cigliano, A.; Dombrowski, F.; Xu, M.; Chen, X.; et al. Loss of Fbxw7 Synergizes with Activated AKT Signaling to Promote C-Myc Dependent Cholangiocarcinogenesis. J. Hepatol. 2019, 71, 742–752. [Google Scholar] [CrossRef]
- D’Artista, L.; Moschopoulou, A.A.; Barozzi, I.; Craig, A.J.; Seehawer, M.; Herrmann, L.; Minnich, M.; Kang, T.-W.; Rist, E.; Henning, M.; et al. MYC Determines Lineage Commitment in Kras Driven Primary Liver Cancer Development. J. Hepatol. 2023, 79, 141–149. [Google Scholar] [CrossRef]
- Li, L.; Qian, M.; Chen, I.; Finkelstein, D.; Onar-thomas, A.; Johnson, M.; Calabrese, C.; Bahrami, A.; López-terrada, D.H.; Yang, J.J.; et al. Acquisition of Cholangiocarcinoma Traits during Advanced Hepatocellular Carcinoma Development in Mice. Am. J. Pathol. 2018, 188, 656–671. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Terakado, Y.; Nakagawa, H.; Hikiba, Y.; Fujii, T.; Matsubara, D.; Noguchi, R.; Zhu, C.; Yamamoto, K.; Kudo, Y.; et al. A Novel Mouse Model of Intrahepatic Cholangiocarcinoma Induced by Liver-Specific Kras Activation and Pten Deletion. Sci. Rep. 2016, 6, 23899. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Debebe, A.; Zeng, N.; Kopp, J.; He, L.; Sander, M.; Stiles, B.L. Transformation of SOX9+ Cells by Pten Deletion Synergizes with Steatotic Liver Injury to Drive Development of Hepatocellular and Cholangiocarcinoma. Sci. Rep. 2021, 11, 11823. [Google Scholar] [CrossRef]
- Li, X.; Tao, J.; Cigliano, A.; Sini, M.; Calderaro, J.; Azoulay, D.; Wang, C.; Liu, Y.; Jiang, L.; Evert, K.; et al. Co-Activation of PIK3CA and Yap Promotes Development of Hepatocellular and Cholangiocellular Tumors in Mouse and Human Liver. Oncotarget 2015, 6, 10102–10115. [Google Scholar] [CrossRef]
- Planas-Paz, L.; Sun, T.; Pikiolek, M.; Cochran, N.R.; Bergling, S.; Orsini, V.; Yang, Z.; Sigoillot, F.; Jetzer, J.; Syed, M.; et al. YAP, but Not RSPO-LGR4/5, Signaling in Biliary Epithelial Cells Promotes a Ductular Reaction in Response to Liver Injury. Cell Stem Cell 2019, 25, 39–53. [Google Scholar] [CrossRef]
- Oertel, M.; Shafritz, D.A. Stem Cells, Cell Transplantation and Liver Repopulation. Biochim. Biophys. Acta 2008, 1782, 61–74. [Google Scholar] [CrossRef]
- Turner, R.; Lozoya, O.; Wang, Y.; Cardinale, V.; Gaudio, E.; Alpini, G.; Mendel, G.; Wauthier, E.; Barbier, C.; Alvaro, D.; et al. Human Hepatic Stem Cell and Maturational Liver Lineage Biology. Hepatology 2011, 53, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, H.; Kuroki, T.; Kitasato, A.; Adachi, T.; Tanaka, T.; Hirabaru, M.; Hirayama, T.; Kuroshima, N.; Hidaka, M.; Soyama, A.; et al. Sox9 Expression in Carcinogenesis and Its Clinical Significance in Intrahepatic Cholangiocarcinoma. Dig. Liver Dis. 2015, 47, 1067–1075. [Google Scholar] [CrossRef]
- Yuan, X.; Li, J.; Coulouarn, C.; Lin, T.; Sulpice, L.; Bergeat, D.; De La Torre, C.; Liebe, R.; Gretz, N.; Ebert, M.P.A.; et al. SOX9 Expression Decreases Survival of Patients with Intrahepatic Cholangiocarcinoma by Conferring Chemoresistance. Br. J. Cancer 2018, 119, 1358–1366. [Google Scholar] [CrossRef]
- Al-Zahrani, K.N.; Abou-Hamad, J.; Pascoal, J.; Labrèche, C.; Garland, B.; Sabourin, L.A. AKT-Mediated Phosphorylation of Sox9 Induces Sox10 Transcription in a Murine Model of HER2-Positive Breast Cancer. Breast Cancer Res. 2021, 23, 55. [Google Scholar] [CrossRef]
- Suryo Rahmanto, A.; Savov, V.; Brunner, A.; Bolin, S.; Weishaupt, H.; Malyukova, A.; Rosén, G.; Čančer, M.; Hutter, S.; Sundström, A.; et al. FBW7 Suppression Leads to SOX9 Stabilization and Increased Malignancy in Medulloblastoma. EMBO J. 2016, 35, 2192–2212. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Liu, W.; Song, R.; Shah, J.J.; Feng, X.; Tsang, C.K.; Morgan, K.M.; Bunting, S.F.; Inuzuka, H.; Zheng, X.F.S.; et al. SOX9 Is Targeted for Proteasomal Degradation by the E3 Ligase FBW7 in Response to DNA Damage. Nucleic Acids Res. 2016, 44, 8855–8869. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domènech Omella, J.; Cortesi, E.E.; Verbinnen, I.; Remmerie, M.; Wu, H.; Cubero, F.J.; Roskams, T.; Janssens, V. A Novel Mouse Model of Combined Hepatocellular-Cholangiocarcinoma Induced by Diethylnitrosamine and Loss of Ppp2r5d. Cancers 2023, 15, 4193. https://doi.org/10.3390/cancers15164193
Domènech Omella J, Cortesi EE, Verbinnen I, Remmerie M, Wu H, Cubero FJ, Roskams T, Janssens V. A Novel Mouse Model of Combined Hepatocellular-Cholangiocarcinoma Induced by Diethylnitrosamine and Loss of Ppp2r5d. Cancers. 2023; 15(16):4193. https://doi.org/10.3390/cancers15164193
Chicago/Turabian StyleDomènech Omella, Judit, Emanuela E. Cortesi, Iris Verbinnen, Michiel Remmerie, Hanghang Wu, Francisco J. Cubero, Tania Roskams, and Veerle Janssens. 2023. "A Novel Mouse Model of Combined Hepatocellular-Cholangiocarcinoma Induced by Diethylnitrosamine and Loss of Ppp2r5d" Cancers 15, no. 16: 4193. https://doi.org/10.3390/cancers15164193
APA StyleDomènech Omella, J., Cortesi, E. E., Verbinnen, I., Remmerie, M., Wu, H., Cubero, F. J., Roskams, T., & Janssens, V. (2023). A Novel Mouse Model of Combined Hepatocellular-Cholangiocarcinoma Induced by Diethylnitrosamine and Loss of Ppp2r5d. Cancers, 15(16), 4193. https://doi.org/10.3390/cancers15164193