
Photodynamic Stromal Depletion in Pancreatic Ductal Adenocarcinoma

Abstract
:Simple Summary
Abstract
1. Introduction
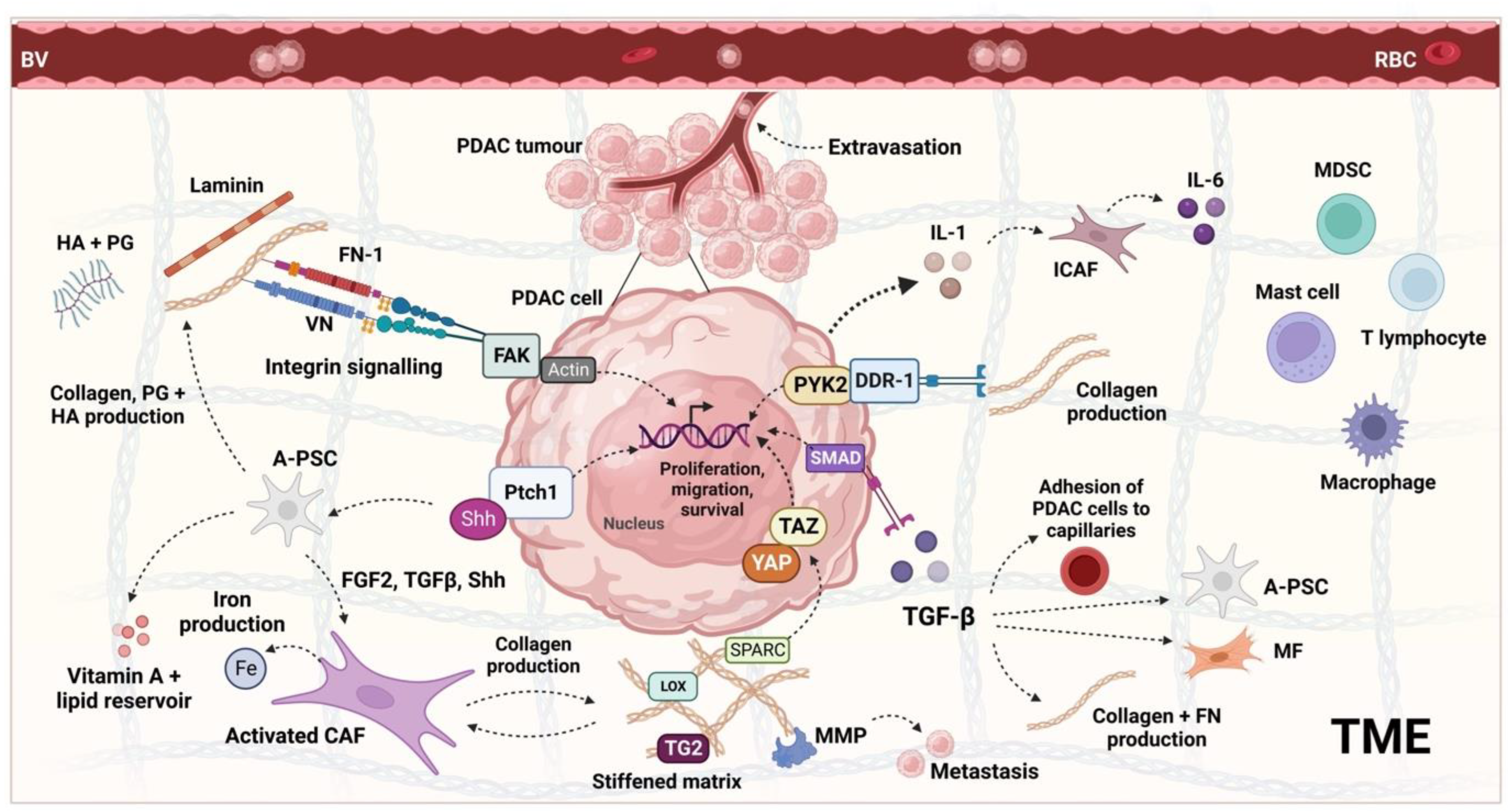
2. The PDAC Tumour Microenvironment and Its Regulation
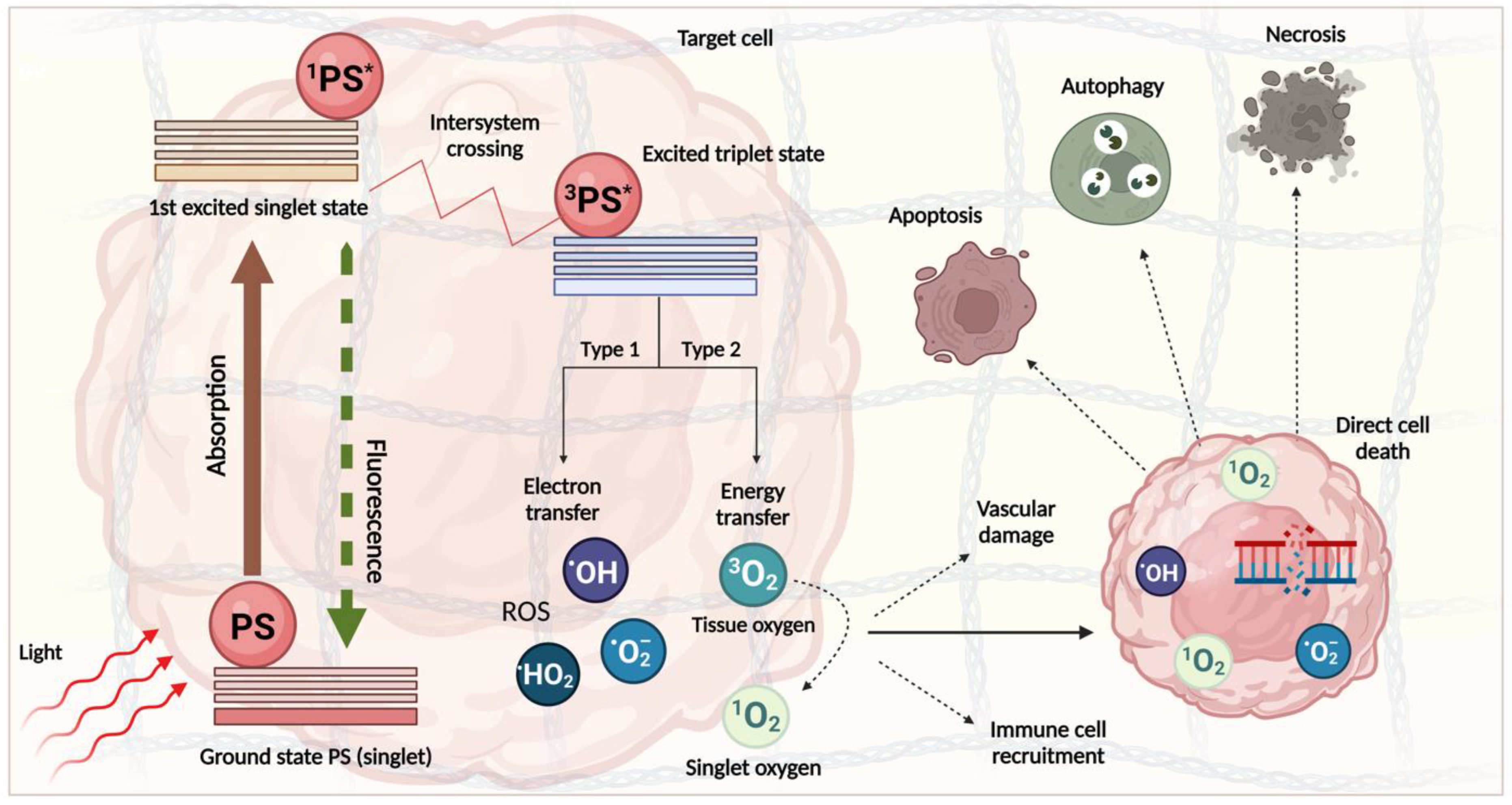
3. PDT Mechanism and Clinical Progress
Clinical Application of PDT for PDAC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | PS | PS Delivery | Light Source Delivery | Targeted or Untargeted PS | PS Dose | Light Dose | Drug–Light Interval | Cell Line or Species | Samples (n) | PDT Effect | Adverse Effects | Outcomes | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Schroder et al., 1988 | 125I-labeled DHE | IV | Bare laser | Untargeted | 4 mg/kg | 40 J/cm2 (fluence) | 3 h | Syrian golden hamster | Distribution study (33), PDT effect study (7) | Necrosis | Duodenal and jejunum perforation (3), death (4), haemorrhage (3), necrosis of liver and spleen (4). | PDT led to necrosis, haemorrhage, and inflammation of the tumours. Of the surviving 2 animals, there were no remaining areas of regular tumour. | [107] |
| Nuutinen et al., 1991 | AIS2Pc | Intra-IVC | Bare fibre on tissue surface | Untargeted | 1–5 μmol kg−1 | 50 J/cm2 (fluence) 50 mW/cm2 (intensity) | 48 h | Female Syrian golden hamster | 12 | Necrosis | At 5 μmol kg−1: duodenal perforations (2), gastric ulcer (2), bile leakage (2), necrosis of liver parenchyma. At 1 μmol kg−1: a duodenal perforation (1). | With 5 μmol kg−1 pancreatic, necrosis was observed. With 1 μmol kg−1, no photodynamic effect was seen in the pancreas (laser placed on pancreas). | [108] |
| Chatlani PT et al., 1992 | AIS2Pc | Intra-IVC | Bare fibre on tissue surface | Untargeted | 5 mg/kg | Normal pancreas: 50, 100, and 200 J/cm2 (fluence). Tumour: 50, 25, and 12.5 J/cm2 (fluence). 25 or 50 mW/cm2 (intensity). | 48 h | Female Syrian golden hamster | N/A | Necrosis | At 200 J/cm2, the normal pancreas demonstrated evidence of damage. In control animals, light doses > 50 J/cm2 led to gastric lesions. | All treated tumours showed evidence of coagulative haemorrhagic necrosis. Areas of necrosis extended beyond 5 mm in diameter (to the end of tumour nodules) when 25 or 50 J/cm2 doses were used. | [109] |
| Evrard S et al., 1994 | Pheophorbide A | IV | Bare fibre on tissue surface | Untargeted | 3 or 9 mg/kg | 35, 75, and 100 J/cm2 (fluence). 120 mW/cm2 (intensity) Halogen light use at 216 or 432 J/cm2 (fluence). | 24 h | Rat | Experimental (9), controls (36) | Necrosis | Duodenum injury | Six rats in experimental group were cured in 120 days. All control rats died in 35 days. | [110] |
| Regula J et al., 1994 | 5-(ALA) | IV or oral | Bare fibre on tissue surface or external irradiation using light-integrating cylindrical applicator | Untargeted | IV (200 mg/kg−1), oral (400 mg/kg−1) | Bare fibre: 50 J/cm2 (fluence) and 50 mW/cm2 (intensity) Cylinder application: 50 J/cm2 (fluence) and 100 mW/cm2 (intensity) | 4 and 5 h | Syrian golden hamster, pancreatic cancer line PC-1 | Experimental (9), controls (4) | Necrosis | Carcinomatosis (10), ascites (10), duodenal infiltration (2), malnutrition (2), and death (8) | Necrosis was evident in all 13 tumours. Smaller tumours showed complete necrosis. PDT-induced necrosis extended from the borders of the tumour. Mean survival time of controls was 42 days: animals treated with 400 mg/kg−1 oral ALA and 50 J/cm2 light using clinical applicator survived longer (p > 0.02). One mouse survived 116 days before being killed. | [111] |
| Mikvy P et al., 1996 | mTHPC | IV | Bare fibre on tissue surface | Untargeted | 1 mg/kg−1 for in vitro studies and 0.1 or 0.3 mg/kg−1 in vivo | 50 J/cm2 (fluence) 50 mW/cm2 (intensity) | 2 or 4 days | Syrian golden hamster | 20 | Necrosis | Free or sealed duodenal perforation (13), partial reversible bile duct obstruction (7) | Maximum necrosis seen 3 days after PDT. Lesions up to 4 mm in pancreas. Fractionating light dose increases lesion size by 30%. | [112] |
| Mikvy P et al., 1997 | mTHPC | Intra-IVC | Bare fibre on tissue surface | Untargeted | 0.1 or 0.3 mg/kg−1 | 50 J/cm2 (fluence) 50 mW/cm2 (intensity) | 2 or 4 days | Syrian golden hamster, pancreatic cancer line PC-1 | 16 | Necrosis | Duodenal perforation (3), bile duct obstruction (4), and duodenal diverticula (2) in experiments with higher dose (0.3 mg/kg−1) and fractionated light delivery | Tumour necrosis was highest 3 days post-PDT (maximum zone 12.4 mm in diameter using light fractionation). Treated tumours were histologically haemorrhagic in the centre and surrounded by inflammatory infiltrate. | [113] |
| Chan H-H et al., 2004 | Porfimer sodium (Photofrin) | IV | Percutaneous light catheters (EUS) | Untargeted | 4.4. mg/kg−1 | 50 J/cm2 (fluence) 0.4 W/cm2 (intensity) | 24 h | Pig (farm, swine) | 3 | Necrosis | Gross ecchymosis on pancreas surface (1), inflammation, slight haemorrhage (3) | Necrosis of 3.6 mm2 can be achieved with 50 J/cm2 light. One hundred percent necrosis could be achieved in the pancreas. EUS-guided, low-dose PDT for ablation of the pancreas is feasible and safe. | [114] |
| Tangutoori et al., 2016 | BPD (Visudyne) | IV | Transcutaneous | Targeted (anti-VEGF mAb bevacizumab) | 0.5 mg/kg | 75 J/cm2 (fluence) 100 mW/cm2 (intensity) | 1 h | AsPC-1, nude mice | Total animals used 54. | Necrosis | Temporary oedema and erythema. Weight loss was minimal, and all groups were within standard limits of toxicity. | Nanoliposomes (nanoPAL) achieved significantly enhanced tumour reduction. No tumour regrowth for 34 days after treatment in all treated mice. Thirty-three percent of nanoPAL mice had complete response. | [115] |
| Li et al., 2017 | 5-ALA (PpIX), Cy5.5 | IV | Direct laser irradiation | Targeted (U11 peptide) | 2 pmol per mouse | 50 mW cm−2 (intensity) | 24 h | PANC1-CTSE, nude mice | Total animals used 96 (24/group) | Apoptosis | None reported | Both PDT and PTT alone can mediate tumour reduction using targeted NPs. PDT + PTT is most effective, as indicated by a higher number of apoptotic cells post-treatment. PDT + PTT leads to a better survival rate (just 1 death over 40 days). | [116] |
| Obaid et al., 2019 | BPD (Visudyne) | IV | Direct NIR irradiation | Targeted (anti-EGFR mAb) | 0.5 mg/kg BPD equivalent | 40 J/cm2 (in vitro), 150 J/cm2 (in vivo) (fluence) 150 mW/cm2 (intensity in vitro) 100 mW/cm2 (intensity in vivo) | 12 h | PaCa-2, PCAF, mice | 24 total | Necrosis | Mice treated with targeted constructs remained healthy following PDT, whereas mice treated with untargeted constructs developed cachexia, weight loss, and moribundity. | Targeted low-dose PDT induced substantial necrosis in tumour tissues (3-fold increase in necrotic area 72 h post-PDT). Targeted PDT reduced tumour collagen density 1.5-fold. | [117] |
| Quilbe et al., 2020 | A novel PS named ‘PS2′ (Pyro-PEG-FA) bound to folic acid (WO2019 016397-A1) | Intraperitoneal injection | Homogenous illumination (in vitro), Extracorporeal fractionation (in vivo) | Targeted (PS-FOL/PS2) | 100 µL of PS solution at 1 mg/mL | 3.6 J/cm2 (in vitro), 29.7 J/cm2 (in vivo) (fluence) 1 mW/cm2 (intensity in vitro) 11 mW/cm2 (intensity in vivo) | N/A | Capan-1, Capan-2, MiapaCa-2, Panc-1, SCID mice | 8 | N/A | None reported | The PS preferentially binds to the membrane of pancreatic cancer cells and is internalised (intracellular labelling detected). Mice subjected to PDT showed tumour growth decrease over time after illumination. PS-FOL/PS2 significantly limits tumour growth in SCID mice. | [118] |
| De Silva et al., 2020 | BPD (Visudyne) | N/A | N/A | Untargeted | N/A | N/A | N/A | C57BL/6 mice | N/A | N/A | N/A | Infiltration of B- and T-cells to the tumour site was observed 1 h–5 days post-PDT. Activated T-cells and DCs were observed in the spleens of PDT-treated animals. | [119] |
| Sun et al., 2021 | Pyropheophorbide A (PPa) | IV | Direct laser irradiation | Targeted (HA) | 5 mg/kg | 200 mW/cm2 (intensity) | 24 h | Panc02 murine pancreatic tumour cells, C57BL/6 mice | Total animals used 25 (5/group) | Apoptosis | None reported | HA targeted Ppa-NPs activate CD8T+ cells in vivo. PDT-mediated tumour volume reductions and survival rates were highest with HA-targeted NPs containing Ppa and JQ1 (a BRD4 inhibiting drug). Apoptotic cell death was observed with NPs following PDT. | [120] |
| Vincent et al., 2021 | BPD (Visudyne) | N/A | Direct laser irradiation | Untargeted | 0.5 mg/kg | 75 J/cm2 (fluence) 100 mW/cm2 (intensity) | 1 h | BxPC-3 human PDAC cells, nude mice | 5 total (2 for PDT experiment) | Necrosis | None reported | PDT priming mediated a 13% collagen reduction and significant regions of necrosis in fresh tissue samples. | [121] |
| Obaid et al., 2022 | BPD (Visudyne) in liposomes (lipidated BPD or BPD-PC) | IV | Bare fibre on tissue surface | Targeted (anti-EGFR mAb) | 0.25, 0.50, or 0.75 mg/kg−1 BPD equivalent | In vitro—20 J cm2. In vivo—0, 50, 100, or 150 J cm2 (fluence) In vivo—100 mW/cm2 (intensity) | 90 min or 12 h | MIA PaCA-2 cell line, patient-derived CAFs, athymic male Swiss nu/nu mice | 4–8 per experiment | N/A | None reported | Targeted liposomes induce 90% tumour growth inhibition at 8.1% of the equivalent dose of nanoliposomal formula. Photoactivation is ineffective without EGFR targeting. Targeted liposomes reduce collagen density by >90% and increase collagen nonalignment by >103-fold. | [122] |
| Liu et al., 2022 | Protoporphyrin IX (PpIx) | Intratumoural injection | Bare fibre on tissue surface | Untargeted | 5.6 mg/kg | 1 W/cm2 (intensity) | 5 h | Panc-1 cells, nude mice | 25 total (5 per group) | Apoptosis | None reported | Significant apoptosis of PSCs was observed in the PDT group. Reduced ECM deposition (including collagen and fibronectin) and downregulated expression of TGF-β and CTGF was observed in the PDT group. PDT resulted in >87% tumour volume reduction. | [123] |
| Yang et al., 2022 | N/A | IV | Direct NIR irradiation | Au nanocage functionalised using collagenase and targeted using membrane coating | 10 mg/kg | N/A | N/A | BxPC3, BALB/c mice, BALB/c nude mice | 6 | Apoptosis, necrosis, ECM degradation | None reported | Targeted and collagenase functionalised NPs exhibited the greatest extent of cell necrosis/apoptosis and highest inhibition of tumour growth. Survival of mice was significantly longer in this group. | [124] |
| Study | PS Used | PS Delivery | Light Source Delivery | Targeted or Untargeted PS | PS Dose | Light Dose | Drug–Light Interval | Disease Staging | Number of Patients | Inclusion Criteria | Exclusion Criteria | PDT Effect | Side Effect | Survival | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bown et al., 2002 | mTHPC | IV | Laparoscopy | Untargeted | 0.15 mg/kg | 20–40 J/cm2 (fluence) 100 mW/cm2 (intensity) | 3 days | Stage 1–3 (UICC TNM) | 16 | Surgical suitability, Karnofsky status > 60% with anticipated survival of at least 3 months, attendance of follow-up | Ampullary cancers and those with cholangiocarcinoma, metastatic disease, previous specific treatment | Necrosis | Early: duodenal necrosis, CBD-duodenal fistula, necrosis around stents and ampulla, ulceration. Late: tumour ingrowth in stent, stenosis | Median 9.5 months | [105] |
| Gerdes et al., 2004 | Porfimer sodium (Photofrin) | IV | Percutaneous or endoscopic | Untargeted | N/A | N/A | 2 days | LA or advanced | 4 | Age > 18, primary carcinoma of pancreas, bile duct, gallbladder, or metastatic bile duct disease, unresectable disease, refusal of surgery if resectable, Karnofsky status 50–100%, bilirubin at least 2 mg/dL | Chemotherapy in last 4 weeks, concurrent RT or brachytherapy to the abdomen, administration of prior or concurrent experimental or investigational drugs | N/A | N/A | No results posted | [125] |
| Huggett et al., 2014 | Benzoporphyrin derivative (Verteporfin) | IV | Laparoscopy | Untargeted | 0.4 mg kg−1 | 5–40 J/cm2 (fluence) 150 mW/cm−1 (intensity) | 60–90 min | LA | 15 | Unsuitable for surgical resection, adequate biliary drainage, EOCG performance status of >2 | Porphyria, LA disease involving > 50% circumference of the duodenum or a major artery within treatment area, metastatic disease | Necrosis | Abdominal pain (3), rise in amylase (1) | Median 15.5 months | [75] |
| Choi J-H et al., 2015 | Chlorin e6 type derivative (Photolon) | IV | EUS | Untargeted | 2.5 mg/kg | 100 J/cm2 of diffuser length (fluence) 300 mW/cm2 (intensity) | 3 h | LA | 4 | LA disease with no metastasis after conventional CRT | Porphyria, major vessel in treatment area, ECOG > 2 | Necrosis | None reported | No long-term follow-up | [106] |
| DeWitt et al., 2019 NCT01770132 | Porfimer sodium (Photofrin) | IV | EUS | Untargeted | 1–2 mg/kg | 50 or 100 J/cm2 (total doses 150 or 300 J/cm2) (fluence) 400 mW/cm2 (intensity) | 40–50 h | T3N0 (1), T3N1 (3), T4N0 (3), T4N1 (5) | 12 | Aged 18–75, unresectable LA disease, Karnofsky status > 70% with anticipated survival of at least 3 months | Metastatic disease, previous CRT, gastric or duodenal ulcers, cystic component > 25% tumour volume, ascites, bowel fistula, portal HT, bulky celiac adenopathy (>2.5 cm diameter), uncorrelated coagulopathy, renal insufficiency, etc. | Necrosis | Sunburn (1), nausea (1), photosensitivity (1), skin hyperpigmentation (1), fatigue (1) | Median 11.5 months | [76] |
| Hanada et al., 2021 | BPD (Verteporfin) | IV | EUS | Untargeted | 0.4 mg/kg | 50 J/cm2 (fluence) 150 mW/cm2 (intensity) | 60–90 min | T3 (5), T2 (2), T1 (1) | 8 | LAPC with adequate biliary drainage | Metastatic disease, disease involving > 50% duodenal or major artery circumference, recent treatment with curative intent | Necrosis | None reported | As of November 2020, 7 patients died with a median survival time of 6.9 months from procedure date. Data collection ongoing | [77] |
| Chandrasekhara et al., 2022 | BPD (Verteporfin) | IV | EUS | Untargeted | 0.4 mg/kg | 50 J/cm2 (fluence) | 1 h | LA or advanced | 30 (still recruiting) | Age > 18, measurable disease defined by RECIST, ECOG of 0, 1, or 2, estimated life expectancy > 12 weeks, adequate biliary drainage | In LA patients, metastatic disease other than lung or liver; lung metastases with greater than 3 lesions or any lesion greater than 5 cm, porphyria, pregnant or breastfeeding, LA disease involving > 50% circumference of duodenum or artery in treatment area, recent treatment with curative intent, history of haemorrhagic diathesis or coagulopathy, other malignancy or systemic disease | Necrosis | None reported (data collection ongoing) | Data collection ongoing | [126] |
4. Role of PDAC Stroma and Implications for PDT
5. Pleiotropic Nature of the PDAC Stroma
6. The Effect of PDT on Stromal Components
6.1. Vascular Remodelling and Pruning
6.2. Targeting the Immune Cell Compartment
6.3. Preclinical Investigation of PDT for Stromal Depletion in PDAC
6.4. Combined Treatment Strategies for PDT-Mediated Stromal Depletion in PDAC
7. Recent Developments and Future Use of PDT for PDAC
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Schawkat, K.; Manning, M.A.; Glickman, J.N.; Mortele, K.J. Pancreatic Ductal Adenocarcinoma and Its Variants: Pearls and Perils. Radiographics 2020, 40, 1219–1239. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Carvajo, M.; Bauden, M.; Andersson, R. Pancreatic cancer stroma: Controversies and current insights. Scand. J. Gastroenterol. 2017, 52, 641–646. [Google Scholar] [CrossRef]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.M.; Thomassian, S.; Gong, J.; Hendifar, A.; Osipov, A. Advances in Pancreatic Ductal Adenocarcinoma Treatment. Cancers 2021, 13, 5510. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Gu, J.; Li, J. Mechanisms of drug resistance of pancreatic ductal adenocarcinoma at different levels. Biosci. Rep. 2020, 40, BSR20200401. [Google Scholar] [CrossRef]
- Braun, R.; Klinkhammer-Schalke, M.; Zeissig, S.R.; van Tol, K.K.; Bolm, L.; Honselmann, K.C.; Petrova, E.; Lapshyn, H.; Deichmann, S.; Abdalla, T.S.A.; et al. Clinical Outcome and Prognostic Factors of Pancreatic Adenosquamous Carcinoma Compared to Ductal Adenocarcinoma—Results from the German Cancer Registry Group. Cancers 2022, 14, 3946. [Google Scholar] [CrossRef]
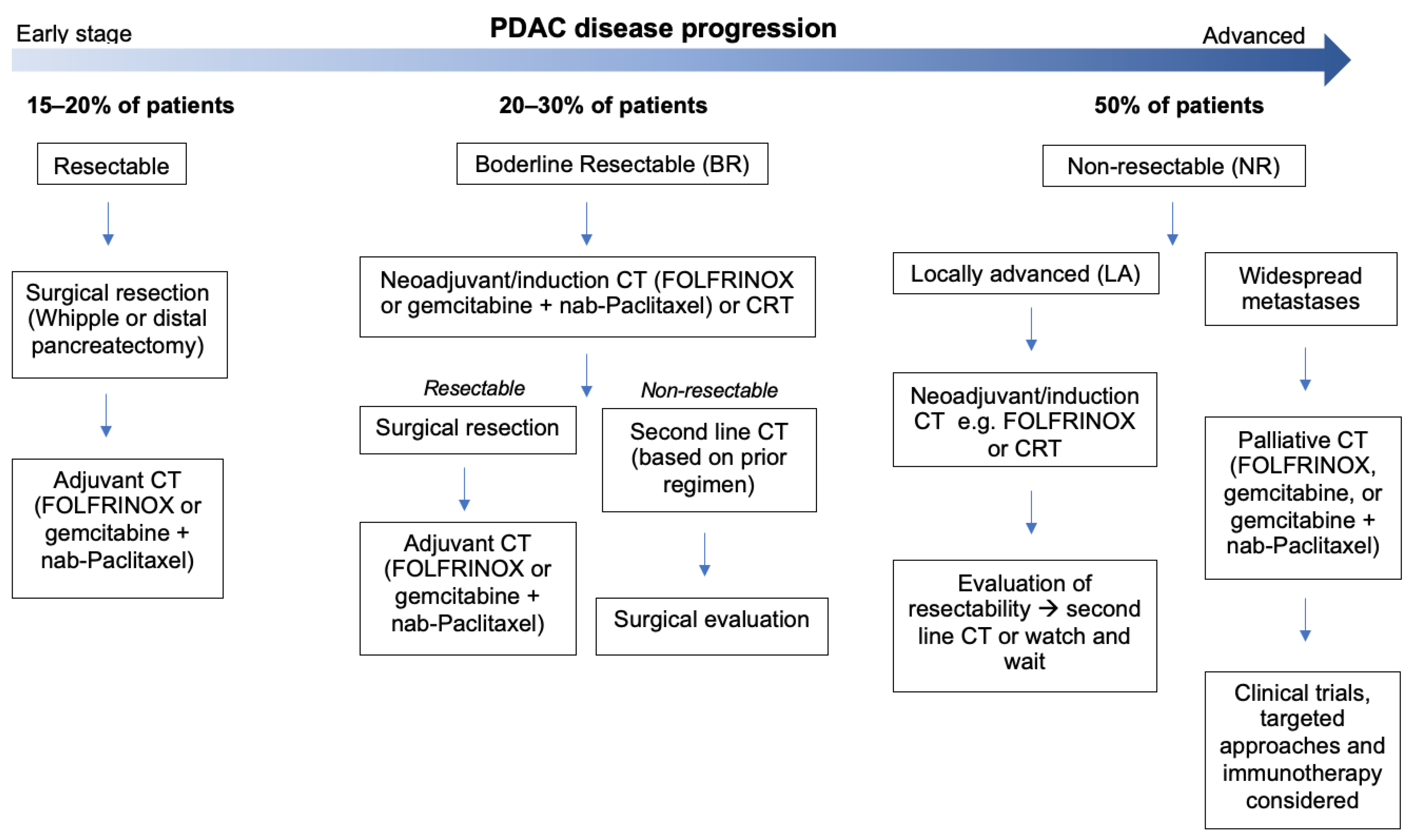
- Evans, D.B.; Ben George, B.; Tsai, S. Non-metastatic Pancreatic Cancer: Resectable, Borderline Resectable, and Locally Advanced-Definitions of Increasing Importance for the Optimal Delivery of Multimodality Therapy. Ann. Surg. Oncol. 2015, 22, 3409–3413. [Google Scholar] [CrossRef] [PubMed]
- Varadhachary, G.R.; Tamm, E.P.; Crane, C.; Evans, D.B.; Wolff, R.A. Borderline resectable pancreatic cancer. Curr. Treat. Opt. Gastroenterol. 2005, 8, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Isaji, S.; Mizuno, S.; Windsor, J.A.; Bassi, C.; Castillo, C.F.-D.; Hackert, T.; Hayasaki, A.; Katz, M.H.; Kim, S.-W.; Kishiwada, M.; et al. International consensus on definition and criteria of borderline resectable pancreatic ductal adenocarcinoma 2017. Pancreatology 2018, 18, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Nießen, A.; Hackert, T. State-of-the-art surgery for pancreatic cancer. Langenbeck’s Arch. Surg. 2021, 407, 443–450. [Google Scholar] [CrossRef]
- Groot, V.P.; Rezaee, N.; Wu, W.; Cameron, J.L.; Fishman, E.K.; Hruban, R.H.; Weiss, M.J.; Zheng, L.; Wolfgang, C.L.; He, J. Patterns, Timing, and Predictors of Recurrence Following Pancreatectomy for Pancreatic Ductal Adenocarcinoma. Ann. Surg. 2018, 267, 936–945. [Google Scholar] [CrossRef]
- Groot, V.P.; Gemenetzis, G.; Blair, A.B.; Rivero-Soto, R.J.; Yu, J.; Javed, A.A.; Burkhart, R.A.; Rinkes, I.H.M.B.; Molenaar, I.Q.; Cameron, J.L.M.; et al. Defining and Predicting Early Recurrence in 957 Patients with Resected Pancreatic Ductal Adenocarcinoma. Ann. Surg. 2019, 269, 1154–1162. [Google Scholar] [CrossRef]
- Parikh, A.A.; Maiga, A.; Bentrem, D.; Squires, M.H., III; Kooby, D.A.; Maithel, S.K.; Weber, S.M.; Cho, C.S.; Katz, M.; Martin, R.C.; et al. Adjuvant Therapy in Pancreas Cancer: Does It Influence Patterns of Recurrence? J. Am. Coll. Surg. 2016, 222, 448–456. [Google Scholar] [CrossRef]
- Shinde, R.S.; Bhandare, M.; Chaudhari, V.; Shrikhande, S.V. Cutting-edge strategies for borderline resectable pancreatic cancer. Ann. Gastroenterol. Surg. 2019, 3, 368–372. [Google Scholar] [CrossRef]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant Chemotherapy with Gemcitabine and Long-term Outcomes among Patients with Resected Pancreatic Cancer. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant Chemotherapy with Gemcitabine vs Observation in Patients Undergoing Curative-Intent Resection of Pancreatic Cancer. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; He, J.; Cameron, J.L.; Makary, M.; Soares, K.; Ahuja, N.; Rezaee, N.; Herman, J.; Zheng, L.; Laheru, D.; et al. The Impact of Postoperative Complications on the Administration of Adjuvant Therapy Following Pancreaticoduode-nectomy for Adenocarcinoma. Ann. Surg. Oncol. 2014, 21, 2873–2881. [Google Scholar] [CrossRef] [PubMed]
- Merkow, R.P.; Bilimoria, K.Y.; Tomlinson, J.S.; Paruch, J.L.; Fleming, J.B.; Talamonti, M.S.; Ko, C.Y.; Bentrem, D.J. Postoperative Complications Reduce Adjuvant Chemotherapy Use in Resectable Pancreatic Cancer. Ann. Surg. 2014, 260, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Åkerberg, D.; Björnsson, B.; Ansari, D. Factors influencing receipt of adjuvant chemotherapy after surgery for pancreatic cancer: A two-center retrospective cohort study. Scand. J. Gastroenterol. 2016, 52, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Labori, K.J.; Katz, M.H.; Tzeng, C.W.; Bjørnbeth, B.A.; Cvancarova, M.; Edwin, B.; Kure, E.H.; Eide, T.J.; Dueland, S.; Buanes, T.; et al. Impact of early disease progression and surgical complications on adjuvant chemotherapy completion rates and survival in patients undergoing the surgery first approach for resectable pancreatic ductal adenocarcinoma—A population-based cohort study. Acta Oncol. 2015, 55, 265–277. [Google Scholar] [CrossRef]
- Tzeng, C.-W.D.; Cao, H.S.T.; Lee, J.E.; Pisters, P.W.; Varadhachary, G.R.; Wolff, R.A.; Abbruzzese, J.L.; Crane, C.H.; Evans, D.B.; Wang, H.; et al. Treatment Sequencing for Resectable Pancreatic Cancer: Influence of Early Metastases and Surgical Complications on Multimodality Therapy Completion and Survival. J. Gastrointest. Surg. 2013, 18, 16–25. [Google Scholar] [CrossRef]
- Muhammadzai, J.; Haider, K.; Moser, M.; Chalchal, H.; Shaw, J.; Gardiner, D.; Dueck, D.-A.; Ahmed, O.; Brunet, B.; Iqbal, M.; et al. Early discontinuation of adjuvant chemotherapy in patients with early-stage pancreatic cancer correlates with inferior survival: A multicenter population-based cohort study. PLoS ONE 2022, 17, e0263250. [Google Scholar] [CrossRef]
- Schwarz, L.; Bruno, M.; Parker, N.H.; Prakash, L.; Mise, Y.; Lee, J.E.; Vauthey, J.-N.; Aloia, T.A.; Conrad, C.; Fleming, J.B.; et al. Active Surveillance for Adverse Events within 90 Days: The Standard for Reporting Surgical Outcomes after Pancreatectomy. Ann. Surg. Oncol. 2015, 22, 3522–3529. [Google Scholar] [CrossRef]
- Polani, F.; Grierson, P.M.; Lim, K.-H. Stroma-targeting strategies in pancreatic cancer: Past lessons, challenges and prospects. World J. Gastroenterol. 2021, 27, 2105–2121. [Google Scholar] [CrossRef]
- Picozzi, V.J.; Stephen, O.-H.; Edwards, A.; Mandelson, M.T.; Dorer, R.; Rocha, F.G.; Alseidi, A.; Biehl, T.; Traverso, W.L.; Helton, W.S.; et al. Five-Year Actual Overall Survival in Resected Pancreatic Cancer: A Contemporary Single-Institution Experience from a Multidisciplinary Perspective. Ann. Surg. Oncol. 2017, 24, 1722–1730. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant Chemotherapy with Fluorouracil Plus Folinic Acid vs Gemcitabine Following Pancreatic Cancer Resection. JAMA 2010, 304, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Sinn, M.; Bahra, M.; Liersch, T.; Gellert, K.; Messmann, H.; Bechstein, W.; Waldschmidt, D.; Jacobasch, L.; Wilhelm, M.; Rau, B.M.; et al. CONKO-005: Adjuvant Chemotherapy with Gemcitabine Plus Erlotinib Versus Gemcitabine Alone in Patients after R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J. Clin. Oncol. 2017, 35, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Chikhladze, S.; Lederer, A.-K.; Kousoulas, L.; Reinmuth, M.; Sick, O.; Fichtner-Feigl, S.; Wittel, U.A. Adjuvant chemotherapy after surgery for pancreatic ductal adenocarcinoma: Retrospective real-life data. World J. Surg. Oncol. 2019, 17, 185. [Google Scholar] [CrossRef]
- Strobel, O.; Lorenz, P.; Hinz, U.M.; Gaida, M.; König, A.-K.; Hank, T.; Niesen, W.; Kaiser, J.; Al-Saeedi, M.; Bergmann, F.; et al. Actual Five-year Survival after Upfront Resection for Pancreatic Ductal Adenocarcinoma. Ann. Surg. 2020, 275, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Moaven, O.; Clark, C.J.; Russell, G.B.M.; Votanopoulos, K.I.; Howerton, R.; Levine, E.A.; Shen, P. Optimal Adjuvant Treatment Approach after Upfront Resection of Pancreatic Cancer. Ann. Surg. 2020, 274, 1058–1066. [Google Scholar] [CrossRef]
- Lim, K.H.; Chung, E.; Khan, A.; Cao, D.; Linehan, D.; Ben-Josef, E.; Wang-Gillam, A. Neoadjuvant Therapy of Pancreatic Cancer: The Emerging Paradigm? Oncologist 2012, 17, 192–200. [Google Scholar] [CrossRef]
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef]
- Reni, M.; Balzano, G.; Zanon, S.; Zerbi, A.; Rimassa, L.; Castoldi, R.; Pinelli, D.; Mosconi, S.; Doglioni, C.; Chiaravalli, M.; et al. Safety and efficacy of preoperative or postoperative chemotherapy for resectable pancreatic adenocarcinoma (PACT-15): A randomised, open-label, phase 2–3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 413–423. [Google Scholar] [CrossRef]
- Heinrich, S.; Schäfer, M.; Weber, A.; Hany, T.F.; Bhure, U.; Pestalozzi, B.C.; Clavien, P.-A.M. Neoadjuvant Chemotherapy Generates a Significant Tumor Response in Resectable Pancreatic Cancer without Increasing Morbidity. Ann. Surg. 2008, 248, 1014–1022. [Google Scholar] [CrossRef]
- Shridhar, R.; Takahashi, C.; Huston, J.; Meredith, K.L. Neoadjuvant therapy and pancreatic cancer: A national cancer database analysis. J. Gastrointest. Oncol. 2019, 10, 663–673. [Google Scholar] [CrossRef]
- Deng, A.; Wang, C.; Cohen, S.J.; Winter, J.M.; Posey, J.; Yeo, C.; Mallick, A.B. Multi-agent neoadjuvant chemotherapy improves survival in early-stage pancreatic cancer: A National Cancer Database analysis. Eur. J. Cancer 2021, 147, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Deig, C.R.; Sutton, T.L.; Beneville, B.; Trone, K.; Stratton, A.; Gunesch, A.N.; Liu, A.I.; Alrohaibani, A.; Mohebnasab, M.; Bassale, S.; et al. Neoadjuvant Therapy Is Associated with Improved Chemotherapy Delivery and Overall Survival Compared to Upfront Resection in Pancreatic Cancer without Increasing Perioperative Complications. Cancers 2022, 14, 609. [Google Scholar] [CrossRef] [PubMed]
- Kamarajah, S.K.; White, S.A.; Naffouje, S.A.; Salti, G.I.; Dahdaleh, F. Adjuvant Chemotherapy Associated with Survival Benefit Following Neoadjuvant Chemotherapy and Pancreatectomy for Pancreatic Ductal Adenocarcinoma: A Population-Based Cohort Study. Ann. Surg. Oncol. 2021, 28, 6790–6802. [Google Scholar] [CrossRef] [PubMed]
- Brunner, T.B.; Scott-Brown, M. The role of radiotherapy in multimodal treatment of pancreatic carcinoma. Radiat. Oncol. 2010, 5, 64. [Google Scholar] [CrossRef]
- Mukherjee, S.; Hurt, C.N.; Bridgewater, J.; Falk, S.; Cummins, S.; Wasan, H.; Crosby, T.; Jephcott, C.; Roy, R.; Radhakrishna, G.; et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): A multicentre, randomised, phase 2 trial. Lancet Oncol. 2013, 14, 317–326. [Google Scholar] [CrossRef]
- Wang, D.; Liu, C.; Zhou, Y.; Yan, T.; Li, C.; Yang, Q.; Xu, Y.; Zhao, L.; Pei, Q.; Tan, F.; et al. Effect of neoadjuvant radiotherapy on survival of non-metastatic pancreatic ductal adenocarcinoma: A SEER database analysis. Radiat. Oncol. 2020, 15, 107. [Google Scholar] [CrossRef]
- Wang, D.; Ge, H.; Tian, M.; Li, C.; Zhao, L.; Pei, Q.; Tan, F.; Li, Y.; Ling, C.; Güngör, C. The Survival Effect of Radiotherapy on Stage IIB/III Pancreatic Cancer Undergone Surgery in Different Age and Tumor Site Groups: A Propensity Scores Matching Analysis Based on SEER Database. Front. Oncol. 2022, 12, 799930. [Google Scholar] [CrossRef]
- Saadat, L.V.; Chou, J.F.; Gonen, M.; Soares, K.C.; Kingham, T.P.; Varghese, A.M.; Jarnagin, W.R.; D’angelica, M.I.; Drebin, J.A.; O’reilly, E.M.; et al. Treatment patterns and survival in patients with early-onset pancreatic cancer. Cancer 2021, 127, 3566–3578. [Google Scholar] [CrossRef]
- Ansari, D.; Althini, C.; Ohlsson, H.; Andersson, R. Early-onset pancreatic cancer: A population-based study using the SEER registry. Langenbeck’s Arch. Surg. 2019, 404, 565–571. [Google Scholar] [CrossRef]
- Winer, L.K.; Dhar, V.K.; Wima, K.; Morris, M.C.; Lee, T.C.; Shah, S.A.; Ahmad, S.A.; Patel, S.H. The Impact of Tumor Location on Resection and Survival for Pancreatic Ductal Adenocarcinoma. J. Surg. Res. 2019, 239, 60–66. [Google Scholar] [CrossRef]
- Takeda, T.; Sasaki, T.; Inoue, Y.; Mie, T.; Furukawa, T.; Kanata, R.; Kasuga, A.; Matsuyama, M.; Ozaka, M.; Takahashi, Y.; et al. Comprehensive comparison of clinicopathological characteristics, treatment, and prognosis of borderline resectable pancreatic cancer according to tumor location. Pancreatology 2020, 20, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.D.; Speers, C.; McGahan, C.E.; Renouf, D.J.; Schaeffer, D.F.; Kennecke, H.F. Prognostic factors and sites of metastasis in unresectable locally advanced pancreatic cancer. Cancer Med. 2015, 4, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Truty, M.J.; Kendrick, M.L.; Nagorney, D.M.; Smoot, R.L.; Cleary, S.P.; Graham, R.P.; Goenka, A.H.; Hallemeier, C.L.; Haddock, M.G.; Harmsen, W.S.; et al. Factors Predicting Response, Perioperative Outcomes, and Survival Following Total Neoadjuvant Therapy for Borderline/Locally Advanced Pancreatic Cancer. Ann. Surg. 2019, 273, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Miquel, M.; Zhang, S.; Pilarsky, C. Pre-clinical Models of Metastasis in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 748631. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic Pancreatic Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2545–2556. [Google Scholar] [CrossRef]
- Thomas, A.G.; Awasthi, N. Targeted therapy for pancreatic cancer: Lessons learned and future opportunities. Dig. Med. Res. 2021, 4, 32. [Google Scholar] [CrossRef]
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat. Cancer 2022, 3, 272–286. [Google Scholar] [CrossRef]
- Grinshpun, A.; Zarbiv, Y.; Roszik, J.; Subbiah, V.; Hubert, A. Beyond KRAS: Practical Molecular Targets in Pancreatic Adenocarcinoma. Case Rep. Oncol. 2019, 12, 7–13. [Google Scholar] [CrossRef]
- Tempero, M.; Oh, D.-Y.; Tabernero, J.; Reni, M.; Van Cutsem, E.; Hendifar, A.; Waldschmidt, D.-T.; Starling, N.; Bachet, J.-B.; Chang, H.-M.; et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine for first-line treatment of patients with metastatic pancreatic adenocarcinoma: Phase III RESOLVE study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Lonardi, S.; Bendell, J.; Sim, H.-W.; Macarulla, T.; Lopez, C.D.; Van Cutsem, E.; Martin, A.J.M.; Park, J.O.; Greil, R.; et al. Randomized Phase III Study of FOLFOX Alone or with Pegilodecakin as Second-Line Therapy in Patients with Metastatic Pancreatic Cancer That Progressed after Gemcitabine (SEQUOIA). J. Clin. Oncol. 2021, 39, 1108–1118. [Google Scholar] [CrossRef]
- Shin, S.; Park, C.M.; Kwon, H.; Lee, K.-H. Erlotinib plus gemcitabine versus gemcitabine for pancreatic cancer: Real-world analysis of Korean national database. BMC Cancer 2016, 16, 443. [Google Scholar] [CrossRef] [PubMed]
- Hammel, P.; Huguet, F.; van Laethem, J.L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouché, O.; Shannon, J.; André, T.; et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients with Locally Advanced Pancreatic Cancer Controlled after 4 Months of Gemcitabine with or without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016, 315, 1844–1853. [Google Scholar] [CrossRef]
- Di Marco, M.; Grassi, E.; Durante, S.; Vecchiarelli, S.; Palloni, A.; Macchini, M.; Casadei, R.; Ricci, C.; Panzacchi, R.; Santini, D.; et al. State of the art biological therapies in pancreatic cancer. World J. Gastrointest. Oncol. 2016, 8, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Safran, H.; Miner, T.; Bahary, N.; Whiting, S.; Lopez, C.D.; Sun, W.; Charpentier, K.; Shipley, J.; Anderson, E.; McNulty, B.; et al. Lapatinib and Gemcitabine for Metastatic Pancreatic Cancer. Am. J. Clin. Oncol. 2011, 34, 50–52. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef]
- Hu, C.; Xia, R.; Zhang, X.; Li, T.; Ye, Y.; Li, G.; He, R.; Li, Z.; Lin, Q.; Zheng, S.; et al. circFARP1 enables cancer-associated fibroblasts to promote gemcitabine resistance in pancreatic cancer via the LIF/STAT3 axis. Mol. Cancer 2022, 21, 24. [Google Scholar] [CrossRef]
- Jimeno, A.; Weiss, G.J.; Miller, W.H.; Gettinger, S.; Eigl, B.J.; Chang, A.L.S.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I Study of the Hedgehog Pathway Inhibitor IPI-926 in Adult Patients with Solid Tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kelley, R.K.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Jiang, B.; Zhou, L.; Lu, J.; Wang, Y.; Liu, C.; You, L.; Guo, J. Stroma-Targeting Therapy in Pancreatic Cancer: One Coin with Two Sides? Front. Oncol. 2020, 10, 576399. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.-C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed]
- Huggett, M.T.; Jermyn, M.; Gillams, A.; Illing, R.; Mosse, S.; Novelli, M.; Kent, E.; Bown, S.G.; Hasan, T.; Pogue, B.W.; et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br. J. Cancer 2014, 110, 1698–1704. [Google Scholar] [CrossRef]
- DeWitt, J.M.; Sandrasegaran, K.; O’Neil, B.; House, M.G.; Zyromski, N.J.; Sehdev, A.; Perkins, S.M.; Flynn, J.; McCranor, L.; Shahda, S. Phase 1 study of EUS-guided photodynamic therapy for locally advanced pancreatic cancer. Gastrointest. Endosc. 2019, 89, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Hanada, Y.; Pereira, S.P.; Pogue, B.; Maytin, E.V.; Hasan, T.; Linn, B.; Mangels-Dick, T.; Wang, K.K. EUS-guided verteporfin photodynamic therapy for pancreatic cancer. Gastrointest. Endosc. 2021, 94, 179–186. [Google Scholar] [CrossRef]
- Karimnia, V.; Rizvi, I.; Slack, F.J.; Celli, J.P. Photodestruction of Stromal Fibroblasts Enhances Tumor Response to PDT in 3D Pancreatic Cancer Coculture Models. Photochem. Photobiol. 2020, 97, 416–426. [Google Scholar] [CrossRef]
- Celli, J.P. Stromal Interactions as Regulators of Tumor Growth and Therapeutic Response: A Potential Target for Photodynamic Therapy? Isr. J. Chem. 2012, 52, 757–766. [Google Scholar] [CrossRef]
- Jafari, R.; Cramer, G.M.; Celli, J.P. Modulation of Extracellular Matrix Rigidity Via Riboflavin-mediated Photocrosslinking Regulates Invasive Motility and Treatment Response in a 3D Pancreatic Tumor Model. Photochem. Photobiol. 2020, 96, 365–372. [Google Scholar] [CrossRef]
- Maneshi, P.; Mason, J.; Dongre, M.; Öhlund, D. Targeting Tumor-Stromal Interactions in Pancreatic Cancer: Impact of Collagens and Mechanical Traits. Front. Cell Dev. Biol. 2021, 9, 787485. [Google Scholar] [CrossRef] [PubMed]
- Korc, M. Pancreatic cancer–associated stroma production. Am. J. Surg. 2007, 194, S84–S86. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Haghnejad, V.; Schaefer, M.; Gauchotte, G.; Caron, B.; Peyrin-Biroulet, L.; Bronowicki, J.-P.; Neuzillet, C.; Lopez, A. The Immune Landscape of Human Pancreatic Ductal Carcinoma: Key Players, Clinical Implications, and Challenges. Cancers 2022, 14, 995. [Google Scholar] [CrossRef]
- Carstens, J.L.; de Sampaio, P.C.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Kpeglo, D.; Hughes, M.D.; Dougan, L.; Haddrick, M.; Knowles, M.A.; Evans, S.D.; Peyman, S.A. Modeling the mechanical stiffness of pancreatic ductal adenocarcinoma. Matrix Biol. Plus 2022, 14, 100109. [Google Scholar] [CrossRef]
- Ogawa, Y.; Masugi, Y.; Abe, T.; Yamazaki, K.; Ueno, A.; Fujii-Nishimura, Y.; Hori, S.; Yagi, H.; Abe, Y.; Kitago, M.; et al. Three Distinct Stroma Types in Human Pancreatic Cancer Identified by Image Analysis of Fibroblast Subpopulations and Collagen. Clin. Cancer Res. 2021, 27, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e18. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Askan, G.; Sahin, I.H.; Chou, J.F.; Yavas, A.; Capanu, M.; Iacobuzio-Donahue, C.A.; Basturk, O.; O’reilly, E.M. Pancreatic cancer stem cells may define tumor stroma characteristics and recurrence patterns in pancreatic ductal adenocarcinoma. BMC Cancer 2021, 21, 385. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Minamide, T.; Yano, T. Role of photodynamic therapy in the treatment of esophageal cancer. Dig. Endosc. 2019, 31, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.; Lee, N.V.; Liu, K.Y.; Poh, C.F. Prospects in the Application of Photodynamic Therapy in Oral Cancer and Premalignant Lesions. Cancers 2016, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Crous, A.; Abrahamse, H. Photodynamic therapy of lung cancer, where are we? Front. Pharmacol. 2022, 13, 932098. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.A.; Evans, D.H.; Abrahamse, H. Photodynamic therapy (PDT): A short review on cellular mechanisms and cancer research applications for PDT. J. Photochem. Photobiol. B 2009, 96, 1–8. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in PDT: Part three—Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagnosis Photodyn. Ther. 2005, 2, 91–106. [Google Scholar] [CrossRef]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef]
- Mroz, P.; Szokalska, A.; Wu, M.X.; Hamblin, M.R. Photodynamic Therapy of Tumors Can Lead to Development of Systemic Antigen-Specific Immune Response. PLoS ONE 2010, 5, e15194. [Google Scholar] [CrossRef]
- Mashayekhi, V.; Op’t Hoog, C.; Oliveira, S. Vascular targeted photodynamic therapy: A review of the efforts towards molecular targeting of tumor vasculature. J. Porphyr. Phthalocyanines 2019, 23, 1229–1240. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin Michael, R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef]
- Liu, K.-H.; Wang, C.-P.; Chang, M.-F.; Chung, Y.-W.; Lou, P.-J.; Lin, J.-H. Molecular characterization of photosensitizer-mediated photodynamic therapy by gene expression profiling. Hum. Exp. Toxicol. 2013, 33, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Kessel, D. Correlation between subcellular localization and photodynamic efficacy. J. Porphyr. Phthalocyanines 2004, 8, 1009–1014. [Google Scholar] [CrossRef]
- Uchoa, A.F.; Oliveira, C.S.; Baptista, M.S. Relationship between structure and photoactivity of porphyrins derived from pro-toporphyrin IX. J. Porphyr. Phthalocyanines 2010, 14, 832–845. [Google Scholar] [CrossRef]
- Zhen, Z.; Tang, W.; Chuang, Y.-J.; Todd, T.; Zhang, W.; Lin, X.; Niu, G.; Liu, G.; Wang, L.; Pan, Z.; et al. Tumor Vasculature Targeted Photodynamic Therapy for Enhanced Delivery of Nanoparticles. ACS Nano 2014, 8, 6004–6013. [Google Scholar] [CrossRef]
- Bown, S.G.; Rogowska, A.Z.; Whitelaw, D.E.; Lees, W.-R.; Lovat, L.B.; Ripley, P.; Jones, L.; Wyld, P.; Gillams, A.; Hatfield, A.W.R. Photodynamic therapy for cancer of the pancreas. Gut 2002, 50, 549–557. [Google Scholar] [CrossRef]
- Choi, J.-H.; Oh, D.; Lee, J.H.; Park, J.-H.; Kim, K.-P.; Lee, S.S.; Lee, Y.-J.; Lim, Y.-S.; Song, T.J.; Lee, S.S.; et al. Initial human experience of endoscopic ultrasound-guided photodynamic therapy with a novel photosensitizer and a flexible laser-light catheter. Endoscopy 2015, 47, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Schroder, T.; Chen, I.-W.; Sperling, M.; Bell, R.H.; Brackett, K.; Joffe, S.N. Hematoporphyrin derivative uptake and photodynamic therapy in pancreatic carcinoma. J. Surg. Oncol. 1988, 38, 4–9. [Google Scholar] [CrossRef]
- Nuutinen, P.; Chatlani, P.; Bedwell, J.; MacRobert, A.; Phillips, D.; Bown, S. Distribution and photodynamic effect of disulphonated aluminium phthalocyanine in the pancreas and adjacent tissues in the Syrian golden hamster. Br. J. Cancer 1991, 64, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Chatlani, P.T.; O Nuutinen, P.J.; Toda, N.; Barr, H.; Macrobert, A.J.; Bedwell, J.; Bown, S.G. Selective necrosis in hamster pancreatic tumours using photodynamic therapy with phthalocyanine photosensitization. Br. J. Surg. 1992, 79, 786–790. [Google Scholar] [CrossRef]
- Evrard, S.; Keller, P.; Hajri, A.; Balboni, G.; Mendoza-Burgos, L.; Damgé, C.; Marescaux, J.; Aprahamian, M. Experimental pancreatic cancer in the rat treated by photodynamic therapy. Br. J. Surg. 1994, 81, 1185–1189. [Google Scholar] [CrossRef]
- Regula, J.; Ravi, B.; Bedwell, J.; MacRobert, A.; Bown, S. Photodynamic therapy using 5-aminolaevulinic acid for experimental pancreatic cancer—Prolonged animal survival. Br. J. Cancer 1994, 70, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Mlkvy, P.; Messmann, H.; Pauer, M.; Stewart, J.; Millson, C.; MacRobert, A.; Bown, S. Distribution and photodynamic effects of meso-tetrahydroxyphenylchlorin (mTHPC) in the pancreas and adjacent tissues in the Syrian golden hamster. Br. J. Cancer 1996, 73, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Mikvy, P.; Messman, H.; MacRobert, A.J.; Pauer, M.; Sams, V.R.; Davies, C.L.; Stewart, J.C.; Bown, S.G. Photodynamic therapy of a transplanted pancreatic cancer model using mTHPC. Br. J. Cancer 1997, 76, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.-H.; Nishioka, N.S.; Mino, M.; Lauwers, G.Y.; Puricelli, W.P.; Collier, K.N.; Brugge, W.R. EUS-guided photodynamic therapy of the pancreas: A pilot study. Gastrointest. Endosc. 2004, 59, 95–99. [Google Scholar] [CrossRef]
- Tangutoori, S.; Spring, B.Q.; Mai, Z.; Palanisami, A.; Mensah, L.B.; Hasan, T. Simultaneous delivery of cytotoxic and biologic therapeutics using nanophotoactivatable liposomes enhances treatment efficacy in a mouse model of pancreatic cancer. Nanomed. Nanotechnol. Biol. Med. 2015, 12, 223–234. [Google Scholar] [CrossRef]
- Li, H.; Wang, P.; Deng, Y.; Zeng, M.; Tang, Y.; Zhu, W.-H.; Cheng, Y. Combination of active targeting, enzyme-triggered release and fluorescent dye into gold nanoclusters for endomicroscopy-guided photothermal/photodynamic therapy to pancreatic ductal adenocarcinoma. Biomaterials 2017, 139, 30–38. [Google Scholar] [CrossRef]
- Obaid, G.; Bano, S.; Mallidi, S.; Broekgaarden, M.; Kuriakose, J.; Silber, Z.; Bulin, A.L.; Wang, Y.; Mai, Z.; Jin, W.; et al. Impacting Pancreatic Cancer Therapy in Heterotypic in Vitro Organoids and in Vivo Tumors with Specificity-Tuned, NIR-Activable Photoimmunonanoconjugates: Towards Conquering Desmoplasia? Nano Lett. 2019, 19, 7573–7587. [Google Scholar] [CrossRef]
- Quilbe, A.; Moralès, O.; Baydoun, M.; Kumar, A.; Mustapha, R.; Murakami, T.; Leroux, B.; de Schutter, C.; Thecua, E.; Ziane, L.; et al. An Efficient Photodynamic Therapy Treatment for Human Pancreatic Adenocarcinoma. J. Clin. Med. 2020, 9, 192. [Google Scholar] [CrossRef]
- De Silva, P.; Saad, M.A.; Camargo, A.P.; Swain, J.; Palanasami, A.; Obaid, G.; Shetty, S.; Hasan, T. Abstract A17: Enhanced immune infiltration and antitumor immune reactivity in response to optical priming in pancreatic cancer. Cancer Immunol. Res. 2020, 8 (Suppl. S3), A17. [Google Scholar] [CrossRef]
- Sun, F.; Zhu, Q.; Li, T.; Saeed, M.; Xu, Z.; Zhong, F.; Song, R.; Huai, M.; Zheng, M.; Xie, C.; et al. Regulating Glucose Metabolism with Prodrug Nanoparticles for Promoting Photoimmunotherapy of Pancreatic Cancer. Adv. Sci. 2021, 8, 2002746. [Google Scholar] [CrossRef]
- Vincent, P.; Bruza, P.; Palisoul, S.M.; Gunn, J.R.; Samkoe, K.S.; Hoopes, P.J.; Hasan, T.; Pogue, B.W. Visualization and quantification of pancreatic tumor stroma in fresh tissue via ultraviolet surface excitation. J. Biomed. Opt. 2021, 26, 016002. [Google Scholar] [CrossRef] [PubMed]
- Obaid, G.; Bano, S.; Thomsen, H.; Callaghan, S.; Shah, N.; Swain, J.W.R.; Jin, W.; Ding, X.; Cameron, C.G.; McFarland, S.A.; et al. Remediating Desmoplasia with EGFR-Targeted Photoactivable Multi-Inhibitor Liposomes Doubles Overall Survival in Pancreatic Cancer. Adv. Sci. 2022, 9, 2104594. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, X.; Chen, F.; Li, H.; Wang, T.; Liu, N.; Sun, K.; Zhou, G.; Tao, K. Modulating cancer-stroma crosstalk by a nanoparticle-based photodynamic method to pave the way for subsequent therapies. Biomaterials 2022, 289, 121813. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-Y.; Zhang, J.-G.; Zhou, Q.-M.; Yu, J.-N.; Lu, Y.-F.; Wang, X.-J.; Zhou, J.-P.; Ding, X.-F.; Du, Y.-Z.; Yu, R.-S. Extracellular matrix modulating enzyme functionalized biomimetic Au nanoplatform-mediated enhanced tumor penetration and synergistic antitumor therapy for pancreatic cancer. J. Nanobiotechnol. 2022, 20, 524. [Google Scholar] [CrossRef]
- CTG Labs—NCBI. Available online: https://beta.clinicaltrials.gov/study/NCT00003923 (accessed on 23 March 2023).
- Ultrasound-Guided Verteporfin Photodynamic Therapy for the Treatment of Unresectable Solid Pancreatic Tumors or Advanced Pancreatic Cancer, VERTPAC-02 Study—Full Text View—ClinicalTrials.gov. clinicaltrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03033225 (accessed on 23 March 2023).
- Nia, H.T.; Liu, H.; Seano, G.; Datta, M.; Jones, D.; Rahbari, N.; Incio, J.; Chauhan, V.P.; Jung, K.; Martin, J.D.; et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat. Biomed. Eng. 2016, 1, 1. [Google Scholar] [CrossRef]
- Gündel, B.; Liu, X.; Löhr, M.; Heuchel, R. Pancreatic Ductal Adenocarcinoma: Preclinical in vitro and ex vivo Models. Front. Cell Dev. Biol. 2021, 9, 741162. [Google Scholar] [CrossRef]
- Voutouri, C.; Polydorou, C.; Papageorgis, P.; Gkretsi, V.; Stylianopoulos, T. Hyaluronan-Derived Swelling of Solid Tumors, the Contribution of Collagen and Cancer Cells, and Implications for Cancer Therapy. Neoplasia 2016, 18, 732–741. [Google Scholar] [CrossRef]
- Boucher, Y.; Jain, R.K. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: Implications for vascular collapse. Cancer Res. 1992, 52, 5110–5114. [Google Scholar]
- DuFort, C.C.; DelGiorno, K.E.; Carlson, M.A.; Osgood, R.J.; Zhao, C.; Huang, Z.; Thompson, C.B.; Connor, R.J.; Thanos, C.D.; Brockenbrough, J.S.; et al. Interstitial Pressure in Pancreatic Ductal Adenocarcinoma Is Dominated by a Gel-Fluid Phase. Biophys. J. 2016, 110, 2106–2119. [Google Scholar] [CrossRef]
- Zhao, T.; Zhang, R.; He, Q.; Zhou, H.; Song, X.; Gong, T.; Zhang, Z. Partial ligand shielding nanoparticles improve pancreatic ductal adenocarcinoma treatment via a multifunctional paradigm for tumor stroma reprogramming. Acta Biomater. 2022, 145, 122–134. [Google Scholar] [CrossRef]
- Yang, H.; Liu, R.; Xu, Y.; Qian, L.; Dai, Z. Photosensitizer Nanoparticles Boost Photodynamic Therapy for Pancreatic Cancer Treatment. Nano-Micro Lett. 2021, 13, 35. [Google Scholar] [CrossRef]
- Shah, V.; Sheppard, B.; Sears, R.; Alani, A.W. Hypoxia: Friend or Foe for drug delivery in Pancreatic Cancer. Cancer Lett. 2020, 492, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Huang, Y.; Clauser, K.R.; Rickelt, S.; Lau, A.N.; Carr, S.A.; Heiden, M.G.V.; Hynes, R.O. Suppression of pancreatic ductal adenocarcinoma growth and metastasis by fibrillar collagens produced selectively by tumor cells. Nat. Commun. 2021, 12, 2328. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.-J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e6. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Shin, K.; Lim, A.; Zhao, C.; Sahoo, D.; Pan, Y.; Spiekerkoetter, E.; Liao, J.C.; Beachy, P.A. Hedgehog Signaling Restrains Bladder Cancer Progression by Eliciting Stromal Production of Urothelial Differentiation Factors. Cancer Cell 2014, 26, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGF-β to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2018, 9, 282–301. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gündel, B.; Li, X.; Liu, J.; Wright, A.; Löhr, M.; Arvidsson, G.; Heuchel, R. 3D heterospecies spheroids of pancreatic stroma and cancer cells demonstrate key phenotypes of pancreatic ductal adenocarcinoma. Transl. Oncol. 2021, 14, 101107. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, C.; Ohuchida, K.; Shinkawa, T.; Okuda, S.; Otsubo, Y.; Okumura, T.; Sagara, A.; Koikawa, K.; Ando, Y.; Shindo, K.; et al. Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett. 2021, 512, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef]
- Helms, E.J.; Berry, M.W.; Chaw, R.C.; DuFort, C.C.; Sun, D.; Onate, M.K.; Oon, C.; Bhattacharyya, S.; Sanford-Crane, H.; Horton, W.; et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer–Associated Fibroblasts. Cancer Discov. 2021, 12, 484–501. [Google Scholar] [CrossRef]
- Hu, B.; Wu, C.; Mao, H.; Gu, H.; Dong, H.; Yan, J.; Qi, Z.; Yuan, L.; Dong, Q.; Long, J. Subpopulations of cancer-associated fibroblasts link the prognosis and metabolic features of pancreatic ductal adenocarcinoma. Ann. Transl. Med. 2022, 10, 262. [Google Scholar] [CrossRef]
- Maurer, H.C.; Holmstrom, S.R.; He, J.; Laise, P.; Su, T.; Ahmed, A.; Hibshoosh, H.; Chabot, J.A.; Oberstein, P.E.; Sepulveda, A.R.; et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 2019, 68, 1034–1043. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.L.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e1993. [Google Scholar] [CrossRef]
- Erkan, M.; Michalski, C.W.; Rieder, S.; Reiser–Erkan, C.; Abiatari, I.; Kolb, A.; Giese, N.A.; Esposito, I.; Friess, H.; Kleeff, J. The Activated Stroma Index Is a Novel and Independent Prognostic Marker in Pancreatic Ductal Adenocarcinoma. Clin. Gastroenterol. Hepatol. 2008, 6, 1155–1161. [Google Scholar] [CrossRef]
- Bolm, L.; Cigolla, S.; Wittel, U.A.; Hopt, U.T.; Keck, T.; Rades, D.; Bronsert, P.; Wellner, U.F. The Role of Fibroblasts in Pancreatic Cancer: Extracellular Matrix Versus Paracrine Factors. Transl. Oncol. 2017, 10, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Karimnia, V.; Slack, F.J.; Celli, J.P. Photodynamic Therapy for Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 4354. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Matuszewska, K.; Pereira, M.; Petrik, D.; Lawler, J.; Petrik, J. Normalizing Tumor Vasculature to Reduce Hypoxia, Enhance Perfusion, and Optimize Therapy Uptake. Cancers 2021, 13, 4444. [Google Scholar] [CrossRef] [PubMed]
- Cavin, S.; Riedel, T.; Rosskopfova, P.; Gonzalez, M.; Baldini, G.; Zellweger, M.; Wagnières, G.; Dyson, P.J.; Ris, H.; Krueger, T.; et al. Vascular-targeted low dose photodynamic therapy stabilizes tumor vessels by modulating pericyte contractility. Lasers Surg. Med. 2019, 51, 550–561. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Y.; Li, Z.; Wang, G.; Liao, A.; Wang, J.; Li, H.; Guo, Z.; Cheng, B.; Zhang, X. Intelligent Nanodelivery System-Generated 1O2 Mediates Tumor Vessel Normalization by Activating Endothelial TRPV4-eNOS Signaling. Small 2022, 18, 2200038. [Google Scholar] [CrossRef]
- Fingar, V.H. Vascular Effects of Photodynamic Therapy. J. Clin. Laser Med. Surg. 1996, 14, 323–328. [Google Scholar] [CrossRef]
- Krammer, B. Vascular effects of photodynamic therapy. Anticancer Res. 2001, 21, 4271–4277. [Google Scholar]
- Suzuki, T.; Tanaka, M.; Sasaki, M.; Ichikawa, H.; Nishie, H.; Kataoka, H. Vascular Shutdown by Photodynamic Therapy Using Talaporfin Sodium. Cancers 2020, 12, 2369. [Google Scholar] [CrossRef]
- Woodhams, J.H.; MacRobert, A.J.; Bown, S.G. The role of oxygen monitoring during photodynamic therapy and its potential for treatment dosimetry. Photochem. Photobiol. Sci. 2007, 6, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Busch, T.M.; Xing, X.; Yu, G.; Yodh, A.; Wileyto, E.P.; Wang, H.-W.; Durduran, T.; Zhu, T.C.; Wang, K.K.-H. Fluence rate-dependent intratumor heterogeneity in physiologic and cytotoxic responses to Photofrin photodynamic therapy. Photochem. Photobiol. Sci. 2009, 8, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; MacRobert, A.; Curnow, A.; Rogowska, A.; Buonaccorsi, G.; Kato, H.; Bown, S. Optimisation of Illumination for Photodynamic Therapy with mTHPC on Normal Colon and a Transplantable Tumour in Rats. Lasers Med. Sci. 2002, 17, 101–109. [Google Scholar] [CrossRef]
- Busch, T.M. Local physiological changes during photodynamic therapy. Lasers Surg. Med. 2006, 38, 494–499. [Google Scholar] [CrossRef]
- Zhang, T.; Jiang, Z.; Chen, L.; Pan, C.; Sun, S.; Liu, C.; Li, Z.; Ren, W.; Wu, A.; Huang, P. PCN-Fe(III)-PTX nanoparticles for MRI guided high efficiency chemo-photodynamic therapy in pancreatic cancer through alleviating tumor hypoxia. Nano Res. 2020, 13, 273–281. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, R.; Yang, H.; Qu, S.; Qian, L.; Dai, Z. Enhancing Photodynamic Therapy Efficacy Against Cancer Metastasis by Ultrasound-Mediated Oxygen Microbubble Destruction to Boost Tumor-Targeted Delivery of Oxygen and Renal-Clearable Photosensitizer Micelles. ACS Appl. Mater. Interfaces 2022, 14, 25197–25208. [Google Scholar] [CrossRef] [PubMed]
- Burdett, E.; Kasper, F.K.; Mikos, A.G.; Ludwig, J.A.; Pradhan, S.; Hassani, I.; Clary, J.M.; Lipke, E.A.; Ghajar, C.M.; Bissell, M.J.; et al. Engineering Tumors: A Tissue Engineering Perspective in Cancer Biology. Tissue Eng. Part B Rev. 2010, 16, 351–359. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Anbil, S.; Bulin, A.-L.; Obaid, G.; Mai, Z.; Baglo, Y.; Rizvi, I.; Hasan, T. Modulation of redox metabolism negates cancer-associated fibroblasts-induced treatment resistance in a heterotypic 3D culture platform of pancreatic cancer. Biomaterials 2019, 222, 119421. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Roy, B.; Anderson, M.; Leggett, C.L.; Levy, M.J.; Pogue, B.; Hasan, T.; Wang, K.K. Verteporfin- and sodium porfimer-mediated photodynamic therapy enhances pancreatic cancer cell death without activating stromal cells in the microenvironment. J. Biomed. Opt. 2019, 24, 118001. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Öhlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Ruth, T.Y.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Anbil, S.; Pigula, M.; Huang, H.-C.; Mallidi, S.; Broekgaarden, M.; Baglo, Y.; De Silva, P.; Simeone, D.M.; Mino-Kenudson, M.; Maytin, E.V.; et al. Vitamin D Receptor Activation and Photodynamic Priming Enables Durable Low-dose Chemotherapy. Mol. Cancer Ther. 2020, 19, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-C.; Li, J.; Zhou, L.; Yang, J.-Y.; Zhang, Z.-G.; Liang, Z.-Y.; Zhou, W.-X.; You, L.; Zhang, T.-P.; Zhao, Y.-P. CXCL12-CXCR7 axis contributes to the invasive phenotype of pancreatic cancer. Oncotarget 2016, 7, 62006–62018. [Google Scholar] [CrossRef]
- Huang, H.-C.; Rizvi, I.; Liu, J.; Anbil, S.; Kalra, A.; Lee, H.; Baglo, Y.; Paz, N.; Hayden, D.; Pereira, S.; et al. Photodynamic Priming Mitigates Chemotherapeutic Selection Pressures and Improves Drug Delivery. Cancer Res. 2017, 78, 558–571. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Encarnación-Rosado, J.; Lin, E.Y.; Sohn, A.S.W.; Zhang, H.; Mancias, J.D.; Kimmelman, A.C. Autophagy supports mitochondrial metabolism through the regulation of iron homeostasis in pancreatic cancer. Sci. Adv. 2023, 9, eadf9284. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients with Metastatic Pancreatic Adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Alkhateeb, A.; Bano, S.; Bulin, A.-L.; Obaid, G.; Rizvi, I.; Hasan, T. Cabozantinib Inhibits Photodynamic Therapy-Induced Auto- and Paracrine MET Signaling in Heterotypic Pancreatic Microtumors. Cancers 2020, 12, 1401. [Google Scholar] [CrossRef]
- Delitto, D.; Vertes-George, E.; Hughes, S.J.; Behrns, K.E.; Trevino, J.G. c-Met signaling in the development of tumorigenesis and chemoresistance: Potential applications in pancreatic cancer. World J. Gastroenterol. 2014, 20, 8458–8470. [Google Scholar] [CrossRef]
- Avan, A.; Quint, K.; Nicolini, F.; Funel, N.; Frampton, A.E.; Maftouh, M.; Pelliccioni, S.; Schuurhuis, G.J.; Peters, G.J.; Giovannetti, E. Enhancement of the Antiproliferative Activity of Gemcitabine by Modulation of c-Met Pathway in Pancreatic Cancer. Curr. Pharm. Des. 2013, 19, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Debefve, E.; Pegaz, B.; Ballini, J.-P.; Konan, Y.; Bergh, H.v.D. Combination therapy using aspirin-enhanced photodynamic selective drug delivery. Vasc. Pharmacol. 2007, 46, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Perentes, J.Y.; Wang, Y.; Wang, X.; Abdelnour, E.; Gonzalez, M.; Decosterd, L.; Wagnieres, G.; Bergh, H.v.D.; Peters, S.; Ris, H.-B.; et al. Low-Dose Vascular Photodynamic Therapy Decreases Tumor Interstitial Fluid Pressure, which Promotes Liposomal Doxorubicin Distribution in a Murine Sarcoma Metastasis Model. Transl. Oncol. 2014, 7, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Zuluaga, M.-F.; Gabriel, D.; Lange, N. Enhanced Prostate Cancer Targeting by Modified Protease Sensitive Photosensitizer Prodrugs. Mol. Pharm. 2012, 9, 1570–1579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lovell, S.; De Vita, E.; Jagtap, P.K.A.; Lucy, D.; Grocin, A.G.; Kjær, S.; Borg, A.; Hennig, J.; Miller, A.K.; et al. A KLK6 Activity-Based Probe Reveals a Role for KLK6 Activity in Pancreatic Cancer Cell Invasion. J. Am. Chem. Soc. 2022, 144, 22493–22504. [Google Scholar] [CrossRef]
- Saha, S.; Xiong, X.; Chakraborty, P.K.; Shameer, K.; Arvizo, R.R.; Kudgus, R.A.; Dwivedi, S.K.D.; Hossen, N.; Gillies, E.M.; Robertson, J.D.; et al. Gold Nanoparticle Reprograms Pancreatic Tumor Microenvironment and Inhibits Tumor Growth. ACS Nano 2016, 10, 10636–10651. [Google Scholar] [CrossRef]
- Karimnia, V.; Huang, L.; Slack, F.; Bhatia, S.; Celli, J. Photodynamic stromal depletion (PSD) improves tumor response to PDT and enhances nanoparticle drug delivery in 3D co-culture models of PDAC. In Optical Methods for Tumor Treatment and Detection: Mechanisms and Techniques in Photodynamic and Photobiomodulation Therapy XXX; Kessel, D.H., Hasan, T., Eds.; SPIE: Bellingham, WA, USA, 2022. [Google Scholar]
- Sun, L.; Wang, J.; Yang, B.; Wang, X.; Yang, G.; Wang, X.; Jiang, Y.; Wang, T.; Jiang, J. Assembled small organic molecules for photodynamic therapy and photothermal therapy. RSC Adv. 2021, 11, 10061–10074. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, X. Semiconducting Perylene Diimide Nanostructure: Multifunctional Phototheranostic Nanoplatform. Acc. Chem. Res. 2019, 52, 1245–1254. [Google Scholar] [CrossRef]
- Yang, Z.; Tian, R.; Wu, J.; Fan, Q.; Yung, B.C.; Niu, G.; Jacobson, O.; Wang, Z.; Liu, G.; Yu, G.; et al. Impact of Semiconducting Perylene Diimide Nanoparticle Size on Lymph Node Mapping and Cancer Imaging. ACS Nano 2017, 11, 4247–4255. [Google Scholar] [CrossRef]
- Shi, X.; Li, Q.; Zhang, C.; Pei, H.; Wang, G.; Zhou, H.; Fan, L.; Yang, K.; Jiang, B.; Wang, F.; et al. Semiconducting polymer nano-radiopharmaceutical for combined radio-photothermal therapy of pancreatic tumor. J. Nanobiotechnol. 2021, 19, 337. [Google Scholar] [CrossRef]
- Udartseva, O.O.; Zhidkova, O.V.; Ezdakova, M.I.; Ogneva, I.V.; Andreeva, E.R.; Buravkova, L.B.; Gollnick, S.O. Low-dose photodynamic therapy promotes angiogenic potential and increases immunogenicity of human mesenchymal stromal cells. J. Photochem. Photobiol. B Biol. 2019, 199, 111596. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lintern, N.; Smith, A.M.; Jayne, D.G.; Khaled, Y.S. Photodynamic Stromal Depletion in Pancreatic Ductal Adenocarcinoma. Cancers 2023, 15, 4135. https://doi.org/10.3390/cancers15164135
Lintern N, Smith AM, Jayne DG, Khaled YS. Photodynamic Stromal Depletion in Pancreatic Ductal Adenocarcinoma. Cancers. 2023; 15(16):4135. https://doi.org/10.3390/cancers15164135
Chicago/Turabian StyleLintern, Nicole, Andrew M. Smith, David G. Jayne, and Yazan S. Khaled. 2023. "Photodynamic Stromal Depletion in Pancreatic Ductal Adenocarcinoma" Cancers 15, no. 16: 4135. https://doi.org/10.3390/cancers15164135
APA StyleLintern, N., Smith, A. M., Jayne, D. G., & Khaled, Y. S. (2023). Photodynamic Stromal Depletion in Pancreatic Ductal Adenocarcinoma. Cancers, 15(16), 4135. https://doi.org/10.3390/cancers15164135