
Challenges in Pharmacological Intervention in Perilipins (PLINs) to Modulate Lipid Droplet Dynamics in Obesity and Cancer

 , ,
, ,  and
and 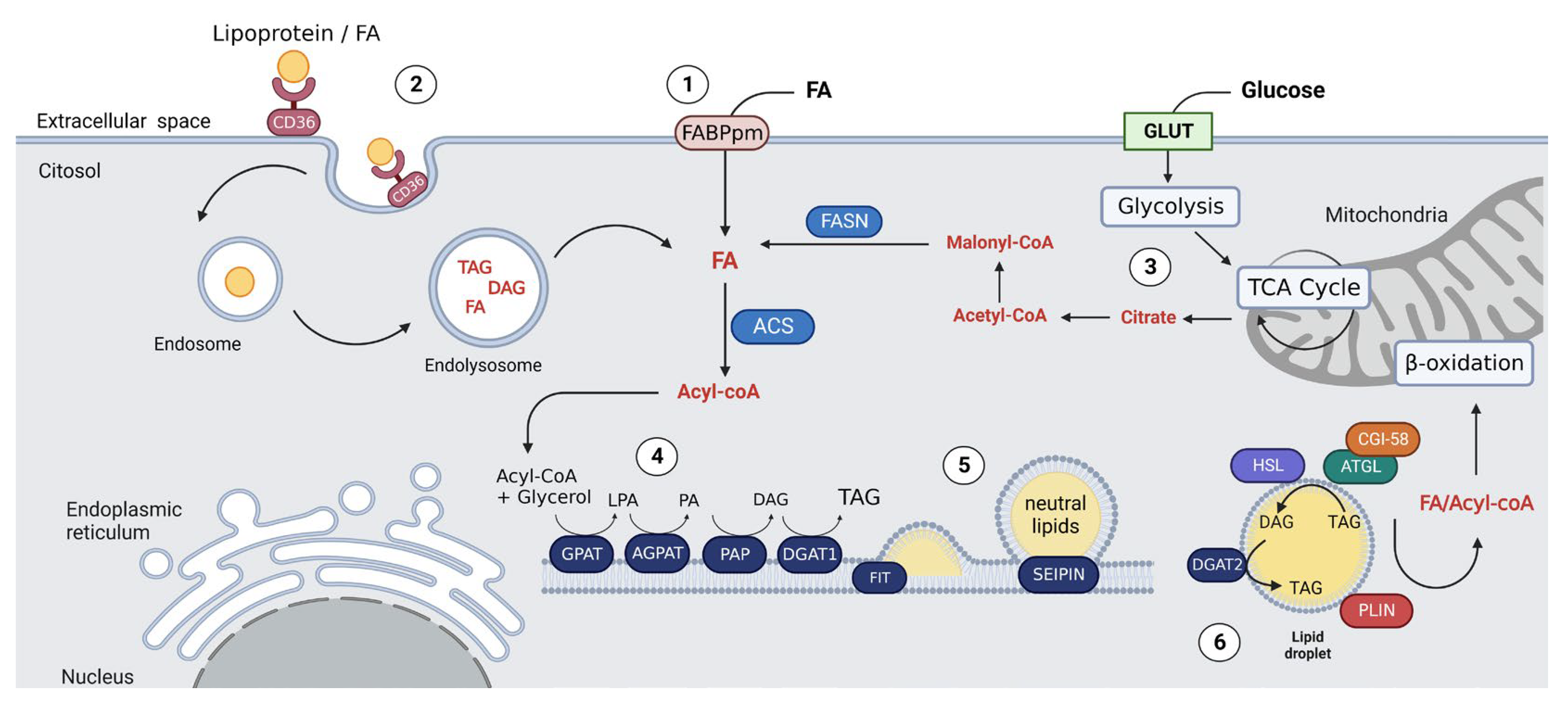{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. LD Biogenesis
3. PLINs as LD Gatekeepers
3.1. Perilipin 1
3.2. Perilipin 2
3.3. Perilipin 3
3.4. Perilipin 4
3.5. Perilipin 5
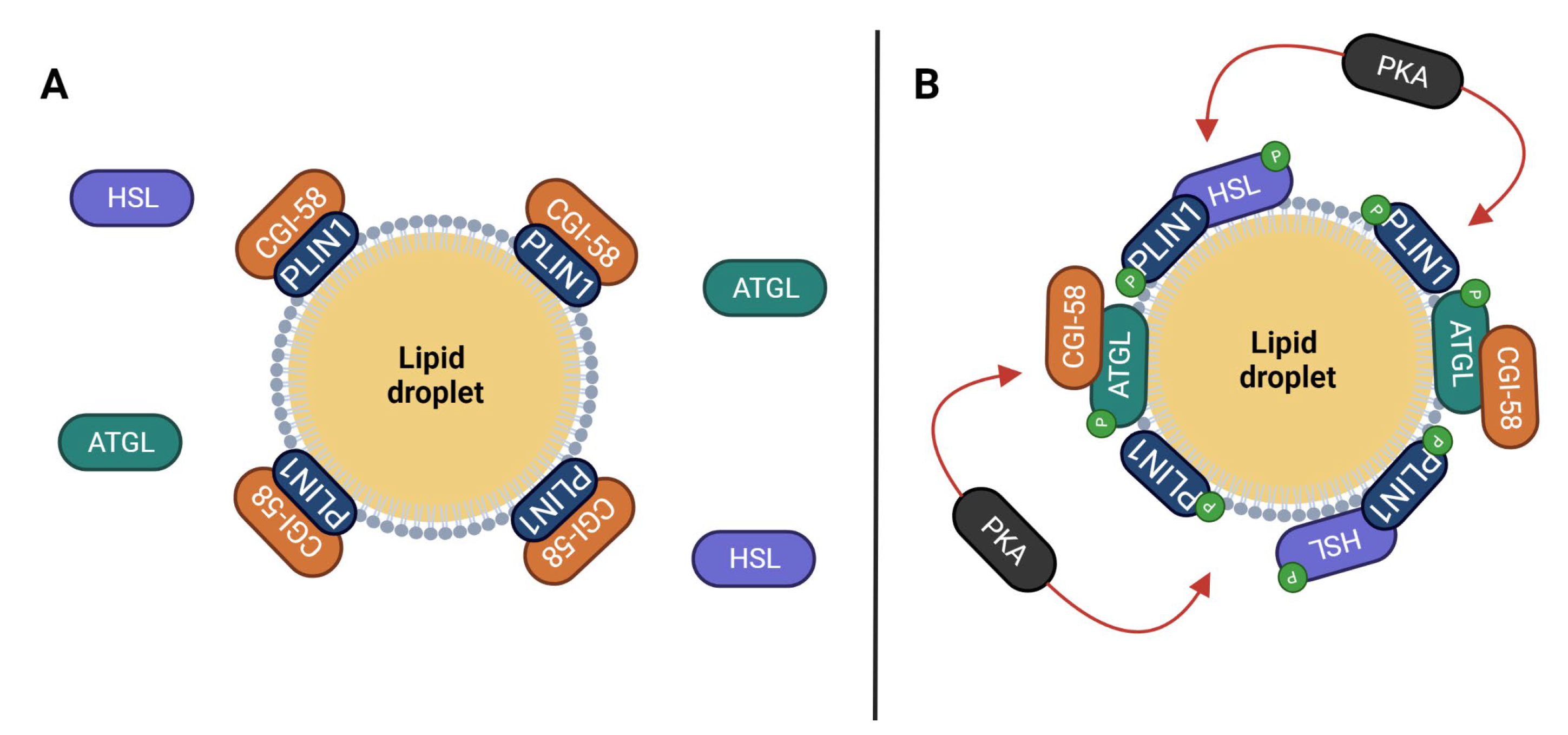
4. Coordination of PLINs’ Function
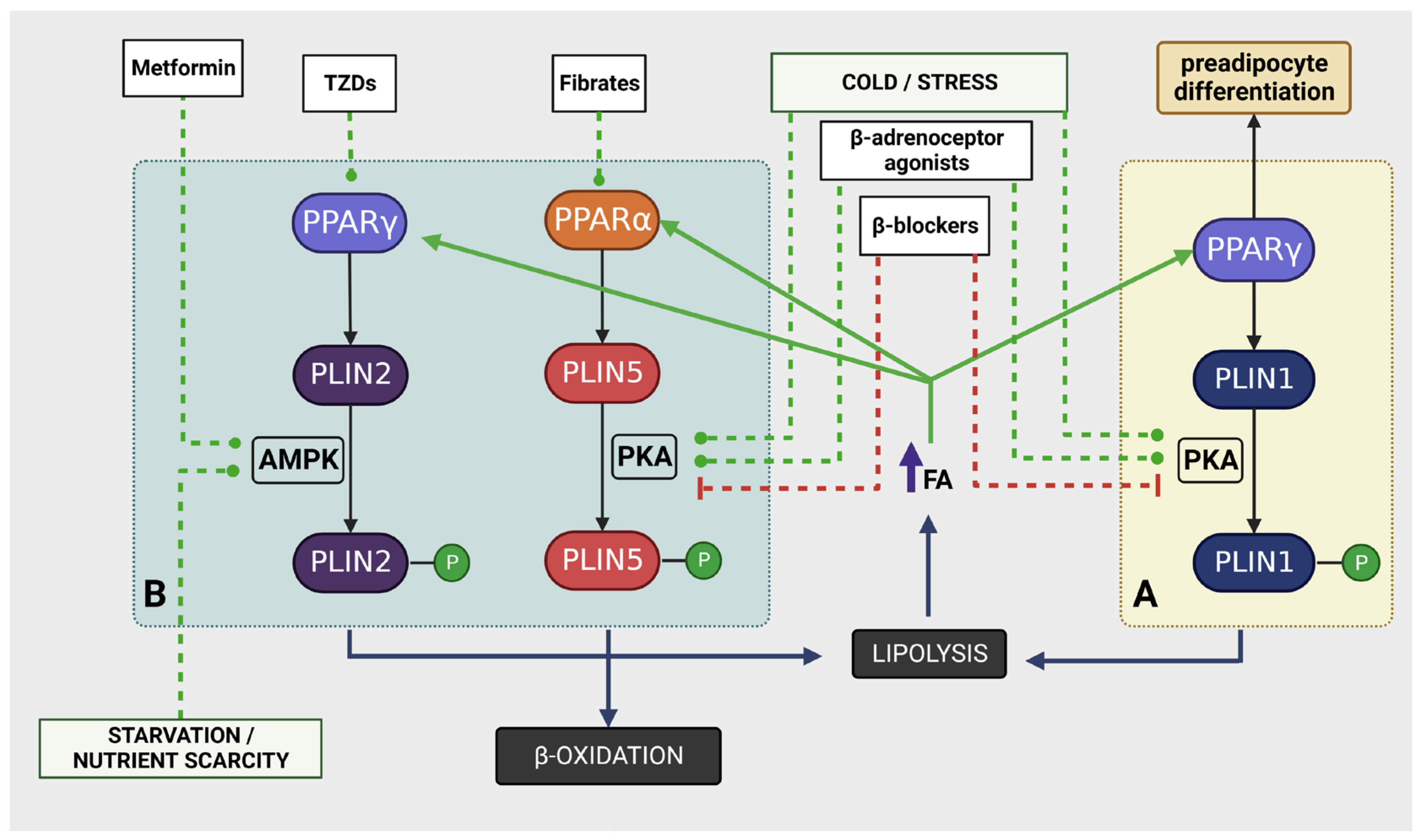
5. Pharmacological Interventions
5.1. PPAR Activators
5.2. AMPK Activators
5.3. SIRT1 Activators
5.4. Modulators of cAMP/PKA Pathway
5.5. Other Drugs
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Royo-Garcia, A.; Courtois, S.; Parejo-Alonso, B.; Espiau-Romera, P.; Sancho, P. Lipid droplets as metabolic determinants for stemness and chemoresistance in cancer. World J. Stem Cells 2021, 13, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Maan, M.; Peters, J.M.; Dutta, M.; Patterson, A.D. Lipid metabolism and lipophagy in cancer. Biochem. Biophys. Res. Commun. 2018, 504, 582–589. [Google Scholar] [CrossRef]
- Matsushita, Y.; Nakagawa, H.; Koike, K. Lipid Metabolism in Oncology: Why It Matters, How to Research, and How to Treat. Cancers 2021, 13, 474. [Google Scholar] [CrossRef]
- Wang, C.W. Lipid droplets, lipophagy, and beyond. Biochim. Biophys. Acta 2016, 1861, 793–805. [Google Scholar] [CrossRef]
- Petan, T. Lipid Droplets in Cancer. Rev. Physiol. Biochem. Pharmacol. 2023, 185, 53–86. [Google Scholar] [CrossRef]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Farese, R.V., Jr.; Walther, T.C. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 2009, 139, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, A.S.; Egan, J.J.; Wek, S.A.; Garty, N.B.; Blanchette-Mackie, E.J.; Londos, C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 1991, 266, 11341–11346. [Google Scholar] [CrossRef]
- Bosch, M.; Pol, A. Eukaryotic lipid droplets: Metabolic hubs, and immune first responders. Trends Endocrinol. Metab. 2022, 33, 218–229. [Google Scholar] [CrossRef]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1260–1272. [Google Scholar] [CrossRef]
- Kimmel, A.R.; Sztalryd, C. The Perilipins: Major Cytosolic Lipid Droplet-Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu. Rev. Nutr. 2016, 36, 471–509. [Google Scholar] [CrossRef]
- Zhang, P.; Meng, L.; Song, L.; Du, J.; Du, S.; Cui, W.; Liu, C.; Li, F. Roles of Perilipins in Diseases and Cancers. Curr. Genom. 2018, 19, 247–257. [Google Scholar] [CrossRef]
- Wilfling, F.; Haas, J.T.; Walther, T.C.; Farese, R.V., Jr. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 2014, 29, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Clim. Assoc. 2010, 121, 206–220. [Google Scholar]
- Hao, J.W.; Wang, J.; Guo, H.; Zhao, Y.Y.; Sun, H.H.; Li, Y.F.; Lai, X.Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Boord, J.B.; Fazio, S.; Linton, M.F. Cytoplasmic fatty acid-binding proteins: Emerging roles in metabolism and atherosclerosis. Curr. Opin. Lipidol. 2002, 13, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.A.; Lee, D.P. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; McFie, P.J.; Banman, S.L.; Brandt, C.; Stone, S.J. Diacylglycerol acyltransferase-2 (DGAT2) and monoacylglycerol acyltransferase-2 (MGAT2) interact to promote triacylglycerol synthesis. J. Biol. Chem. 2014, 289, 28237–28248. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.C.; Farese, R.V., Jr. The life of lipid droplets. Biochim. Biophys. Acta 2009, 1791, 459–466. [Google Scholar] [CrossRef]
- Kadereit, B.; Kumar, P.; Wang, W.J.; Miranda, D.; Snapp, E.L.; Severina, N.; Torregroza, I.; Evans, T.; Silver, D.L. Evolutionarily conserved gene family important for fat storage. Proc. Natl. Acad. Sci. USA 2008, 105, 94–99. [Google Scholar] [CrossRef]
- Wang, G.; Chen, A.; Wu, Y.; Wang, D.; Chang, C.; Yu, G. Fat storage-inducing transmembrane proteins: Beyond mediating lipid droplet formation. Cell Mol. Biol. Lett. 2022, 27, 98. [Google Scholar] [CrossRef]
- Salo, V.T.; Belevich, I.; Li, S.; Karhinen, L.; Vihinen, H.; Vigouroux, C.; Magre, J.; Thiele, C.; Holtta-Vuori, M.; Jokitalo, E.; et al. Seipin regulates ER-lipid droplet contacts and cargo delivery. EMBO J. 2016, 35, 2699–2716. [Google Scholar] [CrossRef]
- Barneda, D.; Christian, M. Lipid droplet growth: Regulation of a dynamic organelle. Curr. Opin. Cell Biol. 2017, 47, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Tan, Y.; Zhao, P.; Ren, Z. SEIPIN: A Key Factor for Nuclear Lipid Droplet Generation and Lipid Homeostasis. Int. J. Mol. Sci. 2020, 21, 8208. [Google Scholar] [CrossRef]
- Rao, M.J.; Goodman, J.M. Seipin: Harvesting fat and keeping adipocytes healthy. Trends Cell Biol. 2021, 31, 912–923. [Google Scholar] [CrossRef]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef]
- D’Souza-Schorey, C.; Chavrier, P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Wolins, N.E.; Brasaemle, D.L.; Bickel, P.E. A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett. 2006, 580, 5484–5491. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Olzmann, J.A. Establishing the lipid droplet proteome: Mechanisms of lipid droplet protein targeting and degradation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1166–1177. [Google Scholar] [CrossRef]
- Schuldiner, M.; Bohnert, M. A different kind of love—Lipid droplet contact sites. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1188–1196. [Google Scholar] [CrossRef]
- Sztalryd, C.; Kimmel, A.R. Perilipins: Lipid droplet coat proteins adapted for tissue-specific energy storage and utilization, and lipid cytoprotection. Biochimie 2014, 96, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.S.; de Mare, S.; Jones, H.A.; Goransson, O.; Lindkvist-Petersson, K. Visualization of lipid directed dynamics of perilipin 1 in human primary adipocytes. Sci. Rep. 2017, 7, 15011. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Omatsu, N.; Morimoto, E.; Nakashima, H.; Ueno, K.; Tanaka, T.; Satouchi, K.; Hirose, F.; Osumi, T. CGI-58 facilitates lipolysis on lipid droplets but is not involved in the vesiculation of lipid droplets caused by hormonal stimulation. J. Lipid Res. 2007, 48, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfacini, D.; Pfeifer, A. GPCR in Adipose Tissue Function-Focus on Lipolysis. Biomedicines 2023, 11, 588. [Google Scholar] [CrossRef] [PubMed]
- Mottagui-Tabar, S.; Ryden, M.; Lofgren, P.; Faulds, G.; Hoffstedt, J.; Brookes, A.J.; Andersson, I.; Arner, P. Evidence for an important role of perilipin in the regulation of human adipocyte lipolysis. Diabetologia 2003, 46, 789–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garaulet, M.; Vera, B.; Bonnet-Rubio, G.; Gomez-Abellan, P.; Lee, Y.C.; Ordovas, J.M. Lunch eating predicts weight-loss effectiveness in carriers of the common allele at PERILIPIN1: The ONTIME (Obesity, Nutrigenetics, Timing, Mediterranean) study. Am. J. Clin. Nutr. 2016, 104, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.E.; Ordovas, J.M. Update on perilipin polymorphisms and obesity. Nutr. Rev. 2012, 70, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Bialesova, L.; Kulyte, A.; Petrus, P.; Sinha, I.; Laurencikiene, J.; Zhao, C.; Wright, K.D.; Arner, P.; Dahlman, I. Epigenetic Regulation of PLIN 1 in Obese Women and its Relation to Lipolysis. Sci. Rep. 2017, 7, 10152. [Google Scholar] [CrossRef] [Green Version]
- Kern, P.A.; Di Gregorio, G.; Lu, T.; Rassouli, N.; Ranganathan, G. Perilipin expression in human adipose tissue is elevated with obesity. J. Clin. Endocrinol. Metab. 2004, 89, 1352–1358. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Souza, S.C.; Endo, M.; Sawada, T.; Perfield, J.W., 2nd; Shimizu, C.; Stancheva, Z.; Nagai, S.; Strissel, K.J.; Yoshioka, N.; et al. Perilipin overexpression in mice protects against diet-induced obesity. J. Lipid Res. 2010, 51, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sullivan, S.; Trujillo, M.; Lee, M.J.; Schneider, S.H.; Brolin, R.E.; Kang, Y.H.; Werber, Y.; Greenberg, A.S.; Fried, S.K. Perilipin expression in human adipose tissues: Effects of severe obesity, gender, and depot. Obes. Res. 2003, 11, 930–936. [Google Scholar] [CrossRef]
- De Oliveira, B.A.P.; de Souza Pinhel, M.A.; Nicoletti, C.F.; de Oliveira, C.C.; Quinhoneiro, D.C.G.; Noronha, N.Y.; Fassini, P.G.; da Silva Junior, W.A.; Junior, W.S.; Nonino, C.B. UCP2 and PLIN1 Expression Affects the Resting Metabolic Rate and Weight Loss on Obese Patients. Obes. Surg. 2017, 27, 343–348. [Google Scholar] [CrossRef]
- Meng, L.X.; Zheng, Y.X.; He, M.L.; Zhou, X.M.; Sun, S.Y.; Ding, Z.J.; Meng, Q.; Li, B.C.; Sun, Y.W. Silencing of perilipin by short hairpin RNA inhibits proliferation and induces apoptosis in liposarcoma cells. Mol. Med. Rep. 2018, 18, 4571–4576. [Google Scholar] [CrossRef] [Green Version]
- Straub, B.K.; Herpel, E.; Singer, S.; Zimbelmann, R.; Breuhahn, K.; Macher-Goeppinger, S.; Warth, A.; Lehmann-Koch, J.; Longerich, T.; Heid, H.; et al. Lipid droplet-associated PAT-proteins show frequent and differential expression in neoplastic steatogenesis. Mod. Pathol. 2010, 23, 480–492. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, C.C.; Mrozinski, J.; Riedel, I.; Heid, H.W.; Moll, R. Perilipin 1 is a highly specific marker for adipocytic differentiation in sarcomas with intermediate sensitivity. J. Cancer Res. Clin. Oncol. 2017, 143, 225–232. [Google Scholar] [CrossRef]
- Kim, S.; Lee, Y.; Koo, J.S. Differential expression of lipid metabolism-related proteins in different breast cancer subtypes. PLoS ONE 2015, 10, e0119473. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Wang, M.; Zhou, L.; Zhang, Y.; Liu, W.; Qin, W.; He, R.; Lu, Y.; Wang, Y.; Chen, X.Z.; et al. Prognostic significance of PLIN1 expression in human breast cancer. Oncotarget 2016, 7, 54488–54502. [Google Scholar] [CrossRef] [Green Version]
- Brasaemle, D.L.; Barber, T.; Wolins, N.E.; Serrero, G.; Blanchette-Mackie, E.J.; Londos, C. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J. Lipid Res. 1997, 38, 2249–2263. [Google Scholar] [CrossRef]
- Imamura, M.; Inoguchi, T.; Ikuyama, S.; Taniguchi, S.; Kobayashi, K.; Nakashima, N.; Nawata, H. ADRP stimulates lipid accumulation and lipid droplet formation in murine fibroblasts. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E775–E783. [Google Scholar] [CrossRef] [Green Version]
- Listenberger, L.L.; Ostermeyer-Fay, A.G.; Goldberg, E.B.; Brown, W.J.; Brown, D.A. Adipocyte differentiation-related protein reduces the lipid droplet association of adipose triglyceride lipase and slows triacylglycerol turnover. J. Lipid Res. 2007, 48, 2751–2761. [Google Scholar] [CrossRef] [Green Version]
- Najt, C.P.; Devarajan, M.; Mashek, D.G. Perilipins at a glance. J. Cell Sci. 2022, 135, jcs259501. [Google Scholar] [CrossRef]
- Patel, S.; Yang, W.; Kozusko, K.; Saudek, V.; Savage, D.B. Perilipins 2 and 3 lack a carboxy-terminal domain present in perilipin 1 involved in sequestering ABHD5 and suppressing basal lipolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 9163–9168. [Google Scholar] [CrossRef]
- Dalen, K.T.; Schoonjans, K.; Ulven, S.M.; Weedon-Fekjaer, M.S.; Bentzen, T.G.; Koutnikova, H.; Auwerx, J.; Nebb, H.I. Adipose tissue expression of the lipid droplet-associating proteins S3-12 and perilipin is controlled by peroxisome proliferator-activated receptor-gamma. Diabetes 2004, 53, 1243–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgkinson, C.P.; Ye, S. Microarray analysis of peroxisome proliferator-activated receptor-gamma induced changes in gene expression in macrophages. Biochem. Biophys. Res. Commun. 2003, 308, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Schadinger, S.E.; Bucher, N.L.; Schreiber, B.M.; Farmer, S.R. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1195–E1205. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ye, H.; Serrero, G. Stimulation of adipose differentiation related protein (ADRP) expression in adipocyte precursors by long-chain fatty acids. J. Cell Physiol. 2000, 182, 297–302. [Google Scholar] [CrossRef]
- Lee, Y.J.; Ko, E.H.; Kim, J.E.; Kim, E.; Lee, H.; Choi, H.; Yu, J.H.; Kim, H.J.; Seong, J.K.; Kim, K.S.; et al. Nuclear receptor PPARgamma-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13656–13661. [Google Scholar] [CrossRef]
- Motomura, W.; Inoue, M.; Ohtake, T.; Takahashi, N.; Nagamine, M.; Tanno, S.; Kohgo, Y.; Okumura, T. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem. Biophys. Res. Commun. 2006, 340, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Sikder, K.; Shukla, S.K.; Patel, N.; Singh, H.; Rafiq, K. High Fat Diet Upregulates Fatty Acid Oxidation and Ketogenesis via Intervention of PPAR-gamma. Cell Physiol. Biochem. 2018, 48, 1317–1331. [Google Scholar] [CrossRef]
- Sharma, A.M.; Staels, B. Review: Peroxisome proliferator-activated receptor gamma and adipose tissue--understanding obesity-related changes in regulation of lipid and glucose metabolism. J. Clin. Endocrinol. Metab. 2007, 92, 386–395. [Google Scholar] [CrossRef]
- Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma: A key regulator of adipogenesis and systemic insulin sensitivity. Eur. J. Med. Res. 1997, 2, 457–464. [Google Scholar]
- Zhang, Y.; Lin, S.; Peng, J.; Liang, X.; Yang, Q.; Bai, X.; Li, Y.; Li, J.; Dong, W.; Wang, Y.; et al. Amelioration of hepatic steatosis by dietary essential amino acid-induced ubiquitination. Mol. Cell 2022, 82, 1528–1542.e1510. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, Y.; Lin, S.; Li, Y.; Zhu, A.J.; Shi, H.; Liu, M. Identification of Ubr1 as an amino acid sensor of steatosis in liver and muscle. J. Cachexia Sarcopenia Muscle 2023, 14, 1454–1467. [Google Scholar] [CrossRef]
- Conte, M.; Vasuri, F.; Trisolino, G.; Bellavista, E.; Santoro, A.; Degiovanni, A.; Martucci, E.; D’Errico-Grigioni, A.; Caporossi, D.; Capri, M.; et al. Increased Plin2 expression in human skeletal muscle is associated with sarcopenia and muscle weakness. PLoS ONE 2013, 8, e73709. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Casey, C.A.; Donohue, T.M., Jr.; Kubik, J.L.; Kumar, V.; Naldrett, M.J.; Woods, N.T.; Frisbie, C.P.; McNiven, M.A.; Thomes, P.G. Lipid droplet membrane proteome remodeling parallels ethanol-induced hepatic steatosis and its resolution. J. Lipid Res. 2021, 62, 100049. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Liu, R.; Lee, J.H.; Li, J.; Yu, R.; Tan, L.; Xia, Y.; Zheng, Y.; Bian, X.L.; Lorenzi, P.L.; Chen, Q.; et al. Choline kinase alpha 2 acts as a protein kinase to promote lipolysis of lipid droplets. Mol. Cell 2021, 81, 2722–2735.e2729. [Google Scholar] [CrossRef]
- Bracken, C.P.; Whitelaw, M.L.; Peet, D.J. The hypoxia-inducible factors: Key transcriptional regulators of hypoxic responses. Cell Mol. Life Sci. 2003, 60, 1376–1393. [Google Scholar] [CrossRef]
- Fong, G.H.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef] [Green Version]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2alpha-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schodel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef]
- DiStefano, M.T.; Danai, L.V.; Roth Flach, R.J.; Chawla, A.; Pedersen, D.J.; Guilherme, A.; Czech, M.P. The Lipid Droplet Protein Hypoxia-inducible Gene 2 Promotes Hepatic Triglyceride Deposition by Inhibiting Lipolysis. J. Biol. Chem. 2015, 290, 15175–15184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povero, D.; Johnson, S.M.; Liu, J. Hypoxia, hypoxia-inducible gene 2 (HIG2)/HILPDA, and intracellular lipolysis in cancer. Cancer Lett. 2020, 493, 71–79. [Google Scholar] [CrossRef]
- Liu, W.; Liu, X.; Liu, Y.; Ling, T.; Chen, D.; Otkur, W.; Zhao, H.; Ma, M.; Ma, K.; Dong, B.; et al. PLIN2 promotes HCC cells proliferation by inhibiting the degradation of HIF1alpha. Exp. Cell Res. 2022, 418, 113244. [Google Scholar] [CrossRef]
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Sun, Q.; Wu, X.; Zhang, Y.; Xing, X.; Lin, K.; Feng, Y.; Wang, M.; Wang, Y.; Wang, R. Hypoxia as a Double-Edged Sword to Combat Obesity and Comorbidities. Cells 2022, 11, 3735. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, J.; Honda, K.; Ono, M.; Sekine, S.; Tanaka, Y.; Kobayashi, M.; Jung, G.; Sakuma, T.; Nakamori, S.; Sata, N.; et al. Identification of adipophilin as a potential plasma biomarker for colorectal cancer using label-free quantitative mass spectrometry and protein microarray. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2195–2203. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Fitchev, P.S.; Cornwell, M.L.; Greenberg, J.; Cabe, M.; Weber, C.R.; Roy, H.K.; Crawford, S.E.; Savkovic, S.D. FOXO3 growth inhibition of colonic cells is dependent on intraepithelial lipid droplet density. J. Biol. Chem. 2013, 288, 16274–16281. [Google Scholar] [CrossRef] [Green Version]
- Pucer, A.; Brglez, V.; Payre, C.; Pungercar, J.; Lambeau, G.; Petan, T. Group X secreted phospholipase A(2) induces lipid droplet formation and prolongs breast cancer cell survival. Mol. Cancer 2013, 12, 111. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Su, L.; Sun, K. Expression status and prognostic value of the perilipin family of genes in breast cancer. Am. J. Transl. Res. 2021, 13, 4450–4463. [Google Scholar]
- Lin, L.C.; Gao, A.C.; Lai, C.H.; Hsieh, J.T.; Lin, H. Induction of neuroendocrine differentiation in castration resistant prostate cancer cells by adipocyte differentiation-related protein (ADRP) delivered by exosomes. Cancer Lett. 2017, 391, 74–82. [Google Scholar] [CrossRef]
- Morrissey, J.J.; Mobley, J.; Figenshau, R.S.; Vetter, J.; Bhayani, S.; Kharasch, E.D. Urine aquaporin 1 and perilipin 2 differentiate renal carcinomas from other imaged renal masses and bladder and prostate cancer. Mayo Clin. Proc. 2015, 90, 35–42. [Google Scholar] [CrossRef]
- Fujimoto, M.; Yoshizawa, A.; Sumiyoshi, S.; Sonobe, M.; Menju, T.; Hirata, M.; Momose, M.; Date, H.; Haga, H. Adipophilin expression in lung adenocarcinoma is associated with apocrine-like features and poor clinical prognosis: An immunohistochemical study of 328 cases. Histopathology 2017, 70, 232–241. [Google Scholar] [CrossRef]
- Meng, X.; Sun, R.; Wang, W.; Zhang, N.; Cao, S.; Liu, B.; Fang, P.; Deng, S.; Yang, S. ADFP promotes cell proliferation in lung adenocarcinoma via Akt phosphorylation. J. Cell Mol. Med. 2021, 25, 827–839. [Google Scholar] [CrossRef]
- Cao, Q.; Ruan, H.; Wang, K.; Song, Z.; Bao, L.; Xu, T.; Xiao, H.; Wang, C.; Cheng, G.; Tong, J.; et al. Overexpression of PLIN2 is a prognostic marker and attenuates tumor progression in clear cell renal cell carcinoma. Int. J. Oncol. 2018, 53, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, J.J.; London, A.N.; Luo, J.; Kharasch, E.D. Urinary biomarkers for the early diagnosis of kidney cancer. Mayo Clin. Proc. 2010, 85, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, J.J.; Mellnick, V.M.; Luo, J.; Siegel, M.J.; Figenshau, R.S.; Bhayani, S.; Kharasch, E.D. Evaluation of Urine Aquaporin-1 and Perilipin-2 Concentrations as Biomarkers to Screen for Renal Cell Carcinoma: A Prospective Cohort Study. JAMA Oncol. 2015, 1, 204–212. [Google Scholar] [CrossRef]
- Morrissey, J.J.; Mobley, J.; Song, J.; Vetter, J.; Luo, J.; Bhayani, S.; Figenshau, R.S.; Kharasch, E.D. Urinary concentrations of aquaporin-1 and perilipin-2 in patients with renal cell carcinoma correlate with tumor size and stage but not grade. Urology 2014, 83, 256.e9–256.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.B.; Morrissey, J.J.; Mobley, J.M.; Figenshau, K.G.; Vetter, J.M.; Bhayani, S.B.; Kharasch, E.D.; Figenshau, R.S. Urinary aquaporin 1 and perilipin 2: Can these novel markers accurately characterize small renal masses and help guide patient management? Int. J. Urol. 2019, 26, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Zhong, L.; Zhou, J.; Lu, M.; Xing, T.; Ma, L.; Shen, J. Data-Independent Acquisition-Based Quantitative Proteomic Analysis Reveals Potential Biomarkers of Kidney Cancer. Proteom. Clin. Appl. 2017, 11, 1700066. [Google Scholar] [CrossRef] [PubMed]
- Tolkach, Y.; Luders, C.; Meller, S.; Jung, K.; Stephan, C.; Kristiansen, G. Adipophilin as prognostic biomarker in clear cell renal cell carcinoma. Oncotarget 2017, 8, 28672–28682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, M.; Huang, Y.; Shioi, K.; Hattori, K.; Murakami, T.; Nakaigawa, N.; Kishida, T.; Nagashima, Y.; Kubota, Y. Expression of adipose differentiation-related protein: A predictor of cancer-specific survival in clear cell renal carcinoma. Clin. Cancer Res. 2007, 13, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Takada, N.; Hirokawa, M.; Ito, A.; Suzuki, A.; Higuchi, M.; Kuma, S.; Hayashi, T.; Daa, T.; Miyauchi, A. Cytoplasmic Lipid Accumulation Characteristic of the Cribriform Variant of Papillary Thyroid Carcinoma. Pathobiology 2017, 84, 251–257. [Google Scholar] [CrossRef]
- Sun, X.; Yang, S.; Feng, X.; Zheng, Y.; Zhou, J.; Wang, H.; Zhang, Y.; Sun, H.; He, C. The modification of ferroptosis and abnormal lipometabolism through overexpression and knockdown of potential prognostic biomarker perilipin2 in gastric carcinoma. Gastric Cancer 2020, 23, 241–259. [Google Scholar] [CrossRef]
- Fujimoto, M.; Matsuzaki, I.; Yamamoto, Y.; Yoshizawa, A.; Warigaya, K.; Iwahashi, Y.; Kojima, F.; Furukawa, F.; Murata, S.I. Adipophilin expression in cutaneous malignant melanoma. J. Cutan. Pathol. 2017, 44, 228–236. [Google Scholar] [CrossRef]
- Fortunato, D.; Giuffrida, M.G.; Cavaletto, M.; Garoffo, L.P.; Dellavalle, G.; Napolitano, L.; Giunta, C.; Fabris, C.; Bertino, E.; Coscia, A.; et al. Structural proteome of human colostral fat globule membrane proteins. Proteomics 2003, 3, 897–905. [Google Scholar] [CrossRef]
- Heid, H.W.; Schnolzer, M.; Keenan, T.W. Adipocyte differentiation-related protein is secreted into milk as a constituent of milk lipid globule membrane. Biochem. J. 1996, 320 Pt 3, 1025–1030. [Google Scholar] [CrossRef] [Green Version]
- Monks, J.; Orlicky, D.J.; Libby, A.E.; Dzieciatkowska, M.; Ladinsky, M.S.; McManaman, J.L. Perilipin-2 promotes lipid droplet-plasma membrane interactions that facilitate apocrine lipid secretion in secretory epithelial cells of the mouse mammary gland. Front. Cell Dev. Biol. 2022, 10, 958566. [Google Scholar] [CrossRef]
- Chong, B.M.; Reigan, P.; Mayle-Combs, K.D.; Orlicky, D.J.; McManaman, J.L. Determinants of adipophilin function in milk lipid formation and secretion. Trends Endocrinol. Metab. 2011, 22, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Karasek, D.; Vaverkova, H.; Frysak, Z.; Orsag, J.; Novotny, D.; Halenka, M.; Slavik, L. Relationship between serum adipocyte fatty acid-binding protein and endothelial/hemostatic markers in dyslipidemic subjects. Neuro Endocrinol. Lett. 2012, 33 (Suppl. 2), 26–31. [Google Scholar]
- Liao, B.; Geng, L.; Zhang, F.; Shu, L.; Wei, L.; Yeung, P.K.K.; Lam, K.S.L.; Chung, S.K.; Chang, J.; Vanhoutte, P.M.; et al. Adipocyte fatty acid-binding protein exacerbates cerebral ischaemia injury by disrupting the blood-brain barrier. Eur. Heart J. 2020, 41, 3169–3180. [Google Scholar] [CrossRef]
- Wei, C.; Liu, Y.; Xing, E.; Ding, Z.; Tian, Y.; Zhao, Z.; Fan, W.; Sun, L. Association Between Novel Pro- and Anti- Inflammatory Adipocytokines in Patients with Acute Coronary Syndrome. Clin. Appl. Thromb. Hemost. 2022, 28, 10760296221128021. [Google Scholar] [CrossRef]
- Orlicky, D.J.; Libby, A.E.; Bales, E.S.; McMahan, R.H.; Monks, J.; La Rosa, F.G.; McManaman, J.L. Perilipin-2 promotes obesity and progressive fatty liver disease in mice through mechanistically distinct hepatocyte and extra-hepatocyte actions. J. Physiol. 2019, 597, 1565–1584. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.Y.; Miyoshi, H.; Nakamura, A.; Greenberg, A.S.; Atsumi, T. Lipid Droplet Protein PLIN1 Regulates Inflammatory Polarity in Human Macrophages and is Involved in Atherosclerotic Plaque Development by Promoting Stable Lipid Storage. J. Atheroscler. Thromb. 2023, 30, 170–181. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, L.; Han, C.; Huang, R.; Ooi, K.; Qian, X.; Ren, X.; Chu, D.; Zhang, H.; Du, D.; et al. PLIN2 Mediates Neuroinflammation and Oxidative/Nitrosative Stress via Downregulating Phosphatidylethanolamine in the Rostral Ventrolateral Medulla of Stressed Hypertensive Rats. J. Inflamm. Res. 2021, 14, 6331–6348. [Google Scholar] [CrossRef]
- Conte, M.; Medici, V.; Malagoli, D.; Chiariello, A.; Cirrincione, A.; Davin, A.; Chikhladze, M.; Vasuri, F.; Legname, G.; Ferrer, I.; et al. Expression pattern of perilipins in human brain during aging and in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12756. [Google Scholar] [CrossRef]
- Bogie, J.F.J.; Grajchen, E.; Wouters, E.; Corrales, A.G.; Dierckx, T.; Vanherle, S.; Mailleux, J.; Gervois, P.; Wolfs, E.; Dehairs, J.; et al. Stearoyl-CoA desaturase-1 impairs the reparative properties of macrophages and microglia in the brain. J. Exp. Med. 2020, 217, e20191660. [Google Scholar] [CrossRef]
- Loix, M.; Wouters, E.; Vanherle, S.; Dehairs, J.; McManaman, J.L.; Kemps, H.; Swinnen, J.V.; Haidar, M.; Bogie, J.F.J.; Hendriks, J.J.A. Perilipin-2 limits remyelination by preventing lipid droplet degradation. Cell Mol. Life Sci. 2022, 79, 515. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhao, Y.; Guo, H.; Li, Q.; Yan, C.; Li, Y.; He, S.; Wang, N.; Wang, Q. Impaired lipophagy induced-microglial lipid droplets accumulation contributes to the buildup of TREM1 in diabetes-associated cognitive impairment. Autophagy 2023, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Franceschi, C.; Sandri, M.; Salvioli, S. Perilipin 2 and Age-Related Metabolic Diseases: A New Perspective. Trends Endocrinol. Metab. 2016, 27, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Wolins, N.E.; Rubin, B.; Brasaemle, D.L. TIP47 associates with lipid droplets. J. Biol. Chem. 2001, 276, 5101–5108. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.M.; Ajjaji, D.; Fleming, K.D.; Borbat, P.P.; Jenkins, M.L.; Moeller, B.E.; Fernando, S.; Bhatia, S.R.; Freed, J.H.; Burke, J.E.; et al. Structural insights into perilipin 3 membrane association in response to diacylglycerol accumulation. Nat. Commun. 2023, 14, 3204. [Google Scholar] [CrossRef]
- Skinner, J.R.; Shew, T.M.; Schwartz, D.M.; Tzekov, A.; Lepus, C.M.; Abumrad, N.A.; Wolins, N.E. Diacylglycerol enrichment of endoplasmic reticulum or lipid droplets recruits perilipin 3/TIP47 during lipid storage and mobilization. J. Biol. Chem. 2009, 284, 30941–30948. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Sohn, J.H.; Han, J.S.; Park, Y.J.; Jeon, Y.G.; Ji, Y.; Dalen, K.T.; Sztalryd, C.; Kimmel, A.R.; Kim, J.B. Perilipin 3 Deficiency Stimulates Thermogenic Beige Adipocytes Through PPARalpha Activation. Diabetes 2018, 67, 791–804. [Google Scholar] [CrossRef]
- Szigeti, A.; Minik, O.; Hocsak, E.; Pozsgai, E.; Boronkai, A.; Farkas, R.; Balint, A.; Bodis, J.; Sumegi, B.; Bellyei, S. Preliminary study of TIP47 as a possible new biomarker of cervical dysplasia and invasive carcinoma. Anticancer Res. 2009, 29, 717–724. [Google Scholar]
- Wang, K.; Ruan, H.; Song, Z.; Cao, Q.; Bao, L.; Liu, D.; Xu, T.; Xiao, H.; Wang, C.; Cheng, G.; et al. PLIN3 is up-regulated and correlates with poor prognosis in clear cell renal cell carcinoma. Urol. Oncol. 2018, 36, 343.e9–343.e19. [Google Scholar] [CrossRef]
- Lung, J.; Hung, M.S.; Wang, T.Y.; Chen, K.L.; Luo, C.W.; Jiang, Y.Y.; Wu, S.Y.; Lee, L.W.; Lin, P.Y.; Chen, F.F.; et al. Lipid Droplets in Lung Cancers Are Crucial for the Cell Growth and Starvation Survival. Int. J. Mol. Sci. 2022, 23, 12533. [Google Scholar] [CrossRef]
- Lamprou, I.; Kakouratos, C.; Tsolou, A.; Pavlidis, P.; Xanthopoulou, E.T.; Nanos, C.; Tsaroucha, A.; Sivridis, E.; Giatromanolaki, A.; Koukourakis, M.I. Lipophagy-Related Protein Perilipin-3 and Resistance of Prostate Cancer to Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2022, 113, 401–414. [Google Scholar] [CrossRef]
- Lamprou, I.; Tsolou, A.; Kakouratos, C.; Mitrakas, A.G.; Xanthopoulou, E.T.; Kassela, K.; Karakasiliotis, I.; Zois, C.E.; Giatromanolaki, A.; Koukourakis, M.I. Suppressed PLIN3 frequently occurs in prostate cancer, promoting docetaxel resistance via intensified autophagy, an event reversed by chloroquine. Med. Oncol. 2021, 38, 116. [Google Scholar] [CrossRef]
- Zhou, L.; Song, Z.; Hu, J.; Liu, L.; Hou, Y.; Zhang, X.; Yang, X.; Chen, K. ACSS3 represses prostate cancer progression through downregulating lipid droplet-associated protein PLIN3. Theranostics 2021, 11, 841–860. [Google Scholar] [CrossRef]
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol. Med. 2019, 92, 435–452. [Google Scholar]
- Wolins, N.E.; Skinner, J.R.; Schoenfish, M.J.; Tzekov, A.; Bensch, K.G.; Bickel, P.E. Adipocyte protein S3-12 coats nascent lipid droplets. J. Biol. Chem. 2003, 278, 37713–37721. [Google Scholar] [CrossRef] [Green Version]
- Scherer, P.E.; Bickel, P.E.; Kotler, M.; Lodish, H.F. Cloning of cell-specific secreted and surface proteins by subtractive antibody screening. Nat. Biotechnol. 1998, 16, 581–586. [Google Scholar] [CrossRef]
- Hsieh, K.; Lee, Y.K.; Londos, C.; Raaka, B.M.; Dalen, K.T.; Kimmel, A.R. Perilipin family members preferentially sequester to either triacylglycerol-specific or cholesteryl-ester-specific intracellular lipid storage droplets. J. Cell Sci. 2012, 125, 4067–4076. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, F.B.; Khor, V.K.; Shen, W.J.; Azhar, S. Cholesterol ester droplets and steroidogenesis. Mol. Cell Endocrinol. 2013, 371, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Chang, B.; Wu, X.; Li, L.; Sleeman, M.; Chan, L. Inactivation of Plin4 downregulates Plin5 and reduces cardiac lipid accumulation in mice. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E770–E779. [Google Scholar] [CrossRef] [Green Version]
- Richardson, K.; Louie-Gao, Q.; Arnett, D.K.; Parnell, L.D.; Lai, C.Q.; Davalos, A.; Fox, C.S.; Demissie, S.; Cupples, L.A.; Fernandez-Hernando, C.; et al. The PLIN4 variant rs8887 modulates obesity related phenotypes in humans through creation of a novel miR-522 seed site. PLoS ONE 2011, 6, e17944. [Google Scholar] [CrossRef] [Green Version]
- Sirois, I.; Aguilar-Mahecha, A.; Lafleur, J.; Fowler, E.; Vu, V.; Scriver, M.; Buchanan, M.; Chabot, C.; Ramanathan, A.; Balachandran, B.; et al. A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability. Mol. Cancer Res. 2019, 17, 2492–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickel, P.E.; Tansey, J.T.; Welte, M.A. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta 2009, 1791, 419–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashani, M.; Witzel, H.R.; Pawella, L.M.; Lehmann-Koch, J.; Schumacher, J.; Mechtersheimer, G.; Schnolzer, M.; Schirmacher, P.; Roth, W.; Straub, B.K. Widespread expression of perilipin 5 in normal human tissues and in diseases is restricted to distinct lipid droplet subpopulations. Cell Tissue Res. 2018, 374, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, A.R.; Sztalryd, C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr. Opin. Lipidol. 2014, 25, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurens, C.; Bourlier, V.; Mairal, A.; Louche, K.; Badin, P.M.; Mouisel, E.; Montagner, A.; Marette, A.; Tremblay, A.; Weisnagel, J.S.; et al. Perilipin 5 fine-tunes lipid oxidation to metabolic demand and protects against lipotoxicity in skeletal muscle. Sci. Rep. 2016, 6, 38310. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Chen, A. Perilipin 5 restores the formation of lipid droplets in activated hepatic stellate cells and inhibits their activation. Lab. Investig. 2016, 96, 791–806. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Xie, Z.; Yuan, Y.; Sui, W.; Wang, C.; Gao, X.; Zhao, Y.; Zhang, F.; Gu, Y.; Hu, P.; et al. Plin5 alleviates myocardial ischaemia/reperfusion injury by reducing oxidative stress through inhibiting the lipolysis of lipid droplets. Sci. Rep. 2017, 7, 42574. [Google Scholar] [CrossRef] [Green Version]
- Pollak, N.M.; Jaeger, D.; Kolleritsch, S.; Zimmermann, R.; Zechner, R.; Lass, A.; Haemmerle, G. The interplay of protein kinase A and perilipin 5 regulates cardiac lipolysis. J. Biol. Chem. 2015, 290, 1295–1306. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xu, W.; Xu, R.; Wang, Z.; Zhang, X.; Wang, P.; Peng, K.; Li, M.; Li, J.; Tan, Y.; et al. Plin5 Bidirectionally Regulates Lipid Metabolism in Oxidative Tissues. Oxid. Med. Cell Longev. 2022, 2022, 4594956. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Vucur, M.; Luedde, T.; Schneiders, S.; Kalampoka, S.; Weiss, T.S.; Weiskirchen, R. Perilipin 5 and Lipocalin 2 Expression in Hepatocellular Carcinoma. Cancers 2019, 11, 385. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Yan, G.; Sun, H.; Zhang, J.; Song, D.; Kong, R.; Yan, P.; Hu, P.; Xie, A.; Wang, S.; et al. Identification of prognostic and metastasis-related alternative splicing signatures in hepatocellular carcinoma. Biosci. Rep. 2020, 40, BSR20201001. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Arimura, N.; Horiba, T.; Imagawa, M.; Shimizu, M.; Sato, R. The peroxisome proliferator-activated receptor gamma regulates expression of the perilipin gene in adipocytes. J. Biol. Chem. 2004, 279, 10070–10076. [Google Scholar] [CrossRef] [Green Version]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Croce, M.A.; Gropler, M.C.; Varma, V.; Yao-Borengasser, A.; Rasouli, N.; Kern, P.A.; et al. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 2006, 55, 3418–3428. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, N.S.; Kumar, N.; Duseja, A. Peroxisome Proliferator-Activated Receptors and Their Agonists in Nonalcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2019, 9, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Burri, L.; Thoresen, G.H.; Berge, R.K. The Role of PPARα Activation in Liver and Muscle. PPAR Res. 2010, 2010, 542359. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Hamza, M.S.; Pott, S.; Vega, V.B.; Thomsen, J.S.; Kandhadayar, G.S.; Ng, P.W.; Chiu, K.P.; Pettersson, S.; Wei, C.L.; Ruan, Y.; et al. De-novo identification of PPARgamma/RXR binding sites and direct targets during adipogenesis. PLoS ONE 2009, 4, e4907. [Google Scholar] [CrossRef] [Green Version]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Giulitti, F.; Petrungaro, S.; Mandatori, S.; Tomaipitinca, L.; de Franchis, V.; D’Amore, A.; Filippini, A.; Gaudio, E.; Ziparo, E.; Giampietri, C. Anti-tumor Effect of Oleic Acid in Hepatocellular Carcinoma Cell Lines via Autophagy Reduction. Front. Cell Dev. Biol. 2021, 9, 629182. [Google Scholar] [CrossRef]
- Azukisawa, S.; Zheng, J.; Guo, X.; Ura, H.; Niida, Y.; Itoh, T.; Yamada, S. The differential expression of perilipin-2 in hepatoblastoma and its association with prognosis. Histol. Histopathol. 2021, 36, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- De Lima-Souza, R.A.; Rodrigues, N.M.; Scarini, J.F.; Silva, M.F.S.; Tincani, A.J.; Egal, E.S.A.; Altemani, A.; Mariano, F.V. Metabolic alterations in carcinoma ex pleomorphic adenoma development of lacrimal glands. Int. Ophthalmol. 2022, 42, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Li, Y.; He, T.; Liao, Y.; Du, Q.; Wang, P.; Qiao, J.; Guo, H. The prognostic miR-532-5p-correlated ceRNA-mediated lipid droplet accumulation drives nodal metastasis of cervical cancer. J. Adv. Res. 2022, 37, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Rios Garcia, M.; Meissburger, B.; Chan, J.; de Guia, R.M.; Mattijssen, F.; Roessler, S.; Birkenfeld, A.L.; Raschzok, N.; Riols, F.; Tokarz, J.; et al. Trip13 Depletion in Liver Cancer Induces a Lipogenic Response Contributing to Plin2-Dependent Mitotic Cell Death. Adv. Sci. 2022, 9, e2104291. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Pati, S.; Irfan, W.; Jameel, A.; Ahmed, S.; Shahid, R.K. Obesity and Cancer: A Current Overview of Epidemiology, Pathogenesis, Outcomes, and Management. Cancers 2023, 15, 485. [Google Scholar] [CrossRef]
- Mukherjee, A.; Bilecz, A.J.; Lengyel, E. The adipocyte microenvironment and cancer. Cancer Metastasis Rev. 2022, 41, 575–587. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- McGraw, D.W.; Liggett, S.B. Molecular mechanisms of beta2-adrenergic receptor function and regulation. Proc. Am. Thorac. Soc. 2005, 2, 292–296, discussion 292–311. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.; Kacprzak, K.; Quintas, C.; Goncalves, J.; Fresco, P. Activation of beta-Adrenoceptors Promotes Lipid Droplet Accumulation in MCF-7 Breast Cancer Cells via cAMP/PKA/EPAC Pathways. Int. J. Mol. Sci. 2023, 24, 767. [Google Scholar] [CrossRef]
- Amaro, F.; Silva, D.; Reguengo, H.; Oliveira, J.C.; Quintas, C.; Vale, N.; Goncalves, J.; Fresco, P. beta-Adrenoceptor Activation in Breast MCF-10A Cells Induces a Pattern of Catecholamine Production Similar to that of Tumorigenic MCF-7 Cells. Int. J. Mol. Sci. 2020, 21, 7968. [Google Scholar] [CrossRef]
- Silva, D.; Quintas, C.; Goncalves, J.; Fresco, P. Contribution of adrenergic mechanisms for the stress-induced breast cancer carcinogenesis. J. Cell Physiol. 2022, 237, 2107–2127. [Google Scholar] [CrossRef]
- Montoya, A.; Amaya, C.N.; Belmont, A.; Diab, N.; Trevino, R.; Villanueva, G.; Rains, S.; Sanchez, L.A.; Badri, N.; Otoukesh, S.; et al. Use of non-selective beta-blockers is associated with decreased tumor proliferative indices in early stage breast cancer. Oncotarget 2017, 8, 6446–6460. [Google Scholar] [CrossRef] [Green Version]
- Gervois, P.; Fruchart, J.C.; Staels, B. Drug Insight: Mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptors. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 145–156. [Google Scholar] [CrossRef]
- Curry, A.M.; White, D.S.; Donu, D.; Cen, Y. Human Sirtuin Regulators: The “Success” Stories. Front. Physiol. 2021, 12, 752117. [Google Scholar] [CrossRef]
- Chen, M.; Huang, L.; Lv, Y.; Li, L.; Dong, Q. Sulforaphane protects against oxidative stress-induced apoptosis via activating SIRT1 in mouse osteoarthritis. Mol. Med. Rep. 2021, 24, 612. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Smith, U. Adrenergic control of lipid metabolism. Acta Med. Scand. Suppl. 1983, 672, 41–44. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [Green Version]
- Lalloyer, F.; Staels, B. Fibrates, glitazones, and peroxisome proliferator-activated receptors. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 894–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [Green Version]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Wang, M.; Yang, K.; Chi, T.; Liao, Z.; Wei, P. PPAR-alpha Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, 599995. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, W. The Journey of Thiazolidinediones as Modulators of PPARs for the Management of Diabetes: A Current Perspective. Curr. Pharm. Des. 2019, 25, 2540–2554. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- American Association of Neurological Surgeons (AANS); American Society of Neuroradiology (ASNR); Cardiovascular and Interventional Radiology Society of Europe (CIRSE); Canadian Interventional Radiology Association (CIRA); Congress of Neurological Surgeons (CNS); European Society of Minimally Invasive Neurological Therapy (ESMINT); European Society of Neuroradiology (ESNR); European Stroke Organization (ESO); Society for Cardiovascular Angiography and Interventions (SCAI); Society of Interventional Radiology (SIR); et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar]
- De la Rosa Rodriguez, M.A.; Kersten, S. Regulation of lipid droplet-associated proteins by peroxisome proliferator-activated receptors. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1212–1220. [Google Scholar] [CrossRef]
- Biondo, L.A.; Teixeira, A.A.S.; de OS Ferreira, K.C.; Neto, J.C.R. Pharmacological Strategies for Insulin Sensitivity in Obesity and Cancer: Thiazolidinediones and Metformin. Curr. Pharm. Des. 2020, 26, 932–945. [Google Scholar] [CrossRef]
- Reka, A.K.; Goswami, M.T.; Krishnapuram, R.; Standiford, T.J.; Keshamouni, V.G. Molecular cross-regulation between PPAR-gamma and other signaling pathways: Implications for lung cancer therapy. Lung Cancer 2011, 72, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Augimeri, G.; Gelsomino, L.; Plastina, P.; Giordano, C.; Barone, I.; Catalano, S.; Ando, S.; Bonofiglio, D. Natural and Synthetic PPARgamma Ligands in Tumor Microenvironment: A New Potential Strategy against Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9721. [Google Scholar] [CrossRef]
- Chi, T.; Wang, M.; Wang, X.; Yang, K.; Xie, F.; Liao, Z.; Wei, P. PPAR-gamma Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, 737776. [Google Scholar] [CrossRef]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef]
- Consoli, A.; Devangelio, E. Thiazolidinediones and inflammation. Lupus 2005, 14, 794–797. [Google Scholar] [CrossRef]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.F.; Yang, S.H.; Lee, Y.H.; Wu, B.C.; Huang, S.C.; Liu, C.M.; Chen, S.L.; Pan, Y.F.; Chou, S.S.; Chou, M.Y.; et al. Aspirin-induced inhibition of adipogenesis was p53-dependent and associated with inactivation of pentose phosphate pathway. Eur. J. Pharmacol. 2014, 738, 101–110. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Huang, Y.S. Aspirin Breaks the Crosstalk between 3T3-L1 Adipocytes and 4T1 Breast Cancer Cells by Regulating Cytokine Production. PLoS ONE 2016, 11, e0147161. [Google Scholar] [CrossRef]
- Roy, S.; Bhowmik, D.R.; Begum, R.; Amin, M.T.; Islam, M.A.; Ahmed, F.; Hossain, M.S. Aspirin attenuates the expression of adhesion molecules, risk of obesity, and adipose tissue inflammation in high-fat diet-induced obese mice. Prostaglandins Other Lipid Mediat. 2022, 162, 106664. [Google Scholar] [CrossRef]
- Di Minno, A.; Porro, B.; Turnu, L.; Manega, C.M.; Eligini, S.; Barbieri, S.; Chiesa, M.; Poggio, P.; Squellerio, I.; Anesi, A.; et al. Untargeted Metabolomics to Go beyond the Canonical Effect of Acetylsalicylic Acid. J. Clin. Med. 2019, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Gu, Q.; Gu, Y.; Yang, H.; Shi, Q. Metformin Enhances Osteogenesis and Suppresses Adipogenesis of Human Chorionic Villous Mesenchymal Stem Cells. Tohoku J. Exp. Med. 2017, 241, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Yerevanian, A.; Soukas, A.A. Metformin: Mechanisms in Human Obesity and Weight Loss. Curr. Obes. Rep. 2019, 8, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Stumvoll, M. Adiponectin—Its role in metabolism and beyond. Horm. Metab. Res. 2002, 34, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2018, 8, 1031–1063. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mao, S.; Chen, S.; Zhang, W.; Liu, C. PPARs-Orchestrated Metabolic Homeostasis in the Adipose Tissue. Int. J. Mol. Sci. 2021, 22, 8974. [Google Scholar] [CrossRef]
- Lee, M.H.; Klein, R.L.; El-Shewy, H.M.; Luttrell, D.K.; Luttrell, L.M. The adiponectin receptors AdipoR1 and AdipoR2 activate ERK1/2 through a Src/Ras-dependent pathway and stimulate cell growth. Biochemistry 2008, 47, 11682–11692. [Google Scholar] [CrossRef] [Green Version]
- Almabouada, F.; Diaz-Ruiz, A.; Rabanal-Ruiz, Y.; Peinado, J.R.; Vazquez-Martinez, R.; Malagon, M.M. Adiponectin receptors form homomers and heteromers exhibiting distinct ligand binding and intracellular signaling properties. J. Biol. Chem. 2013, 288, 3112–3125. [Google Scholar] [CrossRef] [Green Version]
- Vasiliauskaite-Brooks, I.; Sounier, R.; Rochaix, P.; Bellot, G.; Fortier, M.; Hoh, F.; De Colibus, L.; Bechara, C.; Saied, E.M.; Arenz, C.; et al. Structural insights into adiponectin receptors suggest ceramidase activity. Nature 2017, 544, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Kubota, N.; Yano, W.; Kubota, T.; Yamauchi, T.; Itoh, S.; Kumagai, H.; Kozono, H.; Takamoto, I.; Okamoto, S.; Shiuchi, T.; et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007, 6, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Nehme, R.; Diab-Assaf, M.; Decombat, C.; Delort, L.; Caldefie-Chezet, F. Targeting Adiponectin in Breast Cancer. Biomedicines 2022, 10, 2958. [Google Scholar] [CrossRef]
- Zocchi, M.; Della Porta, M.; Lombardoni, F.; Scrimieri, R.; Zuccotti, G.V.; Maier, J.A.; Cazzola, R. A Potential Interplay between HDLs and Adiponectin in Promoting Endothelial Dysfunction in Obesity. Biomedicines 2022, 10, 1344. [Google Scholar] [CrossRef]
- Tanyanskiy, D.A.; Shavva, V.S.; Dizhe, E.B.; Oleinikova, G.N.; Lizunov, A.V.; Nekrasova, E.V.; Mogilenko, D.A.; Larionova, E.E.; Orlov, S.V.; Denisenko, A.D. Adiponectin Stimulates Apolipoprotein A-1 Gene Expression in HepG2 Cells via AMPK, PPARalpha, and LXRs Signaling Mechanisms. Biochemistry 2022, 87, 1252–1259. [Google Scholar] [CrossRef]
- Vales-Villamarin, C.; Lumpuy-Castillo, J.; Gavela-Perez, T.; de Dios, O.; Perez-Nadador, I.; Soriano-Guillen, L.; Garces, C. Sex-Dependent Mediation of Leptin in the Association of Perilipin Polymorphisms with BMI and Plasma Lipid Levels in Children. Nutrients 2022, 14, 3072. [Google Scholar] [CrossRef]
- Byeon, J.S.; Jeong, J.Y.; Kim, M.J.; Lee, S.M.; Nam, W.H.; Myung, S.J.; Kim, J.G.; Yang, S.K.; Kim, J.H.; Suh, D.J. Adiponectin and adiponectin receptor in relation to colorectal cancer progression. Int. J. Cancer 2010, 127, 2758–2767. [Google Scholar] [CrossRef]
- Karnati, H.K.; Panigrahi, M.K.; Li, Y.; Tweedie, D.; Greig, N.H. Adiponectin as a Potential Therapeutic Target for Prostate Cancer. Curr. Pharm. Des. 2017, 23, 4170–4179. [Google Scholar] [CrossRef] [Green Version]
- Engin, A. Adiponectin-Resistance in Obesity. Adv. Exp. Med. Biol. 2017, 960, 415–441. [Google Scholar] [CrossRef]
- Parida, S.; Siddharth, S.; Sharma, D. Adiponectin, Obesity, and Cancer: Clash of the Bigwigs in Health and Disease. Int. J. Mol. Sci. 2019, 20, 2519. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, Y. Adiponectin: A key player in obesity related disorders. Curr. Pharm. Des. 2010, 16, 1896–1901. [Google Scholar] [CrossRef]
- Shehzad, A.; Iqbal, W.; Shehzad, O.; Lee, Y.S. Adiponectin: Regulation of its production and its role in human diseases. Hormones 2012, 11, 8–20. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Rajitha, B.; Aliya, S.; Kotipatruni, R.P.; Madanraj, A.S.; Hammond, A.; Park, D.; Chigurupati, S.; Alam, A.; Pattnaik, S. The role of adiponectin in obesity-associated female-specific carcinogenesis. Cytokine Growth Factor Rev. 2016, 31, 37–48. [Google Scholar] [CrossRef]
- Padmalayam, I.; Suto, M. Role of adiponectin in the metabolic syndrome: Current perspectives on its modulation as a treatment strategy. Curr. Pharm. Des. 2013, 19, 5755–5763. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Prins, J.; Venkatesh, B. Clinical review: Adiponectin biology and its role in inflammation and critical illness. Crit. Care 2011, 15, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, Y.; Wang, Y.; Chao, Y.; Zhang, J.; Jia, Y.; Tie, J.; Hu, D. Regulation of SIRT1 and Its Roles in Inflammation. Front. Immunol. 2022, 13, 831168. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Montejano, V.I.; Saxena, G.; Kusminski, C.M.; Yang, C.; McAfee, J.L.; Hahner, L.; Hoch, K.; Dubinsky, W.; Narkar, V.A.; Bickel, P.E. Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC-1alpha/SIRT1-dependent transcriptional regulation of mitochondrial function. Nat. Commun. 2016, 7, 12723. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Lei, P.; Teng, C.; Sun, Y.; Song, X.; Li, B.; Shan, Y. Targeting PLIN2/PLIN5-PPARgamma: Sulforaphane Disturbs the Maturation of Lipid Droplets. Mol. Nutr. Food Res. 2019, 63, e1900183. [Google Scholar] [CrossRef]
- Hoffmann, E.; Wald, J.; Lavu, S.; Roberts, J.; Beaumont, C.; Haddad, J.; Elliott, P.; Westphal, C.; Jacobson, E. Pharmacokinetics and tolerability of SRT2104, a first-in-class small molecule activator of SIRT1, after single and repeated oral administration in man. Br. J. Clin. Pharmacol. 2013, 75, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.H.; Sethi, G.; Um, J.Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, Y.; Homcy, C.J. The adenylyl cyclases as integrators of transmembrane signal transduction. Circ. Res. 1997, 80, 297–304. [Google Scholar] [CrossRef]
- Vasudevan, N.T.; Mohan, M.L.; Goswami, S.K.; Naga Prasad, S.V. Regulation of beta-adrenergic receptor function: An emphasis on receptor resensitization. Cell Cycle 2011, 10, 3684–3691. [Google Scholar] [CrossRef] [Green Version]
- Collins, S. beta-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored Fat and Energy Expenditure. Front. Endocrinol. 2011, 2, 102. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.I.; Atherton, P.; Reeds, D.N.; Mohammed, B.S.; Rankin, D.; Rennie, M.J.; Mittendorfer, B. Omega-3 polyunsaturated fatty acids augment the muscle protein anabolic response to hyperinsulinaemia-hyperaminoacidaemia in healthy young and middle-aged men and women. Clin. Sci. 2011, 121, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Marchant-Forde, J.N.; Lay, D.C., Jr.; Marchant-Forde, R.M.; McMunn, K.A.; Richert, B.T. The effects of R-salbutamol on growth, carcass measures, and health of finishing pigs. J. Anim. Sci. 2012, 90, 4081–4089. [Google Scholar] [CrossRef]
- Hostrup, M.; Jacobson, G.A.; Jessen, S.; Lemminger, A.K. Anabolic and lipolytic actions of beta(2) -agonists in humans and antidoping challenges. Drug Test. Anal. 2020, 12, 597–609. [Google Scholar] [CrossRef]
- Piribauer, M.; Jiang, L.; Kostov, T.; Parr, M.; Steidel, S.; Bizjak, D.A.; Steinacker, J.M.; Diel, P. Combinatory in vitro effects of the beta2-agonists salbutamol and formoterol in skeletal muscle cells. Toxicol. Lett. 2023, 378, 10–18. [Google Scholar] [CrossRef]
- Re, G.; Badino, P.; Girardi, C.; Di Carlo, F. Effects of a beta 2-agonist (clenbuterol) on cultured human (CG-5) breast cancer cells. Pharmacol. Res. 1992, 26, 377–384. [Google Scholar] [CrossRef]
- Montoya, A.; Varela-Ramirez, A.; Dickerson, E.; Pasquier, E.; Torabi, A.; Aguilera, R.; Nahleh, Z.; Bryan, B. The beta adrenergic receptor antagonist propranolol alters mitogenic and apoptotic signaling in late stage breast cancer. Biomed. J. 2019, 42, 155–165. [Google Scholar] [CrossRef]
- Phadke, S.; Clamon, G. Beta blockade as adjunctive breast cancer therapy: A review. Crit. Rev. Oncol. Hematol. 2019, 138, 173–177. [Google Scholar] [CrossRef]
- Chang, H.; Lee, S.H. Beta-adrenergic receptor blockers and hepatocellular carcinoma survival: A systemic review and meta-analysis. Clin. Exp. Med. 2023, 23, 853–858. [Google Scholar] [CrossRef]
- Kocak, M.Z.; Er, M.; Ugrakli, M.; Hendem, E.; Araz, M.; Eryilmaz, M.K.; Artac, M. Could the concomitant use of beta blockers with bevacizumab improve survival in metastatic colon cancer? Eur. J. Clin. Pharmacol. 2023, 79, 485–491. [Google Scholar] [CrossRef]
- Falcinelli, M.; Al-Hity, G.; Baron, S.; Mampay, M.; Allen, M.C.; Samuels, M.; Jones, W.; Cilibrasi, C.; Flaherty, R.L.; Giamas, G.; et al. Propranolol reduces IFN-gamma driven PD-L1 immunosuppression and improves anti-tumour immunity in ovarian cancer. Brain Behav. Immun. 2023, 110, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.M.; Kerswill, S.A.; Hari, P.; Cole, S.W.; Logan, B.R.; D’Souza, A.; Shah, N.N.; Horowitz, M.M.; Stolley, M.R.; Sloan, E.K.; et al. Repurposing existing medications as cancer therapy: Design and feasibility of a randomized pilot investigating propranolol administration in patients receiving hematopoietic cell transplantation. BMC Cancer 2018, 18, 593. [Google Scholar] [CrossRef] [PubMed]
- Shamim, M.A.; Shahid, A.; Sardar, P.K.; Yeung, S.; Reyes, J.; Kim, J.; Parsa, C.; Orlando, R.; Wang, J.; Kelly, K.M.; et al. Transfersome Encapsulated with the R-carvedilol Enantiomer for Skin Cancer Chemoprevention. Nanomaterials 2023, 13, 929. [Google Scholar] [CrossRef] [PubMed]
- Nimura, S.; Yamaguchi, T.; Ueda, K.; Kadokura, K.; Aiuchi, T.; Kato, R.; Obama, T.; Itabe, H. Olanzapine promotes the accumulation of lipid droplets and the expression of multiple perilipins in human adipocytes. Biochem. Biophys. Res. Commun. 2015, 467, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Cottingham, C.M.; Patrick, T.; Richards, M.A.; Blackburn, K.D. Tricyclic antipsychotics promote adipogenic gene expression to potentiate preadipocyte differentiation in vitro. Hum. Cell 2020, 33, 502–511. [Google Scholar] [CrossRef]
- Sato, T.; Akimoto, N.; Kitamura, K.; Kurihara, H.; Hayashi, N.; Ito, A. Adapalene suppresses sebum accumulation via the inhibition of triacylglycerol biosynthesis and perilipin expression in differentiated hamster sebocytes in vitro. J. Dermatol. Sci. 2013, 70, 204–210. [Google Scholar] [CrossRef]
- Jang, B.C. Artesunate inhibits adipogeneis in 3T3-L1 preadipocytes by reducing the expression and/or phosphorylation levels of C/EBP-alpha, PPAR-gamma, FAS, perilipin A, and STAT-3. Biochem. Biophys. Res. Commun. 2016, 474, 220–225. [Google Scholar] [CrossRef]
- Funk, M.I.; Conde, M.A.; Piwien-Pilipuk, G.; Uranga, R.M. Novel antiadipogenic effect of menadione in 3T3-L1 cells. Chem. Biol. Interact. 2021, 343, 109491. [Google Scholar] [CrossRef]
- Jang, B.C. Tetrandrine has anti-adipogenic effect on 3T3-L1 preadipocytes through the reduced expression and/or phosphorylation levels of C/EBP-alpha, PPAR-gamma, FAS, perilipin A, and STAT-3. Biochem. Biophys. Res. Commun. 2016, 476, 481–486. [Google Scholar] [CrossRef]
- Kovsan, J.; Ben-Romano, R.; Souza, S.C.; Greenberg, A.S.; Rudich, A. Regulation of adipocyte lipolysis by degradation of the perilipin protein: Nelfinavir enhances lysosome-mediated perilipin proteolysis. J. Biol. Chem. 2007, 282, 21704–21711. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Mihalcioiu, M.; Li, L.; Zakikhani, M.; Camirand, A.; Kremer, R. Vitamin D prevents lipid accumulation in murine muscle through regulation of PPARgamma and perilipin-2 expression. J. Steroid Biochem. Mol. Biol. 2018, 177, 116–124. [Google Scholar] [CrossRef]
- Hao, L.; Guo, Y.; Peng, Q.; Zhang, Z.; Ji, J.; Liu, Y.; Xue, Y.; Li, C.; Zheng, K.; Shi, X. Dihydroartemisinin reduced lipid droplet deposition by YAP1 to promote the anti-PD-1 effect in hepatocellular carcinoma. Phytomedicine 2022, 96, 153913. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bombarda-Rocha, V.; Silva, D.; Badr-Eddine, A.; Nogueira, P.; Gonçalves, J.; Fresco, P. Challenges in Pharmacological Intervention in Perilipins (PLINs) to Modulate Lipid Droplet Dynamics in Obesity and Cancer. Cancers 2023, 15, 4013. https://doi.org/10.3390/cancers15154013
Bombarda-Rocha V, Silva D, Badr-Eddine A, Nogueira P, Gonçalves J, Fresco P. Challenges in Pharmacological Intervention in Perilipins (PLINs) to Modulate Lipid Droplet Dynamics in Obesity and Cancer. Cancers. 2023; 15(15):4013. https://doi.org/10.3390/cancers15154013
Chicago/Turabian StyleBombarda-Rocha, Victória, Dany Silva, Allal Badr-Eddine, Patrícia Nogueira, Jorge Gonçalves, and Paula Fresco. 2023. "Challenges in Pharmacological Intervention in Perilipins (PLINs) to Modulate Lipid Droplet Dynamics in Obesity and Cancer" Cancers 15, no. 15: 4013. https://doi.org/10.3390/cancers15154013
APA StyleBombarda-Rocha, V., Silva, D., Badr-Eddine, A., Nogueira, P., Gonçalves, J., & Fresco, P. (2023). Challenges in Pharmacological Intervention in Perilipins (PLINs) to Modulate Lipid Droplet Dynamics in Obesity and Cancer. Cancers, 15(15), 4013. https://doi.org/10.3390/cancers15154013