
Dedifferentiated Chondrosarcoma from Molecular Pathology to Current Treatment and Clinical Trials

 , , ,
, , ,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Epidemiology
Dedifferentiated Chondrosarcoma in Ollier Disease and Mafucci Syndrome
3. Diagnostics
3.1. Location and Metastasis
3.2. Size
3.3. Symptoms
3.4. Methods of Diagnosis
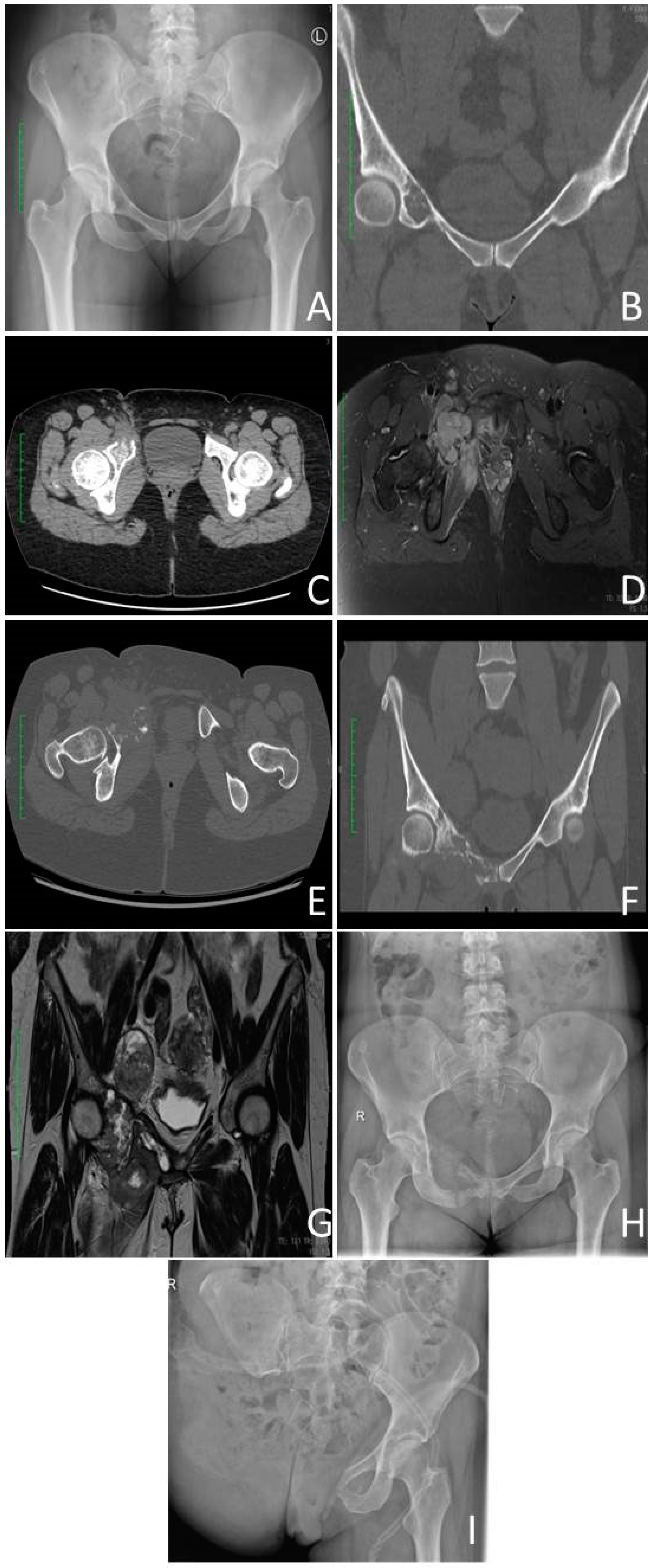
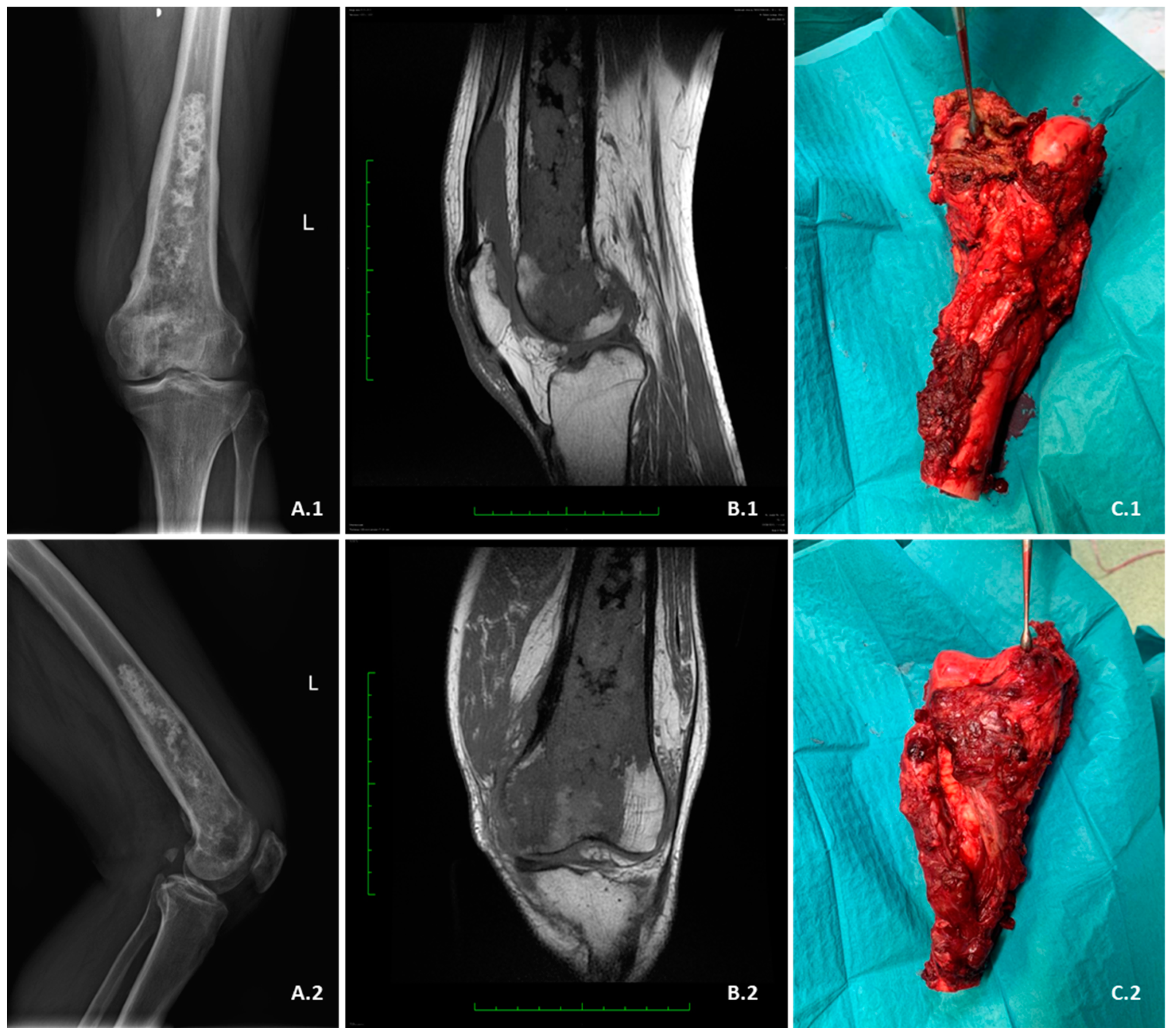
3.4.1. Radiological Criterium

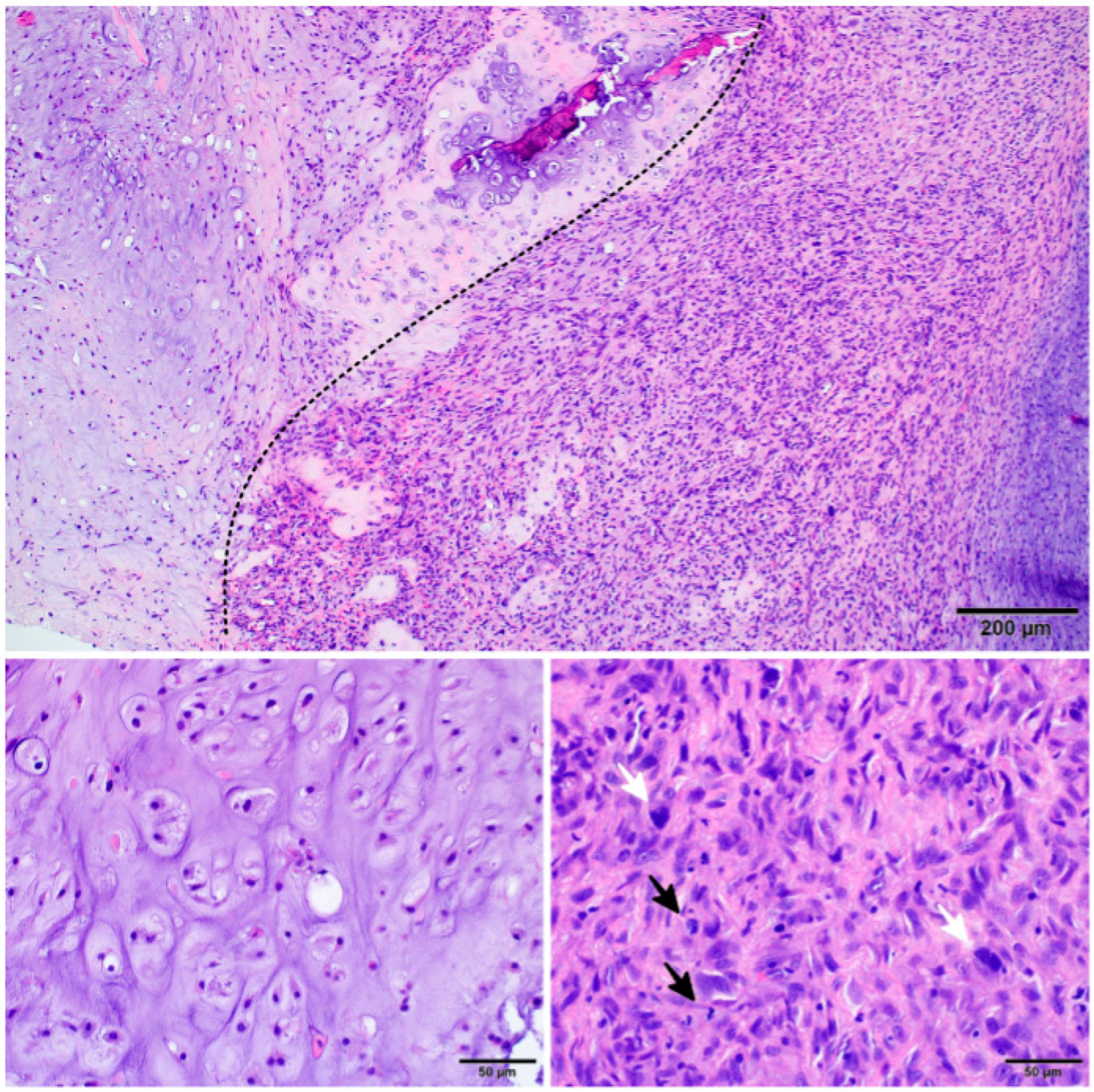
3.4.2. Histopathology
3.4.3. Image-Guided Percutaneous Core–Needle Biopsy
3.5. Staging
4. Pathology
4.1. Low–Grade Component
4.2. High-Grade Component
4.3. Cellular Infiltrates
4.4. Immunohistochemistry (IHC)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein/Subtype | DDCS | CCCS | CCS | MCS |
|---|---|---|---|---|
| S100 | - | + | + | + |
| P53 | + * | + | + ** | + |
| SOX-9 | + | + | + | + |
| Bcl-2 | + | + | + | + |
| IDH1 | + | - | + | - |
| NY-ESO-1 | + | - | + | - |
| Other | CD44, Col1a1, Col2a1, cyclin D1, MDM2, Ki-67, PAI-1, PD-L1, PTHrP, Runx2 | Col2a1, keratine, PTHrP, PDGF, Runx2 | Brachyury, Col2a1, Cox-2, D2-40, Gal-1, MDM2, PTHrP, YKL-40 | CD99, desmin, EMA, MYF4, MYOD1, NKX2.2 |
| References | [6,60,61,63,64,65,67,68,69,70,71] | [54,70,89,93] | [65,70,78,83,94,95,96] | [70,78,89,97,98,99,100] |

4.5. Differential Diagnosis
| References | Type of Sarcoma | S100 | P53 | MDM2 | Ki-67 | Desmin | SMA | EMA | Vimentin | Myosin | h-Caldesmon | Others |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [7,85,86,88,89,90,103] | DDCS | - | + | + | + | - | - | - | ± | ± | ± | CD99 |
| [104,105,106,107,108,109] | OSC | + | + | + | + | ± | ± | - | + | - | - | CD10, CD99, PAX2 |
| [104,110,111,112,113,114] | FS | + | - | + | + | + | + | + | + | + | + | Cd34, CD99 |
| [115,116,117,118] | MFH | - | ± | ± | ± | + | + | ± | + | ± | - | Cd45, CD68 |
| [104,110,119,120,121,122] | RMS | - | - | ± | + | + | - | + | + | + | ± | Myogenin, CDK4, CD56, CD99 |
| [104,110,115,119,123,124] | LMS | - | - | - | ± | + | + | ± | ± | + | + | Calponin, CDK4, CD34 |
5. Genetics
5.1. IDH Mutations
5.2. TP53 Mutations
5.3. Other Mutations
6. Treatment
6.1. Surgical Treatment

6.2. Treatment of Localised Disease
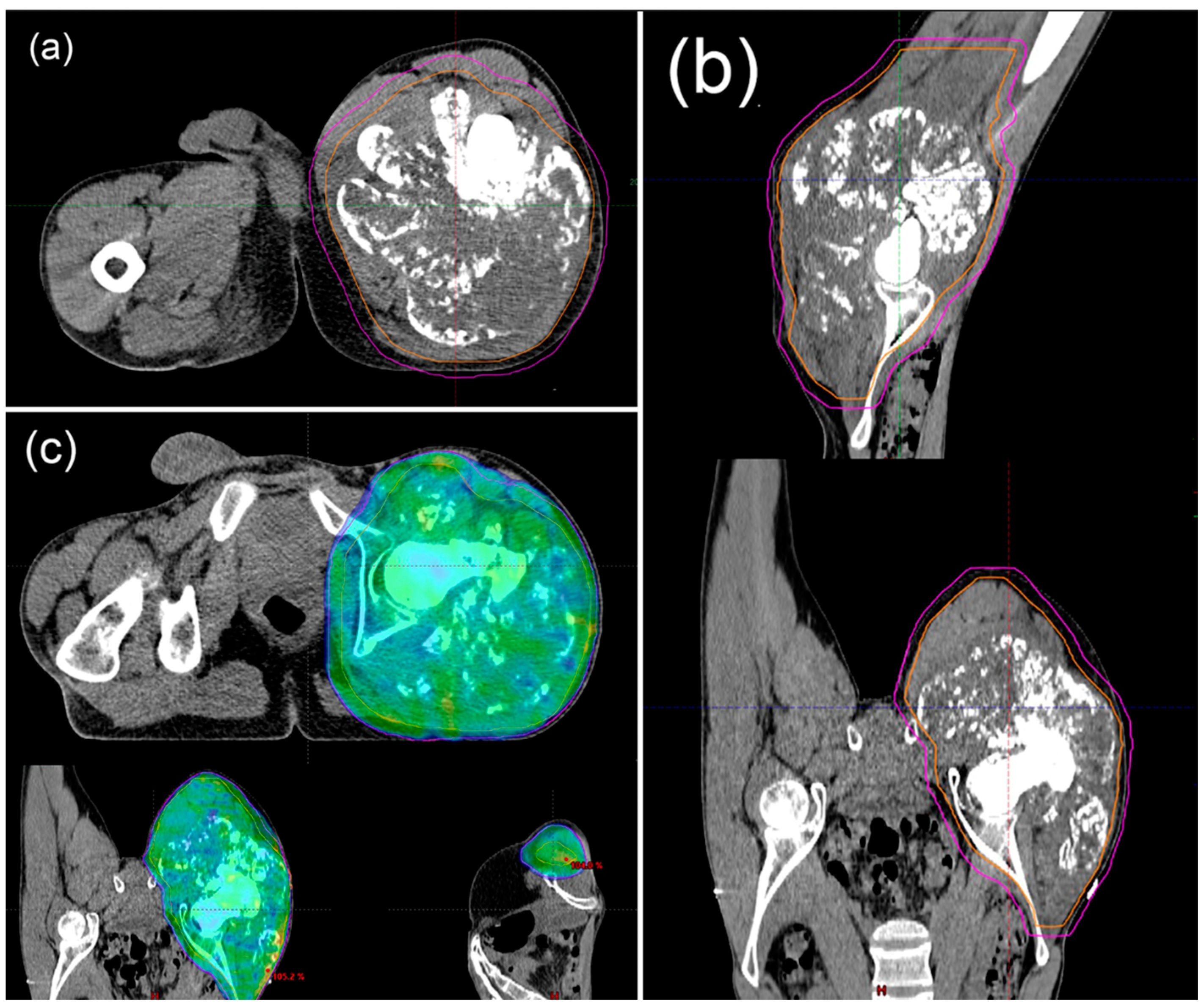
6.3. Ratiotherapy


6.4. Treatment of Metastatic Disease
6.5. Palliative Treatment
6.5.1. Chemotherapy
6.5.2. Immunotherapy
6.5.3. Targeted treatment
7. Clinical Trials
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reith, J.D.; Bauer, T.W.; Fischler, D.F.; Joyce, M.J.; Marks, K.E. Dedifferentiated chondrosarcoma with rhabdomyosarcomatous differentiation. Am. J. Surg. Pathol. 1996, 20, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Wittig, J.C. Dedifferentiated Chondrosarcoma. Available online: https://tumorsurgery.org/tumor-education/bone-tumors/types-of-bone-tumors/dedifferentiated-chondrosarcoma.aspx?fbclid=IwAR08vgOA3CET_NS6UJaczgQK98xoKICoQKeFhBWD6VlXo6XKZgPGdxj_2XI (accessed on 20 February 2023).
- Gelderblom, H.; Hogendoorn, P.C.; Dijkstra, S.D.; van Rijswijk, C.S.; Krol, A.D.; Taminiau, A.H.; Bovée, J.V. The clinical approach towards chondrosarcoma. Oncologist 2008, 13, 320–329. [Google Scholar] [CrossRef]
- Mercuri, M.; Picci, P.; Campanacci, L.; Rulli, E. Dedifferentiated chondrosarcoma. Skelet. Radiol. 1995, 24, 409–416. [Google Scholar] [CrossRef]
- Estrada, E.G.; Ayala, A.G.; Lewis, V.; Czerniak, B. Dedifferentiated chondrosarcoma with a noncartilaginous component mimicking a conventional giant cell tumor of bone. Ann. Diagn. Pathol. 2002, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Rozeman, L.B.; de Bruijn, I.H.B.; Bacchini, P.; Staals, E.L.; Bertoni, F.; Bovée, J.V.M.G.; Hogendoorn, P.C.W. Dedifferentiated peripheral chondrosarcomas: Regulation of EXT-downstream molecules and differentiation-related genes. Mod. Pathol. 2009, 22, 1489–1498. [Google Scholar] [CrossRef]
- Gong, L.H.; Su, Y.B.; Zhang, W.; Liu, W.F.; Dong, R.F.; Sun, X.Q.; Zhang, M.; Ding, Y. Dedifferentiated Central Chondrosarcoma: A Clinical, Histopathological, and Immunohistochemical Analysis of 57 Cases. Front. Med. 2021, 8, 746909. [Google Scholar] [CrossRef]
- Amer, K.M.; Munn, M.; Congiusta, D.; Abraham, J.A.; Basu Mallick, A. Survival and Prognosis of Chondrosarcoma Subtypes: SEER Database Analysis. J. Orthop. Res. 2020, 38, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Gusho, C.A.; Lee, L.; Zavras, A.; Seikel, Z.; Miller, I.; Colman, M.W.; Gitelis, S.; Blank, A.T. Dedifferentiated Chondrosarcoma: A Case Series and Review of the Literature. Orthop. Rev. 2022, 14, 35448. [Google Scholar] [CrossRef]
- Grimer, R.J.; Gosheger, G.; Taminiau, A.; Biau, D.; Matejovsky, Z.; Kollender, Y.; San-Julian, M.; Gherlinzoni, F.; Ferrari, C. Dedifferentiated chondrosarcoma: Prognostic factors and outcome from a European group. Eur. J. Cancer 2007, 43, 2060–2065. [Google Scholar] [CrossRef]
- van Praag Veroniek, V.M.; Rueten-Budde, A.J.; Ho, V.; Dijkstra, P.D.S.; Fiocco, M.; van de Sande, M.A.J. Incidence, outcomes and prognostic factors during 25 years of treatment of chondrosarcomas. Surg. Oncol. 2018, 27, 402–408. [Google Scholar] [CrossRef]
- Hua, K.C.; Hu, Y.C. Treatment method and prognostic factors of chondrosarcoma: Based on Surveillance, Epidemiology, and End Results (SEER) database. Transl. Cancer Res. 2020, 9, 4250–4266. [Google Scholar] [CrossRef]
- Gaillard, F.J.; Kusel, K. Dedifferentiated Chondrosarcoma. Available online: https://radiopaedia.org/articles/6250 (accessed on 20 February 2023).
- Alqubaisi, A.; Oliveira, I.; Singla, N.; Chavda, A.; Khoo, M.; Saifuddin, A. The incidence and diagnostic relevance of pathological fracture in conventional central chondrosarcoma. Skelet. Radiol. 2021, 50, 1131–1140. [Google Scholar] [CrossRef]
- Saifuddin, A.; Mann, B.S.; Mahroof, S.; Pringle, J.A.; Briggs, T.W.; Cannon, S.R. Dedifferentiated chondrosarcoma: Use of MRI to guide needle biopsy. Clin. Radiol. 2004, 59, 268–272. [Google Scholar] [CrossRef]
- Miao, R.; Choy, E.; Raskin, K.A.; Schwab, J.H.; Nielsen, G.P.; Deshpande, V.; Chebib, I.; DeLaney, T.F.; Hornicek, F.J.; Cote, G.M.; et al. Prognostic Factors in Dedifferentiated Chondrosarcoma: A Retrospective Analysis of a Large Series Treated at a Single Institution. Sarcoma 2019, 2019, 9069272. [Google Scholar] [CrossRef] [PubMed]
- Strotman, P.K.; Reif, T.J.; Kliethermes, S.A.; Sandhu, J.K.; Nystrom, L.M. Dedifferentiated chondrosarcoma: A survival analysis of 159 cases from the SEER database (2001–2011). J. Surg. Oncol. 2017, 116, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xi, Y.; Li, M.; Jiao, Q.; Zhang, H.; Yang, Q.; Yao, W. Dedifferentiated chondrosarcoma: Radiological features, prognostic factors and survival statistics in 23 patients. PLoS ONE 2017, 12, e0173665. [Google Scholar] [CrossRef]
- Yokota, K.; Sakamoto, A.; Matsumoto, Y.; Matsuda, S.; Harimaya, K.; Oda, Y.; Iwamoto, Y. Clinical outcome for patients with dedifferentiated chondrosarcoma: A report of 9 cases at a single institute. J. Orthop. Surg. Res. 2012, 7, 38. [Google Scholar] [CrossRef]
- Staals, E.L.; Bacchini, P.; Bertoni, F. Dedifferentiated central chondrosarcoma. Cancer 2006, 106, 2682–2691. [Google Scholar] [CrossRef]
- Johnson, S.; Têtu, B.; Ayala, A.G.; Chawla, S.P. Chondrosarcoma with additional mesenchymal component (dedifferentiated chondrosarcoma). I. A clinicopathologic study of 26 cases. Cancer 1986, 58, 278–286. [Google Scholar] [CrossRef]
- El Abiad, J.M.; Robbins, S.M.; Cohen, B.; Levin, A.S.; Valle, D.L.; Morris, C.D.; de Macena Sobreira, N.L. Natural history of Ollier disease and Maffucci syndrome: Patient survey and review of clinical literature. Am. J. Med. Genet. A 2020, 182, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, H.S.; Zimmerman, N.B.; Simon, M.A.; Wroble, R.R.; Millar, E.A.; Bonfiglio, M. The malignant potential of enchondromatosis. J. Bone Jt. Surg. Am. 1987, 69, 269–274. [Google Scholar] [CrossRef]
- Aycan, O.E.; Sebastiani, E.; Bianchi, G.; Gambarotti, M. Coexistence of secondary chondrosarcoma and lung carcinoma metastasis in the humerus of a patient with Ollier’s disease: A case report. Acta Orthop. Traumatol. Turc. 2019, 53, 68–73. [Google Scholar] [CrossRef]
- Gonzalez, M.R.; Bryce-Alberti, M.; Portmann-Baracco, A.; Inchaustegui, M.L.; Castillo-Flores, S.; Pretell-Mazzini, J. Appendicular dedifferentiated chondrosarcoma: A management and survival study from the SEER database. J. Bone Oncol. 2022, 37, 100456. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.; Liu, S.; Baskovich, B.; Makary, R. A Case Report and Brief Literature Review on Dedifferentiated Chondrosarcoma in Proximal Phalanx: A Rare Location. Cureus 2022, 14, e29105. [Google Scholar] [CrossRef]
- Graham, T.M. IHeartPathology Dedifferentiated Chondrosarcoma. Available online: https://www.iheartpathology.net/ (accessed on 11 April 2019).
- Rozeman, L.B.; Hogendoorn, P.C.; Bovée, J.V. Diagnosis and prognosis of chondrosarcoma of bone. Expert. Rev. Mol. Diagn. 2002, 2, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Malchenko, S.; Seftor, E.A.; Nikolsky, Y.; Hasegawa, S.L.; Kuo, S.; Stevens, J.W.; Poyarkov, S.; Nikolskaya, T.; Kucaba, T.; Wang, M.; et al. Putative multifunctional signature of lung metastases in dedifferentiated chondrosarcoma. Sarcoma 2012, 2012, 820254. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Jiang, Y.Q.; Li, X.L.; Dong, J. Risk Factors for Incidence and Prognosis in Chondrosarcoma Patients with Pulmonary Metastasis at Initial Diagnosis. Med. Sci. Monit. 2019, 25, 10136–10153. [Google Scholar] [CrossRef]
- Hung, Y.P.; Chebib, I.; Bredella, M.A.; Berner, E.A.; Taylor-Black, Q.; Choy, E.; Cote, G.M.; Chen, Y.L.; MacDonald, S.M.; Schwab, J.H.; et al. Prognostic Significance of Percentage and Size of Dedifferentiation in Dedifferentiated Chondrosarcoma. Mod. Pathol. 2023, 36, 100069. [Google Scholar] [CrossRef]
- Badyal, R.K.; Kataria, A.S.; Kaur, M. Primary chondrosarcoma of male breast: A rare case. Indian J. Surg. 2012, 74, 418–419. [Google Scholar] [CrossRef][Green Version]
- Albergo, J.I.; Gaston, C.L.; Jeys, L.M.; Khajuria, A.; Carter, S.R.; Tillman, R.M.; Abudu, A.T.; Grimer, R.J. Management and prognostic significance of pathological fractures through chondrosarcoma of the femur. Int. Orthop. 2015, 39, 943–946. [Google Scholar] [CrossRef]
- Bharath, G.; Burrah, R.; Shivakumar, K.; Manjunath, S.; Bhanumathi, R. Dedifferentiated chondrosarcoma: An aggressive variant of chondrosarcoma. Asian Cardiovasc. Thorac. Ann. 2015, 23, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Douis, H.; Saifuddin, A. The imaging of cartilaginous bone tumours. II. Chondrosarcoma. Skelet. Radiol. 2012, 42, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Littrell, L.A.; Wenger, D.E.; Wold, L.E.; Bertoni, F.; Unni, K.K.; White, L.M.; Kandel, R.; Sundaram, M. Radiographic, CT, and MR Imaging Features of Dedifferentiated Chondrosarcomas: A Retrospective Review of 174 De Novo Cases. RadioGraphics 2004, 24, 1397–1409. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lee, S.K. Classification of Chondrosarcoma: From Characteristic to Challenging Imaging Findings. Cancers 2023, 15, 1703. [Google Scholar] [CrossRef]
- MacSweeney, F.; Darby, A.; Saifuddin, A. Dedifferentiated chondrosarcoma of the appendicular skeleton: MRI-pathological correlation. Skelet. Radiol. 2003, 32, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Tlemsani, C.; Larousserie, F.; De Percin, S.; Audard, V.; Hadjadj, D.; Chen, J.; Biau, D.; Anract, P.; Terris, B.; Goldwasser, F.; et al. Biology and Management of High-Grade Chondrosarcoma: An Update on Targets and Treatment Options. Int. J. Mol. Sci. 2023, 24, 1361. [Google Scholar] [CrossRef]
- Gazendam, A.; Popovic, S.; Parasu, N.; Ghert, M. Chondrosarcoma: A Clinical Review. J. Clin. Med. 2023, 12, 2506. [Google Scholar] [CrossRef]
- Saifuddin, A.; Mitchell, R.; Burnett, S.J.; Sandison, A.; Pringle, J.A. Ultrasound-guided needle biopsy of primary bone tumours. J. Bone Jt. Surg. Br. 2000, 82, 50–54. [Google Scholar] [CrossRef]
- Leddy, L.R.; Holmes, R.E. Chondrosarcoma of bone. Cancer Treat. Res. 2014, 162, 117–130. [Google Scholar] [CrossRef]
- Andreas F Mavrogenis, P.J.P. Bone: Dedifferentiated Chondrosarcoma. Atlas of Genetics and Cytogenetics in Oncology and Haematology. Available online: https://atlasgeneticsoncology.org/solid-tumor/5063/bone-chondrosarcoma (accessed on 3 January 2012).
- Jelinek, J.S.; Murphey, M.D.; Welker, J.A.; Henshaw, R.M.; Kransdorf, M.J.; Shmookler, B.M.; Malawer, M.M. Diagnosis of primary bone tumors with image-guided percutaneous biopsy: Experience with 110 tumors. Radiology 2002, 223, 731–737. [Google Scholar] [CrossRef]
- Altuntas, A.O.; Slavin, J.; Smith, P.J.; Schlict, S.M.; Powell, G.J.; Ngan, S.; Toner, G.; Choong, P.F. Accuracy of computed tomography guided core needle biopsy of musculoskeletal tumours. ANZ J. Surg. 2005, 75, 187–191. [Google Scholar] [CrossRef]
- Omura, M.C.; Motamedi, K.; UyBico, S.; Nelson, S.D.; Seeger, L.L. Revisiting CT-guided percutaneous core needle biopsy of musculoskeletal lesions: Contributors to biopsy success. AJR Am. J. Roentgenol. 2011, 197, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Toki, S.; Sone, M.; Yoshida, A.; Nishisho, T.; Gokita, T.; Kobayashi, E.; Nakatani, F.; Chuman, H.; Sugawara, S.; Arai, Y.; et al. Image-guided core needle biopsy for musculoskeletal lesions. J. Orthop. Sci. 2022, 27, 448–455. [Google Scholar] [CrossRef]
- Dupuy, D.E.; Rosenberg, A.E.; Punyaratabandhu, T.; Tan, M.H.; Mankin, H.J. Accuracy of CT-guided needle biopsy of musculoskeletal neoplasms. AJR Am. J. Roentgenol. 1998, 171, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ozaki, T. New TNM classification (AJCC eighth edition) of bone and soft tissue sarcomas: JCOG Bone and Soft Tissue Tumor Study Group. Jpn. J. Clin. Oncol. 2019, 49, 103–107. [Google Scholar] [CrossRef]
- Sakamoto, A. The molecular pathogenesis of dedifferentiated chondrosarcoma. Indian. J. Orthop. 2014, 48, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Ropke, M.; Boltze, C.; Neumann, H.W.; Roessner, A.; Schneider-Stock, R. Genetic and epigenetic alterations in tumor progression in a dedifferentiated chondrosarcoma. Pathol. Res. Pr. 2003, 199, 437–444. [Google Scholar] [CrossRef]
- Dornauer, K.; Soder, S.; Inwards, C.Y.; Bovee, J.V.; Aigner, T. Matrix biochemistry and cell biology of dedifferentiated chondrosarcomas. Pathol. Int. 2010, 60, 365–372. [Google Scholar] [CrossRef]
- Meister, P.; Konrad, E.A.; Nathrath, W.; Eder, M. Malignant fibrous histiocytoma: Histological patterns and cell types. Pathol. Res. Pr. 1980, 168, 193–212. [Google Scholar] [CrossRef]
- Meijer, D.; de Jong, D.; Pansuriya, T.C.; van den Akker, B.E.; Picci, P.; Szuhai, K.; Bovee, J.V. Genetic characterization of mesenchymal, clear cell, and dedifferentiated chondrosarcoma. Genes. Chromosom. Cancer 2012, 51, 899–909. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Q.; Zhang, S.; Wang, X.; Li, W.; Wen, J.; Huang, X.; Zheng, J.; Huang, G.; Huang, T.; et al. A novel mutated cell line with characteristics of dedifferentiated chondrosarcoma. Int. J. Mol. Med. 2009, 24, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Kozawa, E.; Nishida, Y.; Kawai, A.; Hayakawa, K.; Setsu, N.; Kawashima, H.; Iwata, S.; Tsuchiya, H.; Tsukushi, S.; Takenaka, S.; et al. Clinical features and treatment outcomes of dedifferentiated and grade 3 chondrosarcoma: A multi-institutional study. Cancer Sci. 2022, 113, 2397–2408. [Google Scholar] [CrossRef] [PubMed]
- Simard, F.A.; Richert, I.; Vandermoeten, A.; Decouvelaere, A.V.; Michot, J.P.; Caux, C.; Blay, J.Y.; Dutour, A. Description of the immune microenvironment of chondrosarcoma and contribution to progression. Oncoimmunology 2016, 6, e1265716. [Google Scholar] [CrossRef]
- Singh, A.; Thorpe, S.W.; Darrow, M.; Carr-Ascher, J.R. Case report: Treatment of metastatic dedifferentiated chondrosarcoma with pembrolizumab yields sustained complete response. Front. Oncol. 2022, 12, 991724. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Kim, J.R.; Moon, Y.J.; Kwon, K.S.; Bae, J.S.; Wagle, S.; Kim, K.M.; Park, H.S.; Lee, H.; Moon, W.S.; Chung, M.J.; et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS ONE 2013, 8, e82870. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Shoushtari, A.N.; Agaram, N.P.; Kuk, D.; Qin, L.X.; Carvajal, R.D.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Schwartz, G.K.; et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum. Pathol. 2015, 46, 357–365. [Google Scholar] [CrossRef]
- Kostine, M.; Cleven, A.H.; de Miranda, N.F.; Italiano, A.; Cleton-Jansen, A.M.; Bovee, J.V. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod. Pathol. 2016, 29, 1028–1037. [Google Scholar] [CrossRef]
- Karamchandani, J.R.; Nielsen, T.O.; van de Rijn, M.; West, R.B. Sox10 and S100 in the diagnosis of soft-tissue neoplasms. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Chebib, I.; Hornicek, F.J.; Bredella, M.A.; Deshpande, V.; Nielsen, G.P. Histologic variants of chondrosarcoma. Diagn. Histopath. 2014, 20, 172–180. [Google Scholar] [CrossRef]
- Thoenen, E.; Curl, A.; Iwakuma, T. TP53 in bone and soft tissue sarcomas. Pharmacology 2019, 202, 149–164. [Google Scholar] [CrossRef]
- Terek, R.M.; Healey, J.H.; Garin-Chesa, P.; Mak, S.; Huvos, A.; Albino, A.P. p53 mutations in chondrosarcoma. Diagn. Mol. Pathol. 1998, 7, 51–56. [Google Scholar] [CrossRef]
- Endo, M.; de Graaff, M.A.; Ingram, D.R.; Lim, S.; Lev, D.C.; Briaire-de Bruijn, I.H.; Somaiah, N.; Bovee, J.V.; Lazar, A.J.; Nielsen, T.O. NY-ESO-1 (CTAG1B) expression in mesenchymal tumors. Mod. Pathol. 2015, 28, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.P.; Robbins, P.F.; Raffeld, M.; Aung, P.P.; Tsokos, M.; Rosenberg, S.A.; Miettinen, M.M.; Lee, C.C. NY-ESO-1 expression in synovial sarcoma and other mesenchymal tumors: Significance for NY-ESO-1-based targeted therapy and differential diagnosis. Mod. Pathol. 2012, 25, 854–858. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Tallegas, M.; Miquelestorena-Standley, E.; Labit-Bouvier, C.; Badoual, C.; Francois, A.; Gomez-Brouchet, A.; Aubert, S.; Collin, C.; Tallet, A.; de Pinieux, G. IDH mutation status in a series of 88 head and neck chondrosarcomas: Different profile between tumors of the skull base and tumors involving the facial skeleton and the laryngotracheal tract. Hum. Pathol. 2019, 84, 183–191. [Google Scholar] [CrossRef]
- Tap, W.D.; Villalobos, V.M.; Cote, G.M.; Burris, H.; Janku, F.; Mir, O.; Beeram, M.; Wagner, A.J.; Jiang, L.; Wu, B.; et al. Phase I Study of the Mutant IDH1 Inhibitor Ivosidenib: Safety and Clinical Activity in Patients with Advanced Chondrosarcoma. J. Clin. Oncol. 2020, 38, 1693–1701. [Google Scholar] [CrossRef]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fritchie, K.; Wei, S.; Ali, N.; Curless, K.; Shen, T.; Brini, A.T.; Latif, F.; Sumathi, V.; Siegal, G.P.; et al. Diagnostic utility of IDH1/2 mutations to distinguish dedifferentiated chondrosarcoma from undifferentiated pleomorphic sarcoma of bone. Hum. Pathol. 2017, 65, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Bai, Y.; Chen, J.; Sun, K.; Luo, Y.; Huang, W.; Zhang, H. Clonality analysis and IDH1 and IDH2 mutation detection in both components of dedifferentiated chondrosarcoma, implicated its monoclonal origin. J. Bone Oncol. 2020, 22, 100293. [Google Scholar] [CrossRef]
- Dermawan, J.K.T.; Nafa, K.; Mohanty, A.; Xu, Y.; Rijo, I.; Casanova, J.; Villafania, L.; Benhamida, J.; Kelly, C.M.; Tap, W.D.; et al. Distinct IDH1/2-associated Methylation Profile and Enrichment of TP53 and TERT Mutations Distinguish Dedifferentiated Chondrosarcoma from Conventional Chondrosarcoma. Cancer Res. Commun. 2023, 3, 431–443. [Google Scholar] [CrossRef]
- Mak, I.W.; Singh, S.; Turcotte, R.; Ghert, M. The epigenetic regulation of SOX9 by miR-145 in human chondrosarcoma. J. Cell. Biochem. 2015, 116, 37–44. [Google Scholar] [CrossRef]
- Tang, X.; Lu, X.; Guo, W.; Ren, T.; Zhao, H.; Zhao, F.; Tang, G. Different expression of Sox9 and Runx2 between chondrosarcoma and dedifferentiated chondrosarcoma cell line. Eur. J. Cancer Prev. 2010, 19, 466–471. [Google Scholar] [CrossRef]
- Grote, H.J.; Schneider-Stock, R.; Neumann, W.; Roessner, A. Mutation of p53 with loss of heterozygosity in the osteosarcomatous component of a dedifferentiated chondrosarcoma. Virchows Arch. 2000, 436, 494–497. [Google Scholar] [CrossRef]
- Simms, W.W.; Ordonez, N.G.; Johnston, D.; Ayala, A.G.; Czerniak, B. p53 expression in dedifferentiated chondrosarcoma. Cancer 1995, 76, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Knosel, T.; Werner, M.; Jung, A.; Kirchner, T.; Durr, H.R. Dedifferentiated chondrosarcoma mimicking a giant cell tumor. Is this low grade dedifferentiated chondrosarcoma? Pathol. Res. Pr. 2014, 210, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Bovee, J.V.; Cleton-Jansen, A.M.; Rosenberg, C.; Taminiau, A.H.; Cornelisse, C.J.; Hogendoorn, P.C. Molecular genetic characterization of both components of a dedifferentiated chondrosarcoma, with implications for its histogenesis. J. Pathol. 1999, 189, 454–462. [Google Scholar] [CrossRef]
- Gilbert, A. Chondrosarcoma Resistance to Radiation Therapy: Origins and Potential Therapeutic Solutions. Cancers 2023, 15, 1962. [Google Scholar] [CrossRef] [PubMed]
- van Oosterwijk, J.G.; Meijer, D.; van Ruler, M.A.; van den Akker, B.E.; Oosting, J.; Krenacs, T.; Picci, P.; Flanagan, A.M.; Liegl-Atzwanger, B.; Leithner, A.; et al. Screening for potential targets for therapy in mesenchymal, clear cell, and dedifferentiated chondrosarcoma reveals Bcl-2 family members and TGFbeta as potential targets. Am. J. Pathol. 2013, 182, 1347–1356. [Google Scholar] [CrossRef]
- Franchi, A.; Baroni, G.; Sardi, I.; Giunti, L.; Capanna, R.; Campanacci, D. Dedifferentiated peripheral chondrosarcoma: A clinicopathologic, immunohistochemical, and molecular analysis of four cases. Virchows Arch. 2012, 460, 335–342. [Google Scholar] [CrossRef]
- Makise, N.; Sekimizu, M.; Konishi, E.; Motoi, T.; Kubo, T.; Ikoma, H.; Watanabe, S.I.; Okuma, T.; Hiraoka, N.; Fukayama, M.; et al. H3K27me3 deficiency defines a subset of dedifferentiated chondrosarcomas with characteristic clinicopathological features. Mod. Pathol. 2019, 32, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Zajac, A.E.; Kopec, S.; Szostakowski, B.; Spalek, M.J.; Fiedorowicz, M.; Bylina, E.; Filipowicz, P.; Szumera-Cieckiewicz, A.; Tysarowski, A.; Czarnecka, A.M.; et al. Chondrosarcoma-from Molecular Pathology to Novel Therapies. Cancers 2021, 13, 2390. [Google Scholar] [CrossRef]
- Aigner, T.; Dertinger, S.; Belke, J.; Kirchner, T. Chondrocytic cell differentiation in clear cell chondrosarcoma. Hum. Pathol. 1996, 27, 1301–1305. [Google Scholar] [CrossRef]
- Oakley, G.J.; Fuhrer, K.; Seethala, R.R. Brachyury, SOX-9, and podoplanin, new markers in the skull base chordoma vs chondrosarcoma differential: A tissue microarray-based comparative analysis. Mod. Pathol. 2008, 21, 1461–1469. [Google Scholar] [CrossRef]
- Jeong, W.; Kim, H.J. Biomarkers of chondrosarcoma. J. Clin. Pathol. 2018, 71, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, S.; Christensen, L.H.; Hogdall, E. Markers aiding the diagnosis of chondroid tumors: An immunohistochemical study including osteonectin, bcl-2, cox-2, actin, calponin, D2-40 (podoplanin), mdm-2, CD117 (c-kit), and YKL-40. APMIS 2009, 117, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Syed, M.; Mushtaq, S.; Loya, A.; Hassan, U. NKX3.1 a useful marker for mesenchymal chondrosarcoma: An immunohistochemical study. Ann. Diagn. Pathol. 2021, 50, 151660. [Google Scholar] [CrossRef]
- Kim, M.J.; Cho, K.J.; Ayala, A.G.; Ro, J.Y. Chondrosarcoma: With updates on molecular genetics. Sarcoma 2011, 2011, 405437. [Google Scholar] [CrossRef]
- Folpe, A.L.; Graham, R.P.; Martinez, A.; Schembri-Wismayer, D.; Boland, J.; Fritchie, K.J. Mesenchymal chondrosarcomas showing immunohistochemical evidence of rhabdomyoblastic differentiation: A potential diagnostic pitfall. Hum. Pathol. 2018, 77, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Fanburg-Smith, J.C.; Auerbach, A.; Marwaha, J.S.; Wang, Z.; Santi, M.; Judkins, A.R.; Rushing, E.J. Immunoprofile of mesenchymal chondrosarcoma: Aberrant desmin and EMA expression, retention of INI1, and negative estrogen receptor in 22 female-predominant central nervous system and musculoskeletal cases. Ann. Diagn. Pathol. 2010, 14, 8–14. [Google Scholar] [CrossRef]
- Estrada-Villaseñor, E.; Rico-Martínez, G.; Linares-Gonzalez, L.M. Diagnosis of a dedifferentiated chondrosarcoma of the pelvis by fine needle aspiration. A case report. Acta Cytol. 2010, 54, 217–220. [Google Scholar] [CrossRef]
- Sobti, A.; Agrawal, P.; Agarwala, S.; Agarwal, M. Giant Cell Tumor of Bone—An Overview. Arch. Bone Jt. Surg. 2016, 4, 2–9. [Google Scholar]
- Wick, M.R.; Siegal, G.P.; Mills, S.E.; Thompson, R.C.; Sawhney, D.; Fechner, R.E. Dedifferentiated chondrosarcoma of bone. An immunohistochemical and lectin-histochemical study. Virchows Arch. A Pathol. Anat. Histopathol. 1987, 411, 23–32. [Google Scholar] [CrossRef]
- Bahrami, A.; Truong, L.D.; Ro, J.Y. Undifferentiated tumor: True identity by immunohistochemistry. Arch. Pathol. Lab. Med. 2008, 132, 326–348. [Google Scholar] [CrossRef]
- Yoshida, A.; Ushiku, T.; Motoi, T.; Beppu, Y.; Fukayama, M.; Tsuda, H.; Shibata, T. MDM2 and CDK4 immunohistochemical coexpression in high-grade osteosarcoma: Correlation with a dedifferentiated subtype. Am. J. Surg. Pathol. 2012, 36, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Junior, A.T.; de Abreu Alves, F.; Pinto, C.A.; Carvalho, A.L.; Kowalski, L.P.; Lopes, M.A. Clinicopathological and immunohistochemical analysis of twenty-five head and neck osteosarcomas. Oral. Oncol. 2003, 39, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Al-Khan, A.A.; Gunn, H.J.; Day, M.J.; Tayebi, M.; Ryan, S.D.; Kuntz, C.A.; Saad, E.S.; Richardson, S.J.; Danks, J.A. Immunohistochemical Validation of Spontaneously Arising Canine Osteosarcoma as a Model for Human Osteosarcoma. J. Comp. Pathol. 2017, 157, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Mardanpour, K.; Rahbar, M.; Mardanpour, S. Coexistence of HER2, Ki67, and p53 in Osteosarcoma: A Strong Prognostic Factor. N. Am. J. Med. Sci. 2016, 8, 210–214. [Google Scholar] [CrossRef]
- Barger, A.; Graca, R.; Bailey, K.; Messick, J.; de Lorimier, L.P.; Fan, T.; Hoffmann, W. Use of alkaline phosphatase staining to differentiate canine osteosarcoma from other vimentin-positive tumors. Vet. Pathol. 2005, 42, 161–165. [Google Scholar] [CrossRef]
- Thway, K. Pathology of soft tissue sarcomas. Clin. Oncol. R. Coll. Radiol. 2009, 21, 695–705. [Google Scholar] [CrossRef]
- Augsburger, D.; Nelson, P.J.; Kalinski, T.; Udelnow, A.; Knosel, T.; Hofstetter, M.; Qin, J.W.; Wang, Y.; Gupta, A.S.; Bonifatius, S.; et al. Current diagnostics and treatment of fibrosarcoma -perspectives for future therapeutic targets and strategies. Oncotarget 2017, 8, 104638–104653. [Google Scholar] [CrossRef]
- Folpe, A.L. Fibrosarcoma: A review and update. Histopathology 2014, 64, 12–25. [Google Scholar] [CrossRef]
- Meis-Kindblom, J.M.; Kindblom, L.G.; Enzinger, F.M. Sclerosing epithelioid fibrosarcoma. A variant of fibrosarcoma simulating carcinoma. Am. J. Surg. Pathol. 1995, 19, 979–993. [Google Scholar] [CrossRef]
- Munday, J.S.; Stedman, N.L.; Richey, L.J. Histology and immunohistochemistry of seven ferret vaccination-site fibrosarcomas. Vet. Pathol. 2003, 40, 288–293. [Google Scholar] [CrossRef]
- Oda, Y.; Miyajima, K.; Kawaguchi, K.; Tamiya, S.; Oshiro, Y.; Hachitanda, Y.; Oya, M.; Iwamoto, Y.; Tsuneyoshi, M. Pleomorphic leiomyosarcoma: Clinicopathologic and immunohistochemical study with special emphasis on its distinction from ordinary leiomyosarcoma and malignant fibrous histiocytoma. Am. J. Surg. Pathol. 2001, 25, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Coindre, J.M.; Mariani, O.; Chibon, F.; Mairal, A.; De Saint Aubain Somerhausen, N.; Favre-Guillevin, E.; Bui, N.B.; Stoeckle, E.; Hostein, I.; Aurias, A. Most malignant fibrous histiocytomas developed in the retroperitoneum are dedifferentiated liposarcomas: A review of 25 cases initially diagnosed as malignant fibrous histiocytoma. Mod. Pathol. 2003, 16, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.W.; Fisher, C.; Gatter, K.C. An immunohistochemical study of differentiation in malignant fibrous histiocytoma. Histopathology 1987, 11, 375–383. [Google Scholar] [CrossRef]
- Al-Agha, O.M.; Igbokwe, A.A. Malignant fibrous histiocytoma: Between the past and the present. Arch. Pathol. Lab. Med. 2008, 132, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Coindre, J.M. Immunohistochemistry in the diagnosis of soft tissue tumours. Histopathology 2003, 43, 1–16. [Google Scholar] [CrossRef]
- Machado, I.; Mayordomo-Aranda, E.; Giner, F.; Llombart-Bosch, A. The Role of Immunohistochemistry in Rhabdomyosarcoma Diagnosis Using Tissue Microarray Technology and a Xenograft Model. Fetal Pediatr. Pathol. 2015, 34, 271–281. [Google Scholar] [CrossRef]
- Wachtel, M.; Runge, T.; Leuschner, I.; Stegmaier, S.; Koscielniak, E.; Treuner, J.; Odermatt, B.; Behnke, S.; Niggli, F.K.; Schafer, B.W. Subtype and prognostic classification of rhabdomyosarcoma by immunohistochemistry. J. Clin. Oncol. 2006, 24, 816–822. [Google Scholar] [CrossRef]
- Mentzel, T.; Kuhnen, C. Spindle cell rhabdomyosarcoma in adults: Clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch. 2006, 449, 554–560. [Google Scholar] [CrossRef]
- Carvalho, J.C.; Thomas, D.G.; Lucas, D.R. Cluster analysis of immunohistochemical markers in leiomyosarcoma delineates specific anatomic and gender subgroups. Cancer 2009, 115, 4186–4195. [Google Scholar] [CrossRef]
- Mills, A.M.; Ly, A.; Balzer, B.L.; Hendrickson, M.R.; Kempson, R.L.; McKenney, J.K.; Longacre, T.A. Cell cycle regulatory markers in uterine atypical leiomyoma and leiomyosarcoma: Immunohistochemical study of 68 cases with clinical follow-up. Am. J. Surg. Pathol. 2013, 37, 634–642. [Google Scholar] [CrossRef]
- Lugowska, I.; Teterycz, P.; Mikula, M.; Kulecka, M.; Kluska, A.; Balabas, A.; Piatkowska, M.; Wagrodzki, M.; Pienkowski, A.; Rutkowski, P.; et al. IDH1/2 Mutations Predict Shorter Survival in Chondrosarcoma. J. Cancer 2018, 9, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Sekimizu, M.; Endo, M.; Kobayashi, E.; Iwata, S.; Fukushima, S.; Yoshida, A.; Kitabayashi, I.; Ichikawa, H.; Kawai, A.; et al. Prognostic impact of IDH mutations in chondrosarcoma. J. Orthop. Sci. 2022, 27, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Vuong, H.G.; Ngo, T.N.M.; Dunn, I.F. Prognostic importance of IDH mutations in chondrosarcoma: An individual patient data meta-analysis. Cancer Med. 2021, 10, 4415–4423. [Google Scholar] [CrossRef]
- Li, Y.; Yang, S.; Liu, Y.; Yang, S. Mice with Trp53 and Rb1 deficiency in chondrocytes spontaneously develop chondrosarcoma via overactivation of YAP signaling. Cell Death Dis. 2022, 13, 570. [Google Scholar] [CrossRef] [PubMed]
- Venneker, S.; Kruisselbrink, A.B.; Baranski, Z.; Palubeckaite, I.; Briaire-de Bruijn, I.H.; Oosting, J.; French, P.J.; Danen, E.H.J.; Bovee, J. Beyond the Influence of IDH Mutations: Exploring Epigenetic Vulnerabilities in Chondrosarcoma. Cancers 2020, 12, 3589. [Google Scholar] [CrossRef]
- Nakagawa, M.; Yamaguchi, M.; Endo, M.; Machida, Y.; Hattori, A.; Tanzawa, F.; Tsutsumi, S.; Kitabayashi, I.; Kawai, A.; Nakatani, F. Clinical usefulness of 2-hydroxyglutarate as a biomarker in IDH-mutant chondrosarcoma. J. Bone Oncol. 2022, 34, 100430. [Google Scholar] [CrossRef] [PubMed]
- Bovée, J.V.M.G.; Bloem, J.L.; Flanagan, A.M.; Nielsen, G.P.; Yoshida, A. WHO Classification of Tumours: Soft Tissue and Bone Tumours, 5th ed.; WHO: Geneva, Switzerland, 2020; Volume 3, pp. 370–390. [Google Scholar]
- Nicolle, R.; Ayadi, M.; Gomez-Brouchet, A.; Armenoult, L.; Banneau, G.; Elarouci, N.; Tallegas, M.; Decouvelaere, A.V.; Aubert, S.; Redini, F.; et al. Integrated molecular characterization of chondrosarcoma reveals critical determinants of disease progression. Nat. Commun. 2019, 10, 4622. [Google Scholar] [CrossRef]
- Miwa, S.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Therapeutic Targets and Emerging Treatments in Advanced Chondrosarcoma. Int. J. Mol. Sci. 2022, 23, 1096. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef]
- Sandberg, A.A. Genetics of chondrosarcoma and related tumors. Curr. Opin. Oncol. 2004, 16, 342–354. [Google Scholar] [CrossRef]
- Lucas, C.G.; Grenert, J.P.; Horvai, A. Targeted Next-Generation Sequencing Identifies Molecular and Genetic Events in Dedifferentiated Chondrosarcoma. Arch. Pathol. Lab. Med. 2021, 145, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Nazeri, E.; Gouran Savadkoohi, M.; Majidzadeh, A.K.; Esmaeili, R. Chondrosarcoma: An overview of clinical behavior, molecular mechanisms mediated drug resistance and potential therapeutic targets. Crit. Rev. Oncol. Hematol. 2018, 131, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, Y.; Chaturvedi, V.; Hayden, D.; Nazeer, T.; Johnson, M.; Johnston, D.A.; Ordez, N.G.; Ayala, A.G.; Czerniak, B. Altered p53 is associated with aggressive behavior of chondrosarcoma. Cancer 1998, 83, 2324–2334. [Google Scholar] [CrossRef]
- Suzuki, H.; Zhou, X.; Yin, J.; Lei, J.; Jiang, H.Y.; Suzuki, Y.; Chan, T.; Hannon, G.J.; Mergner, W.J.; Abraham, J.M.; et al. Intragenic mutations of CDKN2B and CDKN2A in primary human esophageal cancers. Hum. Mol. Genet. 1995, 4, 1883–1887. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, P.S.; Behjati, S.; Cooke, S.L.; Van Loo, P.; Wedge, D.C.; Pillay, N.; Marshall, J.; O’Meara, S.; Davies, H.; Nik-Zainal, S.; et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat. Genet. 2013, 45, 923–926. [Google Scholar] [CrossRef]
- Amary, M.F.; Ye, H.; Forbes, G.; Damato, S.; Maggiani, F.; Pollock, R.; Tirabosco, R.; Flanagan, A.M. Isocitrate dehydrogenase 1 mutations (IDH1) and p16/CDKN2A copy number change in conventional chondrosarcomas. Virchows Arch. 2015, 466, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.A. Chondrosarcoma: Biology, genetics, and epigenetics. F1000Research 2018, 7, 1826. [Google Scholar] [CrossRef]
- Gao, L.; Hong, X.; Guo, X.; Cao, D.; Gao, X.; DeLaney, T.F.; Gong, X.; Chen, R.; Ni, J.; Yao, Y.; et al. Targeted next-generation sequencing of dedifferentiated chondrosarcoma in the skull base reveals combined TP53 and PTEN mutations with increased proliferation index, an implication for pathogenesis. Oncotarget 2016, 7, 43557–43569. [Google Scholar] [CrossRef]
- O’Malley, D.P.; Opheim, K.E.; Barry, T.S.; Chapman, D.B.; Emond, M.J.; Conrad, E.U.; Norwood, T.H. Chromosomal changes in a dedifferentiated chondrosarcoma: A case report and review of the literature. Cancer Genet. Cytogenet. 2001, 124, 105–111. [Google Scholar] [CrossRef]
- Kattepur, A.K.; Jones, R.L.; Gulia, A. Dedifferentiated chondrosarcoma: Current standards of care. Future Oncol. 2021, 17, 4983–4991. [Google Scholar] [CrossRef]
- Stevenson, J.D.; Laitinen, M.K.; Parry, M.C.; Sumathi, V.; Grimer, R.J.; Jeys, L.M. The role of surgical margins in chondrosarcoma. Eur. J. Surg. Oncol. 2018, 44, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Deloin, X.; Dumaine, V.; Biau, D.; Karoubi, M.; Babinet, A.; Tomeno, B.; Anract, P. Pelvic chondrosarcomas: Surgical treatment options. Orthop. Traumatol. Surg. Res. 2009, 95, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Bruns, J.; Fiedler, W.; Werner, M.; Delling, G. Dedifferentiated chondrosarcoma—A fatal disease. J. Cancer Res. Clin. Oncol. 2005, 131, 333–339. [Google Scholar] [CrossRef]
- Sambri, A.; Tuzzato, G.; Donati, D.M.; De Paolis, M.; Bianchi, G. Pathological fracture does not affect prognosis in dedifferentiated chondrosarcoma of the limbs. J. Orthop. Sci. 2021, 26, 473–477. [Google Scholar] [CrossRef]
- Walter, S.G.; Knöll, P.; Eysel, P.; Quaas, A.; Gaisendrees, C.; Nißler, R.; Hieggelke, L. Molecular In-Depth Characterization of Chondrosarcoma for Current and Future Targeted Therapies. Cancers 2023, 15, 2556. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, L.; Vanel, D.; Leclere, J. Imaging of chondrosarcomas. Cancer Imaging 2003, 4, 36–38. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.D.; Anderson, M.S.; Epperly, S.C.; Zuckerman, L.M. Successful treatment of a dedifferentiated chondrosarcoma of the proximal humerus with a hemicortical articular surface sparing allograft: A case report. Int. J. Surg. Case Rep. 2020, 72, 590–595. [Google Scholar] [CrossRef]
- Davies, B.W.; Prescott, C.R.; Said, S.A.; Campana, J.; Attie-Castro, F.A.; Velasco, E.C.A.A.; Durairaj, V.D. Radiation-induced dedifferentiated chondrosarcoma with orbital invasion. Ophthalmic Plast. Reconstr. Surg. 2014, 30, 205–208. [Google Scholar] [CrossRef]
- Coskun, H.S.; Erdogan, F.; Buyukceran, I.; Dabak, N. Evaluation of prognostic factors affecting survival in chondrosarcoma treatment and comparison with literature. Jt. Dis. Relat. Surg. 2022, 33, 440–448. [Google Scholar] [CrossRef]
- Kremenevski, N.; Schlaffer, S.M.; Coras, R.; Kinfe, T.M.; Graillon, T.; Buchfelder, M. Skull Base Chordomas and Chondrosarcomas. Neuroendocrinology 2020, 110, 836–847. [Google Scholar] [CrossRef]
- Harwood, A.R.; Krajbich, J.I.; Fornasier, V.L. Radiotherapy of chondrosarcoma of bone. Cancer 1980, 45, 2769–2777. [Google Scholar] [CrossRef] [PubMed]
- Krochak, R.; Harwood, A.R.; Cummings, B.J.; Quirt, I.C. Results of radical radiation for chondrosarcoma of bone. Radiother. Oncol. 1983, 1, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Lex, J.R.; Evans, S.; Stevenson, J.D.; Parry, M.; Jeys, L.M.; Grimer, R.J. Dedifferentiated chondrosarcoma of the pelvis: Clinical outcomes and current treatment. Clin. Sarcoma Res. 2018, 8, 23. [Google Scholar] [CrossRef]
- Dickey, I.D.; Rose, P.S.; Fuchs, B.; Wold, L.E.; Okuno, S.H.; Sim, F.H.; Scully, S.P. Dedifferentiated chondrosarcoma: The role of chemotherapy with updated outcomes. J. Bone Jt. Surg. Am. 2004, 86, 2412–2418. [Google Scholar] [CrossRef]
- MacDonald, I.J.; Lin, C.Y.; Kuo, S.J.; Su, C.M.; Tang, C.H. An update on current and future treatment options for chondrosarcoma. Expert. Rev. Anticancer. 2019, 19, 773–786. [Google Scholar] [CrossRef]
- van Maldegem, A.; Conley, A.P.; Rutkowski, P.; Patel, S.R.; Lugowska, I.; Desar, I.M.E.; Bovée, J.; Gelderblom, H. Outcome of First-Line Systemic Treatment for Unresectable Conventional, Dedifferentiated, Mesenchymal, and Clear Cell Chondrosarcoma. Oncologist 2019, 24, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Bui, N.; Dietz, H.; Farag, S.; Hirbe, A.C.; Wagner, M.J.; Van Tine, B.A.; Ganjoo, K.; Jones, R.L.; Keedy, V.L.; Davis, E.J. A Retrospective Multi-Institutional Cohort Analysis of Clinical Characteristics and Outcomes in Dedifferentiated Chondrosarcoma. Cancers 2023, 15, 2617. [Google Scholar] [CrossRef]
- Frassica, F.J.; Unni, K.K.; Beabout, J.W.; Sim, F.H. Dedifferentiated chondrosarcoma. A report of the clinicopathological features and treatment of seventy-eight cases. J. Bone Jt. Surg. Am. 1986, 68, 1197–1205. [Google Scholar] [CrossRef]
- Sheth, D.S.; Yasko, A.W.; Johnson, M.E.; Ayala, A.G.; Murray, J.A.; Romsdahl, M.M. Chondrosarcoma of the pelvis. Prognostic factors for 67 patients treated with definitive surgery. Cancer 1996, 78, 745–750. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Sun, T.; Lin, P.P.; Deavers, M.; Harun, N.; Lewis, V.O. Does ifosfamide therapy improve survival of patients with dedifferentiated chondrosarcoma? Clin. Orthop. Relat. Res. 2014, 472, 983–989. [Google Scholar] [CrossRef]
- Hompland, I.; Ferrari, S.; Bielack, S.; Palmerini, E.; Hall, K.S.; Picci, P.; Hecker-Nolting, S.; Donati, D.M.; Blattmann, C.; Bjerkehagen, B.; et al. Outcome in dedifferentiated chondrosarcoma for patients treated with multimodal therapy: Results from the EUROpean Bone Over 40 Sarcoma Study. Eur. J. Cancer 2021, 151, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Mir, O.; Cioffi, A.; Palmerini, E.; Piperno-Neumann, S.; Perrin, C.; Chaigneau, L.; Penel, N.; Duffaud, F.; Kurtz, J.E.; et al. Advanced chondrosarcomas: Role of chemotherapy and survival. Ann. Oncol. 2013, 24, 2916–2922. [Google Scholar] [CrossRef] [PubMed]
- Cranmer, L.D.; Chau, B.; Mantilla, J.G.; Loggers, E.T.; Pollack, S.M.; Kim, T.S.; Kim, E.Y.; Kane, G.M.; Thompson, M.J.; Harwood, J.L.; et al. Is Chemotherapy Associated with Improved Overall Survival in Patients with Dedifferentiated Chondrosarcoma? A SEER Database Analysis. Clin. Orthop. Relat. Res. 2022, 480, 748–758. [Google Scholar] [CrossRef]
- Mitchell, A.D.; Ayoub, K.; Mangham, D.C.; Grimer, R.J.; Carter, S.R.; Tillman, R.M. Experience in the treatment of dedifferentiated chondrosarcoma. J. Bone Jt. Surg. Br. 2000, 82, 55–61. [Google Scholar] [CrossRef]
- Streitbuerger, A.; Ahrens, H.; Gosheger, G.; Henrichs, M.; Balke, M.; Dieckmann, R.; Hardes, J. The treatment of locally recurrent chondrosarcoma: Is extensive further surgery justified? J. Bone Jt. Surg. Br. 2012, 94, 122–127. [Google Scholar] [CrossRef]
- Iseulys, R.; Anne, G.B.; Corinne, B.; Gonzague, D.B.P.; Marie, K.; Jean-Yves, B.; Aurelie, D. The immune landscape of chondrosarcoma reveals an immunosuppressive environment in the dedifferentiated subtypes and exposes CSFR1+ macrophages as a promising therapeutic target. J. Bone Oncol. 2020, 20, 100271. [Google Scholar] [CrossRef]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef]
- Wagner, M.J.; Ricciotti, R.W.; Mantilla, J.; Loggers, E.T.; Pollack, S.M.; Cranmer, L.D. Response to PD1 inhibition in conventional chondrosarcoma. J. Immunother. Cancer 2018, 6, 94. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef]
- Palmerini, E.; Lopez-Pousa, A.; Grignani, G.; Redondo, A.; Hindi, N.; Stacchiotti, S.; Sebio, A.; Lopez-Martin, J.A.; Morales, C.M.V.; Martinez-Trufero, J.; et al. IMMUNOSARC: A collaborative Spanish (GEIS) and Italian (ISG) sarcoma groups phase I/II trial of sunitinib and nivolumab in advanced soft tissue and bone sarcoma: Results from the phase II part, bone sarcoma cohort. J. Clin. Oncol. 2020, 38, 11522. [Google Scholar] [CrossRef]
- Zhang, Y.X.; van Oosterwijk, J.G.; Sicinska, E.; Moss, S.; Remillard, S.P.; van Wezel, T.; Buhnemann, C.; Hassan, A.B.; Demetri, G.D.; Bovee, J.V.; et al. Functional profiling of receptor tyrosine kinases and downstream signaling in human chondrosarcomas identifies pathways for rational targeted therapy. Clin. Cancer Res. 2013, 19, 3796–3807. [Google Scholar] [CrossRef]
- Polychronidou, G.; Karavasilis, V.; Pollack, S.M.; Huang, P.H.; Lee, A.; Jones, R.L. Novel therapeutic approaches in chondrosarcoma. Future Oncol. 2017, 13, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Schrage, Y.M.; Briaire-de Bruijn, I.H.; de Miranda, N.F.; van Oosterwijk, J.; Taminiau, A.H.; van Wezel, T.; Hogendoorn, P.C.; Bovee, J.V. Kinome profiling of chondrosarcoma reveals SRC-pathway activity and dasatinib as option for treatment. Cancer Res. 2009, 69, 6216–6222. [Google Scholar] [CrossRef] [PubMed]
- Bernstein-Molho, R.; Kollender, Y.; Issakov, J.; Bickels, J.; Dadia, S.; Flusser, G.; Meller, I.; Sagi-Eisenberg, R.; Merimsky, O. Clinical activity of mTOR inhibition in combination with cyclophosphamide in the treatment of recurrent unresectable chondrosarcomas. Cancer Chemother. Pharm. 2012, 70, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Micaily, I.; Roche, M.; Ibrahim, M.Y.; Martinez-Outschoorn, U.; Mallick, A.B. Metabolic Pathways and Targets in Chondrosarcoma. Front. Oncol. 2021, 11, 772263. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Stacchiotti, S.; Boglione, A.; Ferraresi, V.; Frustaci, S.; Comandone, A.; Casali, P.G.; Ferrari, S.; Aglietta, M. A phase 2 trial of imatinib mesylate in patients with recurrent nonresectable chondrosarcomas expressing platelet-derived growth factor receptor-alpha or -beta: An Italian Sarcoma Group study. Cancer 2011, 117, 826–831. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Bolejack, V.; Choy, E.; Ganjoo, K.N.; Staddon, A.P.; Chow, W.A.; Tawbi, H.A.; Samuels, B.L.; Patel, S.R.; von Mehren, M.; et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer 2017, 123, 90–97. [Google Scholar] [CrossRef]
- Albarran, V.; Villamayor, M.L.; Chamorro, J.; Rosero, D.I.; Pozas, J.; San Roman, M.; Calvo, J.C.; Perez de Aguado, P.; Moreno, J.; Guerrero, P.; et al. Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas. Int. J. Mol. Sci. 2022, 23, 13784. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, W.; Wang, Y.; Jin, R.; Wang, Y.; Guo, H.; Tang, Y.; Yao, X. Recent advances of IDH1 mutant inhibitor in cancer therapy. Front. Pharmacol. 2022, 13, 982424. [Google Scholar] [CrossRef]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef]
- Lin, Z.S.; Chung, C.C.; Liu, Y.C.; Chang, C.H.; Liu, H.C.; Liang, Y.Y.; Huang, T.L.; Chen, T.M.; Lee, C.H.; Tang, C.H.; et al. EZH2/hSULF1 axis mediates receptor tyrosine kinase signaling to shape cartilage tumor progression. Elife 2023, 12, e79432. [Google Scholar] [CrossRef] [PubMed]
- M.D. Anderson Cancer Center. LN-145 or LN-145-S1 in Treating Patients with Relapsed or Refractory Ovarian Cancer, Triple Negative Breast Cancer (TNBC), Anaplastic Thyroid Cancer, Osteosarcoma, or Other Bone and Soft Tissue Sarcomas. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03449108 (accessed on 27 July 2023).
- Gettinger, S.; Kluger, H.; Schoenfeld, A.; Warner, A.B.; He, K.; Sukari, A.; Thomas, S.S.; Spéville, B.D.d.; Lee, S.; Haefliger, S.; et al. 187TiP Phase II, multicenter study of autologous tumor infiltrating lymphocytes (TIL, LN 144/LN-145/LN-145-S1) in patients with solid tumours. J. Thorac. Oncol. 2021, 04, 16. [Google Scholar] [CrossRef]
- Biermann, J.S.; Chow, W.; Reed, D.R.; Lucas, D.; Adkins, D.R.; Agulnik, M.; Benjamin, R.S.; Brigman, B.; Budd, T.; Curry, W.T.; et al. NCCN Guidelines Insights: Bone Cancer, Version 2.2017. J. Natl. Compr. Canc. Netw. 2017, 15, 155–167. [Google Scholar] [CrossRef]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef] [PubMed]
| Cytostatics | Dosing | Cycles | Reference |
|---|---|---|---|
| DOX | 60–75 mg/m2 | PRC—3–4 cycles POC-1 9 cycles ** | [166] |
| CP | 100–120 mg/m2 | ||
| IF | 10 g/m2 | ||
| DOX | 60 mg/m2 (24 h iv infusion) | AC—9 cycles | [165] |
| CP | 100 mg/m2 (48–72 h of iv infusion) | PC—3 cycles | |
| IF | 6 g/m2 (3 g/m2 per day, 1–2 h i.v. infusion) | POC—6 or 11 cycles *** | |
| MTX * | 8 g/m2 (4 h i.v. infusion) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zając, W.; Dróżdż, J.; Kisielewska, W.; Karwowska, W.; Dudzisz-Śledź, M.; Zając, A.E.; Borkowska, A.; Szumera-Ciećkiewicz, A.; Szostakowski, B.; Rutkowski, P.; et al. Dedifferentiated Chondrosarcoma from Molecular Pathology to Current Treatment and Clinical Trials. Cancers 2023, 15, 3924. https://doi.org/10.3390/cancers15153924
Zając W, Dróżdż J, Kisielewska W, Karwowska W, Dudzisz-Śledź M, Zając AE, Borkowska A, Szumera-Ciećkiewicz A, Szostakowski B, Rutkowski P, et al. Dedifferentiated Chondrosarcoma from Molecular Pathology to Current Treatment and Clinical Trials. Cancers. 2023; 15(15):3924. https://doi.org/10.3390/cancers15153924
Chicago/Turabian StyleZając, Weronika, Julia Dróżdż, Weronika Kisielewska, Weronika Karwowska, Monika Dudzisz-Śledź, Agnieszka E. Zając, Aneta Borkowska, Anna Szumera-Ciećkiewicz, Bartłomiej Szostakowski, Piotr Rutkowski, and et al. 2023. "Dedifferentiated Chondrosarcoma from Molecular Pathology to Current Treatment and Clinical Trials" Cancers 15, no. 15: 3924. https://doi.org/10.3390/cancers15153924
APA StyleZając, W., Dróżdż, J., Kisielewska, W., Karwowska, W., Dudzisz-Śledź, M., Zając, A. E., Borkowska, A., Szumera-Ciećkiewicz, A., Szostakowski, B., Rutkowski, P., & Czarnecka, A. M. (2023). Dedifferentiated Chondrosarcoma from Molecular Pathology to Current Treatment and Clinical Trials. Cancers, 15(15), 3924. https://doi.org/10.3390/cancers15153924