
Clinical Applications of Immunotherapy for Recurrent Glioblastoma in Adults
 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
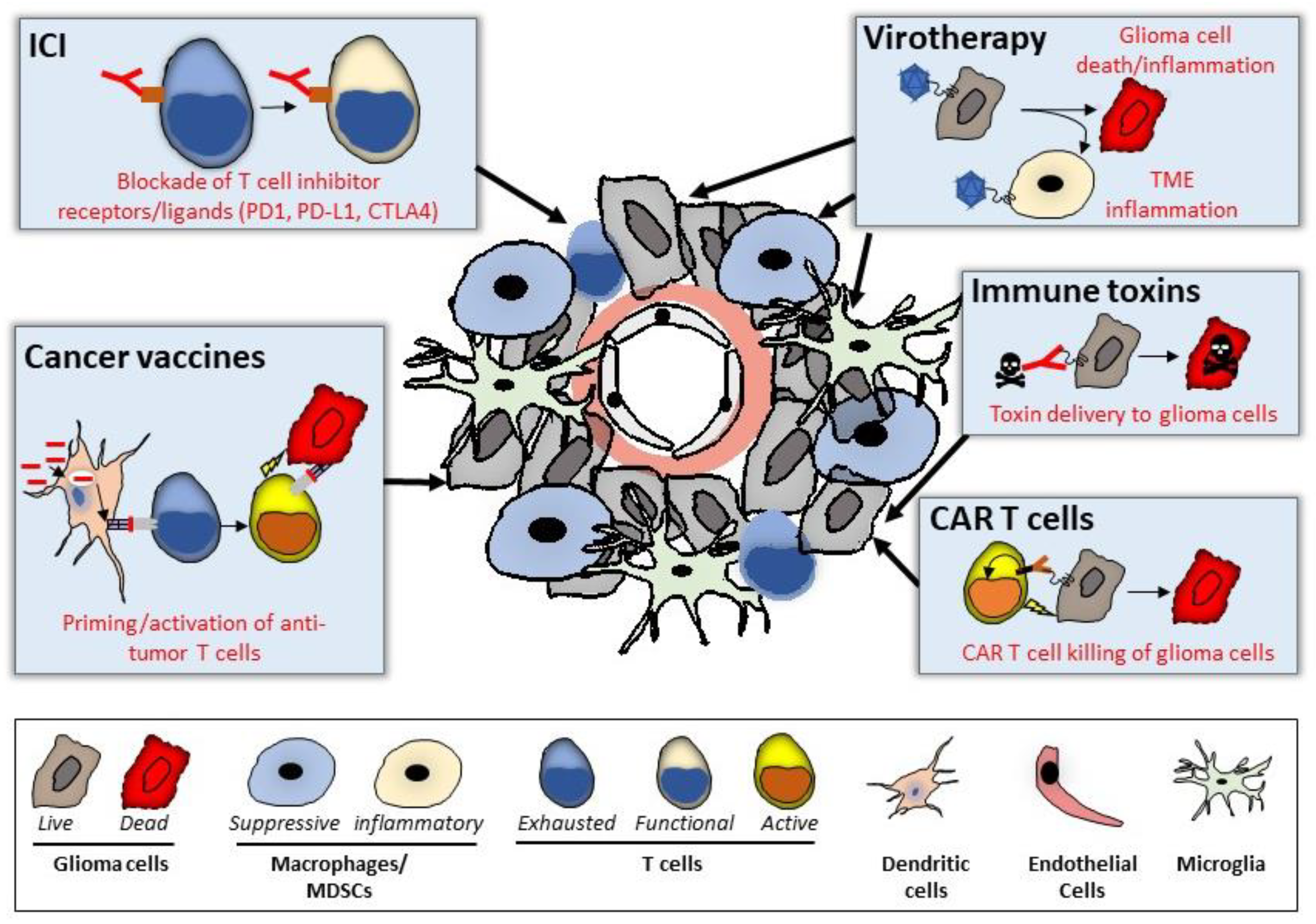
3.1. Immune Checkpoint Blockade
3.1.1. PD-1 Inhibition
3.1.2. PD-L1 Inhibition
3.1.3. CTLA-4 Inhibition
3.1.4. Ongoing Studies
3.2. Virotherapy
3.2.1. Herpes Simplex Virus-1
- HSV G207
- HSV 1716
- HSV G47Δ
3.2.2. Poliovirus
3.2.3. Parvovirus
3.2.4. Adenovirus
3.2.5. Newcastle Virus
3.2.6. Reovirus
3.2.7. Retrovirus
3.2.8. Ongoing Studies
3.3. CAR-Modified T Cell (CART)
3.3.1. CART-EGFRvIII
3.3.2. CART-IL13Rα2
3.3.3. EphA2-Redirected CAR T-Cells
3.3.4. Autologous CMV-Specific T-Cell Therapy
3.3.5. HER2-Specific CART
3.3.6. Ongoing Studies
3.4. Immunotoxins
3.4.1. MDNA55
3.4.2. Depatuxizumab Mofodin
3.4.3. Pseudomonas Exotoxin A
3.4.4. Ongoing Studies
3.5. Cancer Vaccines
3.5.1. ERC1671
3.5.2. Dendritic Cell Vaccines
3.5.3. Wilms Tumor 1 (WT1) Peptide Vaccination
3.5.4. Personalized Peptide Vaccination (PPV)
3.5.5. Heat–Shock Protein Peptide Complex-96
3.5.6. Survivin Long Peptide Vaccine (SurVaxM)
3.5.7. EO2401
3.5.8. VXM01
3.5.9. Ongoing Studies
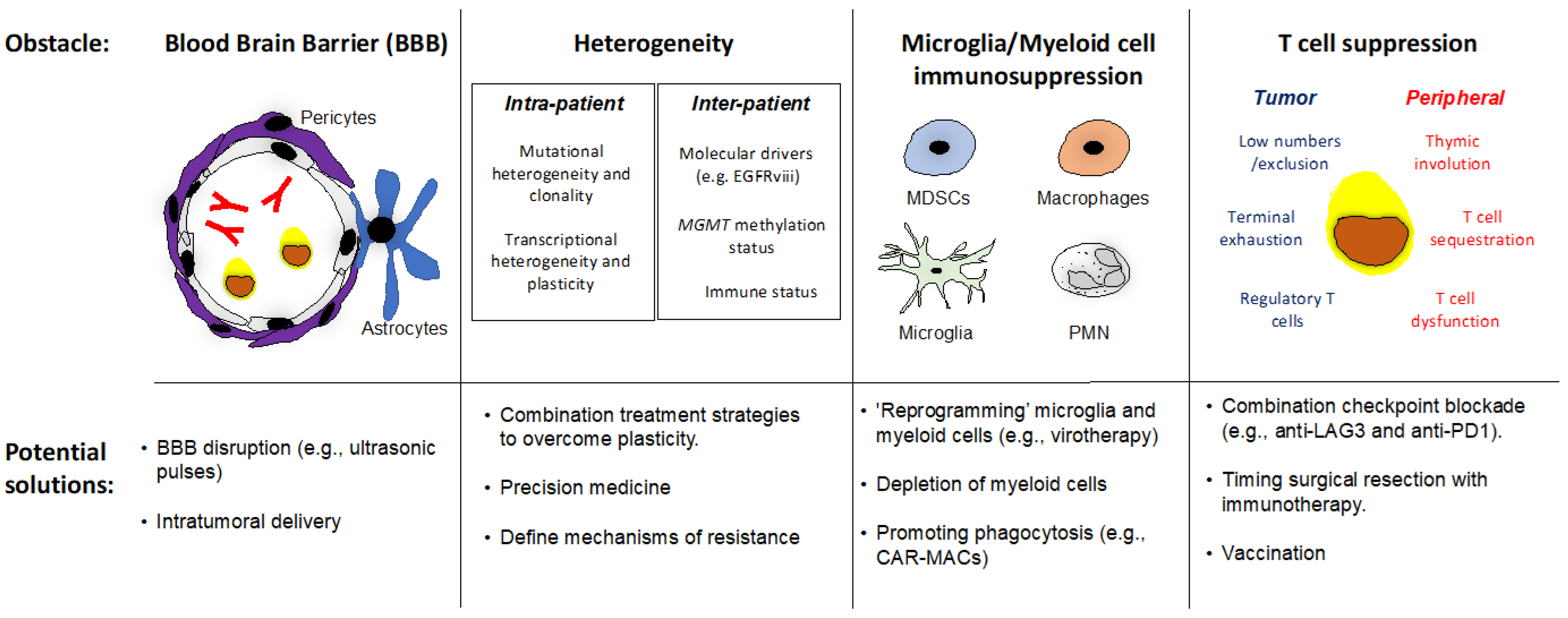
3.6. Future Directions: Overcoming Obstacles to Successful Cancer Immunotherapy in rGBM
3.6.1. The Blood–Brain Barrier
3.6.2. Intra-Patient and Inter-Patient Heterogeneity
3.6.3. Microglia/Myeloid Cell Immunosuppression
3.6.4. T Cell Suppression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23, III1–III105. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Lakomy, R.; Kazda, T.; Selingerova, I.; Poprach, A.; Pospisil, P.; Belanova, R.; Fadrus, P.; Vybihal, V.; Smrcka, M.; Jancalek, R.; et al. Real-World Evidence in Glioblastoma: Stupp’s Regimen after a Decade. Front. Oncol. 2020, 10, 840. [Google Scholar] [CrossRef]
- Kelly, C.; Majewska, P.; Ioannidis, S.; Raza, M.H.; Williams, M. Estimating progression-free survival in patients with glioblastoma using routinely collected data. Neuro Oncol. 2017, 135, 621–627. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Van Linde, M.E.; Brahm, C.G.; Hamer, P.C.D.W.; Reijneveld, J.C.; Bruynzeel, A.M.; Wagemakers, M.; Van Der Weide, H.L.; Enting, R.H.; Verheul, H.; Walenkamp, A.M.E. Treatment outcome of patients with recurrent glioblastoma multiforme: A retrospective multicenter analysis. Neuro Oncol. 2017, 135, 183–192. [Google Scholar] [CrossRef]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.E.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.J.; Buter, J.; Honkoop, A.H.; Boerman, D.; De Vos, F.; et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef]
- Knudsen, A.M.; Halle, B.; Cédile, O.; Burton, M.; Baun, C.; Thisgaard, H.; Anand, A.; Hubert, C.; Thomassen, M.; Michaelsen, S.R.; et al. Surgical resection of glioblastomas induces pleiotrophin-mediated self-renewal of glioblastoma stem cells in recurrent tumors. Neuro Oncol. 2022, 24, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Blobner, J.; Tonn, J.-C. Resection of glioma—Feeding the beast? Neuro Oncol. 2022, 24, 1088–1089. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Anghileri, E.; Patanè, M.; Di Ianni, N.; Sambruni, I.; Maffezzini, M.; Milani, M.; Maddaloni, L.; Pollo, B.; Eoli, M.; Pellegatta, S. Deciphering the Labyrinthine System of the Immune Microenvironment in Recurrent Glioblastoma: Recent Original Advances and Lessons from Clinical Immunotherapeutic Approaches. Cancers 2021, 13, 6156. [Google Scholar] [CrossRef] [PubMed]
- Murota, Y.; Tabu, K.; Taga, T. Cancer Stem Cell-Associated Immune Microenvironment in Recurrent Glioblastomas. Cells 2022, 11, 2054. [Google Scholar] [CrossRef]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Front. Immunol. 2020, 11, 835. [Google Scholar] [CrossRef]
- Rahman, M.; Kresak, J.; Yang, C.; Huang, J.; Hiser, W.; Kubilis, P.; Mitchell, D. Analysis of immunobiologic markers in primary and recurrent glioblastoma. Neuro Oncol. 2018, 137, 249–257. [Google Scholar] [CrossRef]
- Magri, S.; Musca, B.; Bonaudo, C.; Tushe, A.; Russo, M.G.; Masetto, E.; Zagonel, V.; Lombardi, G.; Della Puppa, A.; Mandruzzato, S. Sustained Accumulation of Blood-Derived Macrophages in the Immune Microenvironment of Patients with Recurrent Glioblastoma after Therapy. Cancers 2021, 13, 6178. [Google Scholar] [CrossRef]
- Mohme, M.; Schliffke, S.; Maire, C.L.; Rünger, A.; Glau, L.; Mende, K.C.; Matschke, J.; Gehbauer, C.; Akyüz, N.; Zapf, S.; et al. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-infiltrating Lymphocytes. Clin. Cancer Res. 2018, 24, 4187–4200. [Google Scholar] [CrossRef]
- Cao, T.Q.; Wainwright, D.A.; Lee-Chang, C.; Miska, J.; Sonabend, A.M.; Heimberger, A.B.; Lukas, R.V. Next Steps for Immunotherapy in Glioblastoma. Cancers 2022, 14, 4023. [Google Scholar] [CrossRef]
- Chan, H.Y.; Choi, J.; Jackson, C.; Lim, M. Combination immunotherapy strategies for glioblastoma. Neuro Oncol. 2021, 151, 375–391. [Google Scholar] [CrossRef] [PubMed]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.; Fecci, P.E. Immune suppression in gliomas. Neuro Oncol. 2021, 151, 3–12. [Google Scholar] [CrossRef] [PubMed]
- WTomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neuro Oncol. 2020, 151, 41–53. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Horn, L.; Borghaei, H.; Spigel, D.R.; Steins, M.; Ready, N.; Chow, L.Q.M.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Phase III, randomized trial (CheckMate 057) of nivolumab (NIVO) versus docetaxel (DOC) in advanced non-squamous cell (non-SQ) non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2015, 33, 18. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs. Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Kim, T.M.; Frenel, J.; Simonelli, M.; Lopez, J.; Subramaniam, D.S.; Siu, L.L.; Wang, H.; Krishnan, S.; Stein, K.; et al. Treatment with pembrolizumab in programmed death ligand 1–Positive recurrent glioblastoma: Results from the multicohort phase 1 KEYNOTE-028 trial. Cancer 2021, 127, 1620–1629. [Google Scholar] [CrossRef]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Sahebjam, S.; Forsyth, P.A.; Tran, N.D.; Arrington, J.A.; Macaulay, R.; Etame, A.B.; Walko, C.M.; Boyle, T.; Peguero, E.N.; Jaglal, M.; et al. Hypofractionated stereotactic re-irradiation with pembrolizumab and bevacizumab in patients with recurrent high-grade gliomas: Results from a phase I study. Neuro Oncol. 2021, 23, 677–686. [Google Scholar] [CrossRef]
- Ahern, E.; Solomon, B.J.; Hui, R.; Pavlakis, N.; O’Byrne, K.; Hughes, B.G.M. Neoadjuvant immunotherapy for non-small cell lung cancer: Right drugs, right patient, right time? J. Immunother. Cancer 2021, 9, e002248. [Google Scholar] [CrossRef]
- Leidner, R.; Crittenden, M.; Young, K.; Xiao, H.; Wu, Y.; Couey, M.A.; Patel, A.A.; Cheng, A.C.; Watters, A.L.; Bifulco, C.; et al. Neoadjuvant immunoradiotherapy results in high rate of complete pathological response and clinical to pathological downstaging in locally advanced head and neck squamous cell carcinoma. J. Immunother. Cancer 2021, 9, e002485. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.-Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016, 18, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Lukas, R.V.; Rodon, J.; Becker, K.; Wong, E.T.; Shih, K.; Touat, M.; Fassò, M.; Osborne, S.; Molinero, L.; O’Hear, C.; et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. Neuro Oncol. 2018, 140, 317–328. [Google Scholar] [CrossRef]
- Nayak, L.; Standifer, N.; Dietrich, J.; Clarke, J.L.; Dunn, G.P.; Lim, M.; Cloughesy, T.; Gan, H.K.; Flagg, E.; George, E.; et al. Circulating Immune Cell and Outcome Analysis from the Phase II Study of PD-L1 Blockade with Durvalumab for Newly Diagnosed and Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Awada, G.; Ben Salama, L.; De Cremer, J.; Schwarze, J.K.; Fischbuch, L.; Seynaeve, L.; Du Four, S.; Vanbinst, A.-M.; Michotte, A.; Everaert, H.; et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: A stratified, open-label, single-center phase 2 clinical trial (GliAvAx). J. Immunother. Cancer 2020, 8, e001146. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: A phase I clinical trial. J. Immunother. Cancer 2021, 9, e002296. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Gelb, A.B.; Chen, C.C.; Rao, G.; Reardon, D.A.; Wen, P.Y.; Bi, W.L.; Peruzzi, P.; Amidei, C.; Triggs, D.; et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial. Neuro Oncol. 2022, 24, 951–963. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11, 505. [Google Scholar] [CrossRef]
- Markovitz, N.S.; Roizman, B. Replication-competent herpes simplex viral vectors for cancer therapy. Adv. Virus Res. 2000, 55, 409–424. [Google Scholar] [CrossRef]
- Estevez-Ordonez, D.; Chagoya, G.; Salehani, A.; Atchley, T.J.; Laskay, N.M.; Parr, M.S.; Elsayed, G.A.; Mahavadi, A.K.; Rahm, S.P.; Friedman, G.K.; et al. Immunovirotherapy for the Treatment of Glioblastoma and Other Malignant Gliomas. Neurosurg. Clin. N. Am. 2021, 32, 265–281. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Andreansky, S.S.; He, B.; Gillespie, G.Y.; Soroceanu, L.; Markert, J.; Chou, J.; Roizman, B.; Whitley, R.J. The application of genetically engineered herpes simplex viruses to the treatment of experimental brain tumors. Proc. Natl. Acad. Sci. USA 1996, 93, 11313–11318. [Google Scholar] [CrossRef]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of Herpes Simplex Virus-1 Neurovirulence to γ 1 34.5, a Gene Nonessential for Growth in Culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Hoffman, S.E.; Chen, J.A.; Gupta, S.; Kappel, A.D.; Smith, T.R.; Chiocca, E.A. The Current Landscape of Oncolytic Herpes Simplex Viruses as Novel Therapies for Brain Malignancies. Viruses 2021, 13, 1158. [Google Scholar] [CrossRef] [PubMed]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi–mutated herpes simplex virus–1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib Trial of Mutant Herpes Simplex Virus G207 Inoculated Pre-and Post-tumor Resection for Recurrent GBM. Mol. Ther. 2009, 17, 199–207. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.-C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A Phase 1 Trial of Oncolytic HSV-1, G207, Given in Combination with Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef]
- McKie, E.A.; MacLean, A.R.; Lewis, A.D.; Cruickshank, G.; Rampling, R.P.; Barnett, S.C.; Kennedy, P.; Brown, S.M. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours—Evaluation of a potentially effective clinical therapy. Br. J. Cancer 1996, 74, 745–752. [Google Scholar] [CrossRef]
- Rampling, R.; Cruickshank, G.; Papanastassiou, V.; Nicoll, J.; Hadley, D.; Brennan, D.; Petty, R.; MacLean, A.; Harland, J.; McKie, E.; et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000, 7, 859–866. [Google Scholar] [CrossRef]
- Papanastassiou, V.; Rampling, R.; Fraser, M.; Petty, R.; Hadley, D.; Nicoll, J.; Harland, J.; Mabbs, R.; Brown, M. The potential for efficacy of the modified (ICP 34.5−) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: A proof of principle study. Gene Ther. 2002, 9, 398–406. [Google Scholar] [CrossRef]
- Harrow, S.; Papanastassiou, V.; Harland, J.; Mabbs, R.; Petty, R.; Fraser, M.; Hadley, D.; Patterson, J.; Brown, S.M.; Rampling, R. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Ther. 2004, 11, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Takeshima, Y.; Todo, T. Triple-mutated oncolytic herpes virus for treating both fast- and slow-growing tumors. Cancer Sci. 2021, 112, 3293–3301. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Gromeier, M.; Alexander, L.; Wimmer, E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc. Natl. Acad. Sci. USA 1996, 93, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Lachmann, S.; Rosenfeld, M.R.; Gutin, P.H.; Wimmer, E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc. Natl. Acad. Sci. USA 2000, 97, 6803–6808. [Google Scholar] [CrossRef]
- Merrill, M.K.; Dobrikova, E.Y.; Gromeier, M. Cell-Type-Specific Repression of Internal Ribosome Entry Site Activity by Double-Stranded RNA-Binding Protein 76. J. Virol. 2006, 80, 3147–3156. [Google Scholar] [CrossRef]
- Brown, M.C.; Mosaheb, M.M.; Mohme, M.; McKay, Z.P.; Holl, E.K.; Kastan, J.P.; Yang, Y.; Beasley, G.M.; Hwang, E.S.; Ashley, D.M.; et al. Viral infection of cells within the tumor microenvironment mediates antitumor immunotherapy via selective TBK1-IRF3 signaling. Nat. Commun. 2021, 12, 1858. [Google Scholar] [CrossRef]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen–Specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Friedman, H.; Landi, D.; Friedman, A.; Ashley, D.M.; Curry, W.; Butowski, N.; Sloan, A.; Wen, P.; et al. IMMU-26. Safety and efficacy of pvsripo in recurrent glioblastoma: Long-term follow-up and initial multicenter results. Neuro Oncol. 2021, 23, vi97. [Google Scholar] [CrossRef]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A Phase I Open-Label, Dose-Escalation, Multi-Institutional Trial of Injection with an E1B-Attenuated Adenovirus, ONYX-015, into the Peritumoral Region of Recurrent Malignant Gliomas, in the Adjuvant Setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Clise-Dwyer, K.; Ruisaard, K.E.; Fan, X.; Tian, W.; Gumin, J.; Lamfers, M.L.; Kleijn, A.; Lang, F.F.; Yung, W.-K.A.; et al. Delta-24-RGD Oncolytic Adenovirus Elicits Anti-Glioma Immunity in an Immunocompetent Mouse Model. PLoS ONE 2014, 9, e97407. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Miller, C.R.; Ji, Y.; Gomez-Manzano, C.; Bharara, S.; McMurray, J.S.; Lang, F.F.; Wong, F.; Sawaya, R.; Yung, W.K.A.; et al. Δ24-hyCD adenovirus suppresses glioma growth in vivo by combining oncolysis and chemosensitization. Cancer Gene Ther. 2005, 12, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Ene, C.I.; Fueyo, J.; Lang, F.F. Delta-24 adenoviral therapy for glioblastoma: Evolution from the bench to bedside and future considerations. Neurosurg. Focus 2021, 50, E6. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Van Putten, E.H.; Kleijn, A.; van Beusechem, V.W.; Noske, D.; Lamers, C.H.; de Goede, A.L.; Idema, S.; Hoefnagel, D.; Kloezeman, J.J.; Fueyo, J.; et al. Convection Enhanced Delivery of the Oncolytic Adenovirus Delta24-RGD in Patients with Recurrent GBM: A Phase I Clinical Trial Including Correlative Studies. Clin. Cancer Res. 2022, 28, 1572–1585. [Google Scholar] [CrossRef]
- Lang, F.F.; Tran, N.D.; Puduvalli, V.K.; Elder, J.B.; Fink, K.L.; Conrad, C.A.; Yung, W.K.A.; Penas-Prado, M.; Gomez-Manzano, C.; Peterkin, J.; et al. Phase 1b open-label randomized study of the oncolytic adenovirus DNX-2401 administered with or without interferon gamma for recurrent glioblastoma. J. Clin. Oncol. 2017, 35, 2002. [Google Scholar] [CrossRef]
- Schirrmacher, V. Fifty Years of Clinical Application of Newcastle Disease Virus: Time to Celebrate! Biomedicines 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.H.; Bonsanto, M.M.; Beckhove, P.; Brysch, M.; Geletneky, K.; Ahmadi, R.; Schuele-Freyer, R.; Kremer, P.; Ranaie, G.; Matejic, D.; et al. Antitumor Vaccination of Patients with Glioblastoma Multiforme: A Pilot Study to Assess Feasibility, Safety, and Clinical Benefit. J. Clin. Oncol. 2004, 22, 4272–4281. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II Trial of Intravenous NDV-HUJ Oncolytic Virus in Recurrent Glioblastoma Multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, P.; Roldán, G.; George, D.; Wallace, C.; Palmer, C.A.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A Phase I Trial of Intratumoral Administration of Reovirus in Patients with Histologically Confirmed Recurrent Malignant Gliomas. Mol. Ther. 2008, 16, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Kicielinski, K.P.; Chiocca, E.A.; Yu, J.S.; Gill, G.M.; Coffey, M.; Markert, J.M. Phase 1 Clinical Trial of Intratumoral Reovirus Infusion for the Treatment of Recurrent Malignant Gliomas in Adults. Mol. Ther. 2014, 22, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef]
- Perez, O.D.; Logg, C.R.; Hiraoka, K.; Diago, O.; Burnett, R.; Inagaki, A.; Jolson, D.; Amundson, K.; Buckley, T.; Lohse, D.; et al. Design and Selection of Toca 511 for Clinical Use: Modified Retroviral Replicating Vector with Improved Stability and Gene Expression. Mol. Ther. 2012, 20, 1689–1698. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.-J.; et al. Effect of Vocimagene Amiretrorepvec in Combination with Flucytosine vs. Standard of Care on Survival Following Tumor Resection in Patients with Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1936–1946. [Google Scholar] [CrossRef]
- Aghi, M.; Vogelbaum, M.A.; Kalkanis, S.N.; Bota, D.; Piccioni, D.; Elder, B.; Engh, J.; Kaptain, G.J.; Landolfi, J.; Cloughesy, T.F. OS09.6 Long-term follow-up data from 126 patients with recurrent high grade glioma from three Phase 1 trials of Toca 511 and Toca FC: Update and justification for a Phase 2/3 trial. Neuro Oncol. 2017, 19 (Suppl. 3), iii19. [Google Scholar] [CrossRef][Green Version]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of rQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Ther. Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef]
- Ghonime, M.G.; Cassady, K.A. Combination Therapy Using Ruxolitinib and Oncolytic HSV Renders Resistant MPNSTs Susceptible to Virotherapy. Cancer Immunol. Res. 2018, 6, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.C.; Parker, J.N.; Gillespie, G.Y.; Lakeman, F.D.; Meleth, S.; Markert, J.M.; Cassady, K.A. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther. 2007, 14, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Cassady, K.A.; Bauer, D.F.; Roth, J.; Chambers, M.R.; Shoeb, T.; Coleman, J.; Prichard, M.; Gillespie, G.Y.; Markert, J.M. Pre-clinical Assessment of C134, a Chimeric Oncolytic Herpes Simplex Virus, in Mice and Non-human Primates. Mol. Ther. Oncolytics 2017, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.-M.; Guz-Montgomery, K.; Saha, D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells 2020, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M.; Calcedo, R.; Wilson, J.M.; Lin-Gibson, S.; Rogers, K.C.; et al. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef]
- Zadeh, G.; Lang, F.; Daras, M.; Cloughesy, T.; Colman, H.; Ong, S.; Ramakrishna, R.; Vogelbaum, M.; Groves, M.; Nassiri, F.; et al. ATIM-24. Interim results of a phase II multicenter stydy of the conditionally replicative oncolytic adenovirus DNX-2401 with pembrolizumab (KEYTRUDA) for recurrent glioblastoma; captive study (KEYNOTE-192). Neuro Oncol. 2018, 20, vi6. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Somerville, R.P.; Lu, T.; Yang, J.C.; Sherry, R.M.; Feldman, S.A.; McIntyre, L.; Bot, A.; Rossi, J.; Lam, N.; et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol. Ther. 2017, 25, 2245–2253. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Gilham, D.E.; Debets, R.; Pule, M.; Hawkins, R.E.; Abken, H. CAR–T cells and solid tumors: Tuning T cells to challenge an inveterate foe. Trends Mol. Med. 2012, 18, 377–384. [Google Scholar] [CrossRef]
- Li, G.; Wong, A.J. EGF receptor variant III as a target antigen for tumor immunotherapy. Expert Rev. Vaccines 2008, 7, 977–985. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Suki, D.; Yang, D.; Shi, W.; Aldape, K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 2005, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- O’rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa098. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor–transduced T Cells Targeting EGFRvIII in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol. 2022, 24, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Ieguchi, K.; Maru, Y. Roles of EphA1/A2 and ephrin-A1 in cancer. Cancer Sci. 2019, 110, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Debinski, W. The EphA2 Receptor and EphrinA1 Ligand in Solid Tumors: Function and Therapeutic Targeting. Mol. Cancer Res. 2008, 6, 1795–1806. [Google Scholar] [CrossRef]
- Lin, Q.; Ba, T.; Ho, J.; Chen, D.; Cheng, Y.; Wang, L.; Xu, G.; Xu, L.; Zhou, Y.; Wei, Y.; et al. First-in-Human Trial of EphA2-Redirected CAR T-Cells in Patients with Recurrent Glioblastoma: A Preliminary Report of Three Cases at the Starting Dose. Front. Oncol. 2021, 11, 694941. [Google Scholar] [CrossRef]
- Cobbs, C.S.; Soroceanu, L.; Denham, S.; Zhang, W.; Kraus, M.H. Modulation of Oncogenic Phenotype in Human Glioma Cells by Cytomegalovirus IE1–Mediated Mitogenicity. Cancer Res 2008, 68, 724–730. [Google Scholar] [CrossRef]
- Schuessler, A.; Smith, C.; Beagley, L.; Boyle, G.M.; Rehan, S.; Matthews, K.; Jones, L.; Crough, T.; Dasari, V.; Klein, K.; et al. Autologous T-cell Therapy for Cytomegalovirus as a Consolidative Treatment for Recurrent Glioblastoma. Cancer Res. 2014, 74, 3466–3476. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-Specific T Cells Target Primary Glioblastoma Stem Cells and Induce Regression of Autologous Experimental Tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Ellingson, B.M.; Sampson, J.; Achrol, A.S.; Aghi, M.K.; Bankiewicz, K.; Wang, C.; Bexon, M.; Brem, S.; Brenner, A.; Chowdhary, S.; et al. Modified RANO, immunotherapy RANO, and standard RANO response to convection-enhanced delivery of IL4R-targeted immunotoxin MDNA55 in recurrent glioblastoma. Clin. Cancer Res. 2021, 27, 3916–3925. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Wang, S.; Chawla, S.; Abdullah, K.; Desai, A.; Maloney, E.; Brem, S. Multiparametric MRI assessment of response to convection-enhanced intratumoral delivery of MDNA55, an interleukin-4 receptor targeted immunotherapy, for recurrent glioblastoma. Surg. Neurol. Int. 2021, 12, 337. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Van Den Bent, M.; Gan, H.K.; Reardon, D.A.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients withEGFR-amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro Oncol. 2019, 21, 106–114. [Google Scholar] [CrossRef]
- Bent, M.V.D.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro Oncol. 2020, 22, 684–693. [Google Scholar] [CrossRef]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs. Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010, 12, 871–881. [Google Scholar] [CrossRef]
- Chandramohan, V.; Pegram, C.N.; Piao, H.; Szafranski, S.E.; Kuan, C.-T.; Pastan, I.H.; Bigner, D.D. Production and quality control assessment of a GLP-grade immunotoxin, D2C7-(scdsFv)-PE38KDEL, for a phase I/II clinical trial. Appl. Microbiol. Biotechnol. 2016, 101, 2747–2766. [Google Scholar] [CrossRef]
- Bota, D.A.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.-T.; Fu, B.D.; Hsu, F.P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: Interim results and correlations with CD4+T-lymphocyte counts. CNS Oncol. 2018, 7, CNS22. [Google Scholar] [CrossRef]
- Eoli, M.; Corbetta, C.; Anghileri, E.; Di Ianni, N.; Milani, M.; Cuccarini, V.; Musio, S.; Paterra, R.; Frigerio, S.; Nava, S.; et al. Expansion of effector and memory T cells is associated with increased survival in recurrent glioblastomas treated with dendritic cell immunotherapy. Neuro Oncol. Adv. 2019, 1, vdz022. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Omofoye, O.A.; Rudnick, J.D.; Kim, S.; Tighiouart, M.; Phuphanich, S.; Wang, H.; Mazer, M.; Ganaway, T.; Chu, R.M.; et al. A Phase I Study of Autologous Dendritic Cell Vaccine Pulsed with Allogeneic Stem-like Cell Line Lysate in Patients with Newly Diagnosed or Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination with Extension of Survival among Patients with Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Hashiba, T.; Izumoto, S.; Kagawa, N.; Suzuki, T.; Hashimoto, N.; Maruno, M.; Yoshimine, T. Expression of WT1 Protein and Correlation with Cellular Proliferation in Glial Tumors. Neurol. Med. Chir. 2007, 47, 165–170. [Google Scholar] [CrossRef]
- Izumoto, S.; Tsuboi, A.; Oka, Y.; Suzuki, T.; Hashiba, T.; Kagawa, N.; Hashimoto, N.; Maruno, M.; Elisseeva, O.A.; Shirakata, T.; et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J. Neurosurg. 2008, 108, 963–971. [Google Scholar] [CrossRef]
- Narita, Y.; Arakawa, Y.; Yamasaki, F.; Nishikawa, R.; Aoki, T.; Kanamori, M.; Nagane, M.; Kumabe, T.; Hirose, Y.; Ichikawa, T.; et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro Oncol. 2018, 21, 348–359. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex–96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro Oncol. 2014, 16, 274–279. [Google Scholar] [CrossRef]
- Satoh, K.; Kaneko, K.; Hirota, M.; Masamune, A.; Satoh, A.; Shimosegawa, T. Expression of Survivin Is Correlated with Cancer Cell Apoptosis and Is Involved in the Development of Human Pancreatic Duct Cell Tumors. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2001, 92, 271–278. [Google Scholar] [CrossRef]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef]
- Wick, W.; Idbaih, A.; Tabatabai, G.; Vieito, M.; Stradella, A.; Ghiringhelli, F.; Burger, M.C.; Mildenberger, I.; Herrlinger, U.; Renovanz, M.; et al. EO2401, a novel microbiome-derived therapeutic vaccine for patients with recurrent glioblastoma: ROSALIE study. J. Clin. Oncol. 2022, 40, 2034. [Google Scholar] [CrossRef]
- Wick, W.; Wick, A.; Sahm, F.; Riehl, D.; von Deimling, A.; Bendszus, M.; Kickingereder, P.; Beckhove, P.; Winnenthal, F.H.S.; Jungk, C.; et al. ATIM-35. VXM01 Phase I Study in patients with progressive glioblastioma—Final results. Neuro Oncol. 2018, 20 (Suppl. 6), vi9. [Google Scholar] [CrossRef]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, J.; Kwidzinski, E.; Brandt, C.; Mahlo, J.; Richter, D.; Bechmann, I. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J. Leukoc. Biol. 2006, 80, 797–801. [Google Scholar] [CrossRef]
- Cserr, H.F.; Knopf, P.M. Cervical lymphatics, the blood-brain barrier and the immunoreactivity of the brain: A new view. Immunol. Today 1992, 13, 507–512. [Google Scholar] [CrossRef]
- Bradbury, M.W.; Westrop, R.J. Factors influencing exit of substances from cerebrospinal fluid into deep cervical lymph of the rabbit. J. Physiol. 1983, 339, 519–534. [Google Scholar] [CrossRef]
- Bradbury, M.W.; Cole, D.F. The role of the lymphatic system in drainage of cerebrospinal fluid and aqueous humour. J. Physiol. 1980, 299, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood–Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; Alexe, G.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Muscat, A.M.; Wong, N.C.; Drummond, K.J.; Algar, E.M.; Khasraw, M.; Verhaak, R.; Field, K.; Rosenthal, M.A.; Ashley, D.M. The evolutionary pattern of mutations in glioblastoma reveals therapy-mediated selection. Oncotarget 2017, 9, 7844–7858. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Anderson, K.J.; Courtois, E.T.; Gujar, A.D.; Barthel, F.P.; Varn, F.S.; Luo, D.; Seignon, M.; Yi, E.; Kim, H.; et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat. Genet. 2021, 53, 1456–1468. [Google Scholar] [CrossRef]
- El-Sayes, N.; Vito, A.; Mossman, K. Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy. Cancers 2021, 13, 806. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jiménez-Sánchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235.e21. [Google Scholar] [CrossRef]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Kornete, M.; Joyce, J.A.; Escobar, G.; Escobar, A.; Ascui, G.; Tempio, F.I.; Ortiz, M.C.; Pérez, C.A.; López, M.N.; et al. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy 2019, 11, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Brown, M.C.; Zhang, G.; Stevenson, K.; Mohme, M.; Kornahrens, R.; Bigner, D.D.; Ashley, D.M.; López, G.Y.; Gromeier, M. Polio virotherapy targets the malignant glioma myeloid infiltrate with diffuse microglia activation engulfing the CNS. Neuro Oncol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.; McDowall, C.; Sanchez-Perez, L.; Osorio, C.; Duncker, P.C.; Briley, A.; Swartz, A.M.; Herndon, J.E.; Yu, Y.-R.A.; McLendon, R.E.; et al. Immunotoxin-αCD40 therapy activates innate and adaptive immunity and generates a durable antitumor response in glioblastoma models. Sci. Transl. Med. 2023, 15, eabn5649. [Google Scholar] [CrossRef]
- Georgoudaki, A.-M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Sengupta, S.; Han, Y.; Lesniak, M.S. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol. 2011, 13, 1308–1323. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Ayasoufi, K.; Pfaller, C.K.; Evgin, L.; Khadka, R.H.; Tritz, Z.P.; Goddery, E.N.; Fain, C.E.; Yokanovich, L.T.; Himes, B.T.; Jin, F.; et al. Brain cancer induces systemic immunosuppression through release of non-steroid soluble mediators. Brain 2020, 143, 3629–3652. [Google Scholar] [CrossRef]
- Lorrey, S.J.; Polania, J.W.; Wachsmuth, L.P.; Hoyt-Miggelbrink, A.; Tritz, Z.P.; Edwards, R.; Wolf, D.M.; Johnson, A.J.; Fecci, P.E.; Ayasoufi, K. Systemic immune derangements are shared across various CNS pathologies and reflect novel mechanisms of immune privilege. Neuro Oncol. Adv. 2023, 5, vdad035. [Google Scholar] [CrossRef] [PubMed]
- Advani, S.; Sibley, G.; Song, P.; Hallahan, D.; Kataoka, Y.; Roizman, B.; Weichselbaum, R. Enhancement of replication of genetically engineered herpes simplex viruses by ionizing radiation: A new paradigm for destruction of therapeutically intractable tumors. Gene Ther. 1998, 5, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.D.; Kataoka, Y.; Advani, S.; Chung, S.M.; Arani, R.B.; Gillespie, G.Y.; Whitley, R.J.; Markert, J.M.; Roizman, B.; Weichselbaum, R.R. Ionizing radiation improves survival in mice bearing intracranial high- grade gliomas injected with genetically modified herpes simplex virus. Clin. Cancer Res. 1999, 5, 1517–1522. Available online: https://profiles.wustl.edu/en/publications/ionizing-radiation-improves-survival-in-mice-bearing-intracranial (accessed on 10 June 2023). [PubMed]
- Advani, S.J.; Markert, J.M.; Sood, R.F.; Samuel, S.; Gillespie, G.Y.; Shao, M.Y.; Roizman, B.; Weichselbaum, R.R. Increased oncolytic efficacy for high-grade gliomas by optimal integration of ionizing radiation into the replicative cycle of HSV-1. Gene Ther. 2011, 18, 1098–1102. [Google Scholar] [CrossRef]
- Mezhir, J.J.; Advani, S.J.; Smith, K.D.; Darga, T.E.; Poon, A.P.; Schmidt, H.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Ionizing Radiation Activates Late Herpes Simplex Virus 1 Promoters via the p38 Pathway in Tumors Treated with Oncolytic Viruses. Cancer Res 2005, 65, 9479–9484. [Google Scholar] [CrossRef][Green Version]
- Advani, S.J.; Mezhir, J.J.; Roizman, B.; Weichselbaum, R.R. ReVOLT: Radiation-enhanced viral oncolytic therapy. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 637–646. [Google Scholar] [CrossRef]
- Herrera, F.G.; Bourhis, J.; Coukos, G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J. Clin. 2017, 67, 65–85. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Therapy | Highest Phase Achieved | Trial | |
|---|---|---|---|
| Immune Checkpoint Blockade | PD-1 | III | CheckMate 143 |
| PD-L1 | II | NCT02336165, GliAvAx | |
| CTLA-4 | I | CheckMate 143 | |
| Herpes Simplex Virus | G207 | IIb | NCT00028158 |
| 1716 | I | NCT02031965 | |
| G47Δ | II | UMIN000015995 | |
| Poliovirus | PVSRIPO | II | NCT02986178 |
| Parvovirus | H-1PV | I | ParvOryx01 |
| Adenovirus | ONYX-015 | I | Chiocca et al. [45] |
| Delta24-RGD | Ib | Lang et al. [75] | |
| Newcastle Virus | NDV-HUJ | II | Freeman et al. [83] |
| Reovirus | Reolysin | I | 2011-005635-10 |
| Retrovirus | Toca 511 | III | NCT02414165) |
| CART | EGFRvIII | I | NCT02209376, NCT01454596 |
| IL13Rα2 | III | Kunwar et al. [118] | |
| EphA2-Redirected | Pilot | NCT03423992 | |
| Autologous CMV | I | ACTRN12609000338268 | |
| HER2 | I | NCT01109095 | |
| Immunotoxins | MDNA55 | II | MDNA55-05 |
| Depatux-M | II | INTELLANCE 2/EORTC 1410 | |
| Pseudomonas exotoxin A | III | PRECISE Trial NCT 00076986 | |
| Cancer Cell Vaccine | ERC1671 | II | NCT01903330 |
| Dendritic cell | III | NCT00045968 | |
| WT1 | II | Izumoto et al. [25] | |
| PPV | III | Narita et al. [126] | |
| Heat-shock protein peptide complex-96 | II | NCT00293423 | |
| SurVaxM | I | NCT01250470 | |
| EO2401 | II | NCT04116658 | |
| VXM01 | I | NCT02718443 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivet, M.M.; Brown, M.C.; Reitman, Z.J.; Ashley, D.M.; Grant, G.A.; Yang, Y.; Markert, J.M. Clinical Applications of Immunotherapy for Recurrent Glioblastoma in Adults. Cancers 2023, 15, 3901. https://doi.org/10.3390/cancers15153901
Olivet MM, Brown MC, Reitman ZJ, Ashley DM, Grant GA, Yang Y, Markert JM. Clinical Applications of Immunotherapy for Recurrent Glioblastoma in Adults. Cancers. 2023; 15(15):3901. https://doi.org/10.3390/cancers15153901
Chicago/Turabian StyleOlivet, Meagan Mandabach, Michael C. Brown, Zachary J. Reitman, David M. Ashley, Gerald A. Grant, Yuanfan Yang, and James M. Markert. 2023. "Clinical Applications of Immunotherapy for Recurrent Glioblastoma in Adults" Cancers 15, no. 15: 3901. https://doi.org/10.3390/cancers15153901
APA StyleOlivet, M. M., Brown, M. C., Reitman, Z. J., Ashley, D. M., Grant, G. A., Yang, Y., & Markert, J. M. (2023). Clinical Applications of Immunotherapy for Recurrent Glioblastoma in Adults. Cancers, 15(15), 3901. https://doi.org/10.3390/cancers15153901