
Transcriptomic Maps of Colorectal Liver Metastasis: Machine Learning of Gene Activation Patterns and Epigenetic Trajectories in Support of Precision Medicine

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Gene Expression Data of Colon Liver Metastases (CRLM)
2.2. SOM Portrayal and Downstream Functional Analysis
2.3. Prognostic Maps
2.4. oposSOM Browser of Livermet-Transcriptomes
3. Results
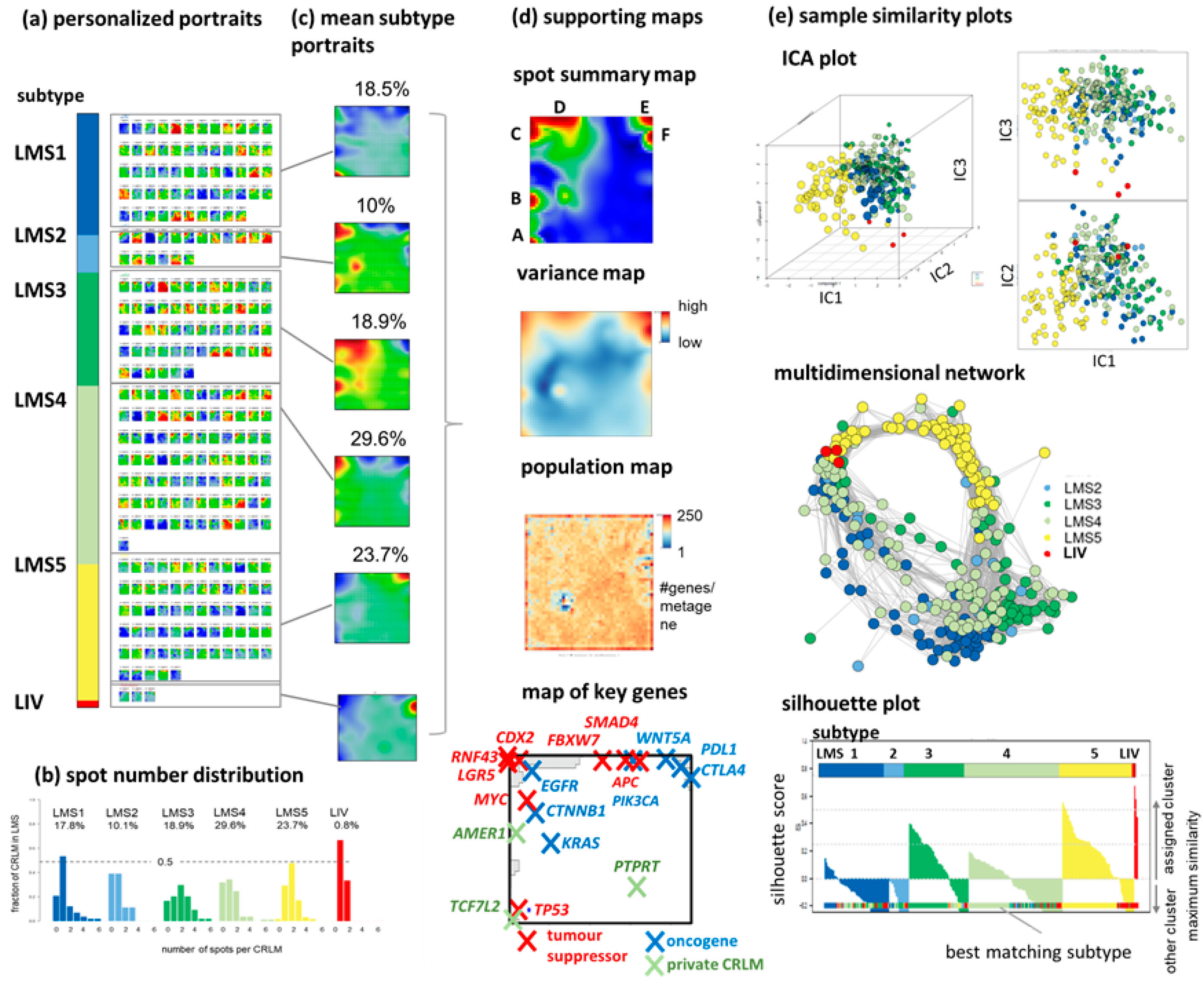
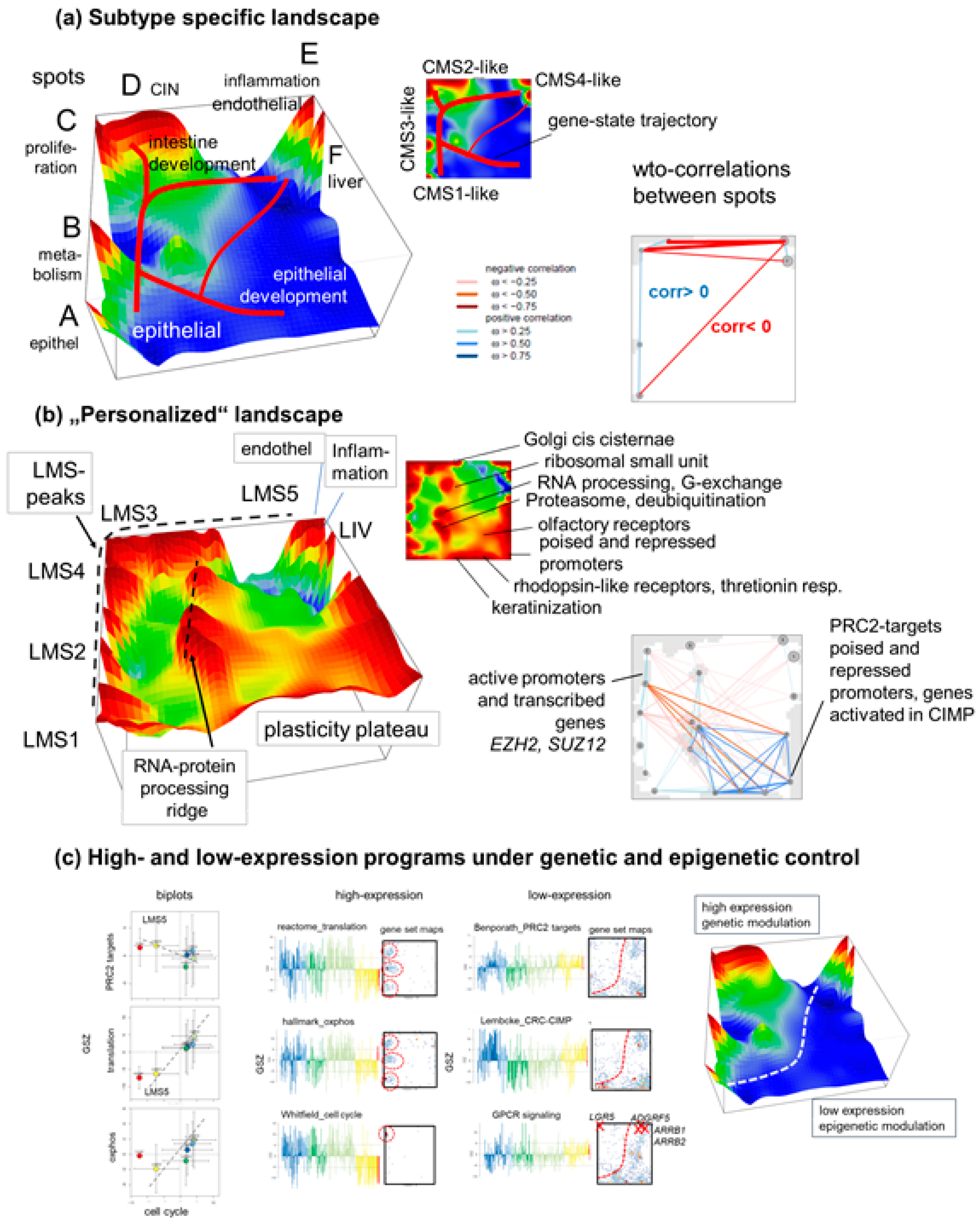
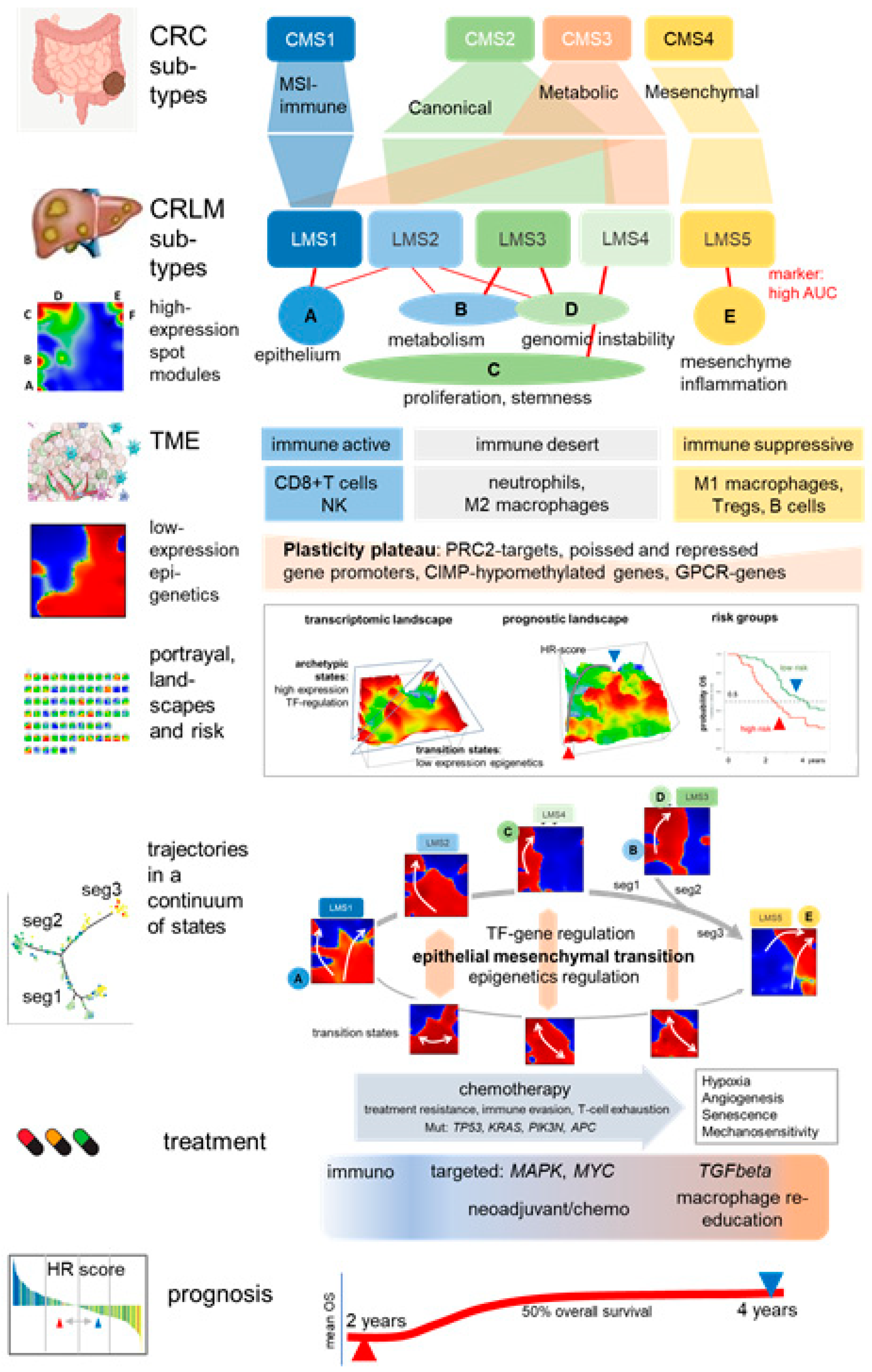
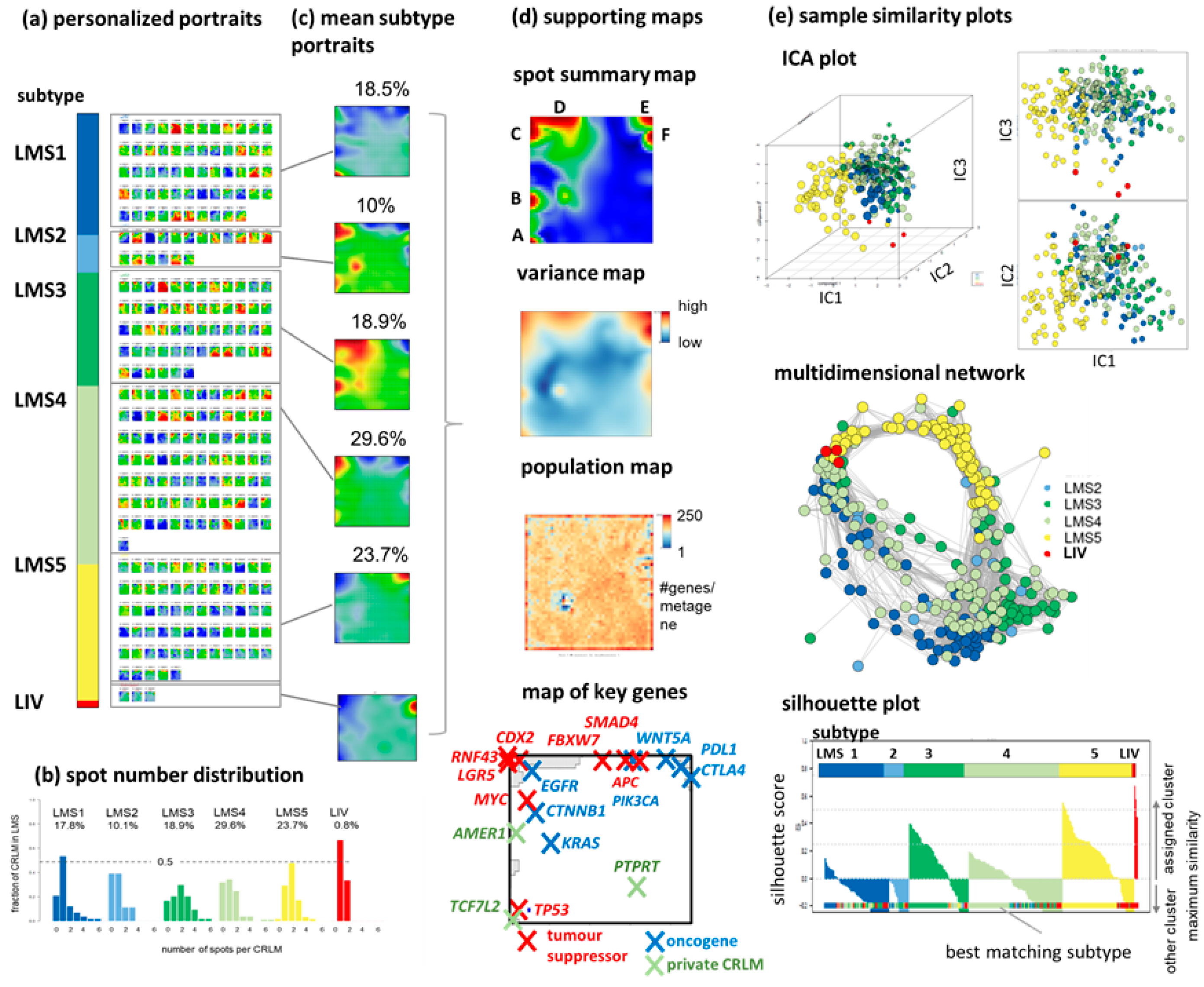
3.1. SOM Portrayal Deciphers Liver Metastases Subtypes (LMS)
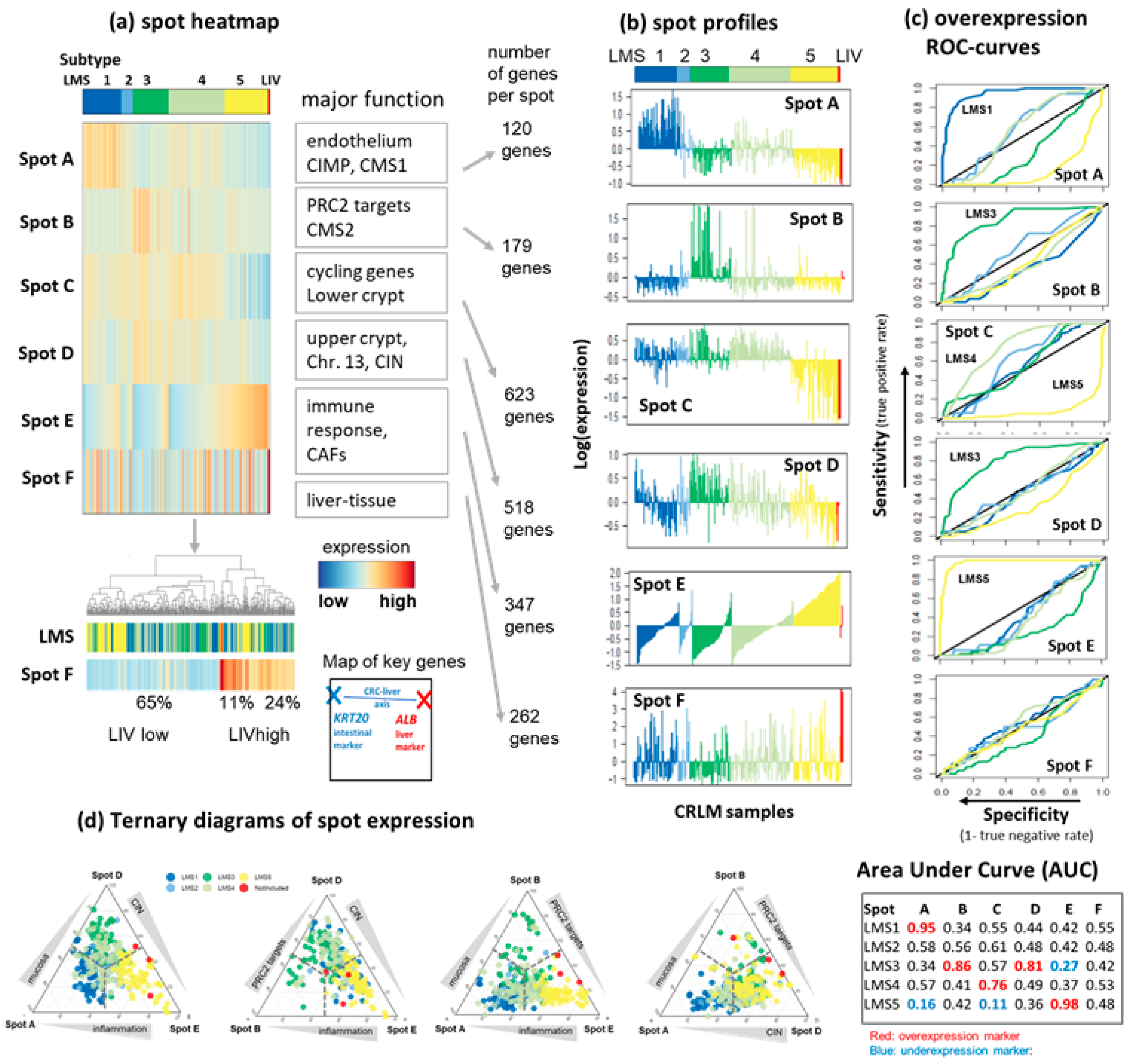
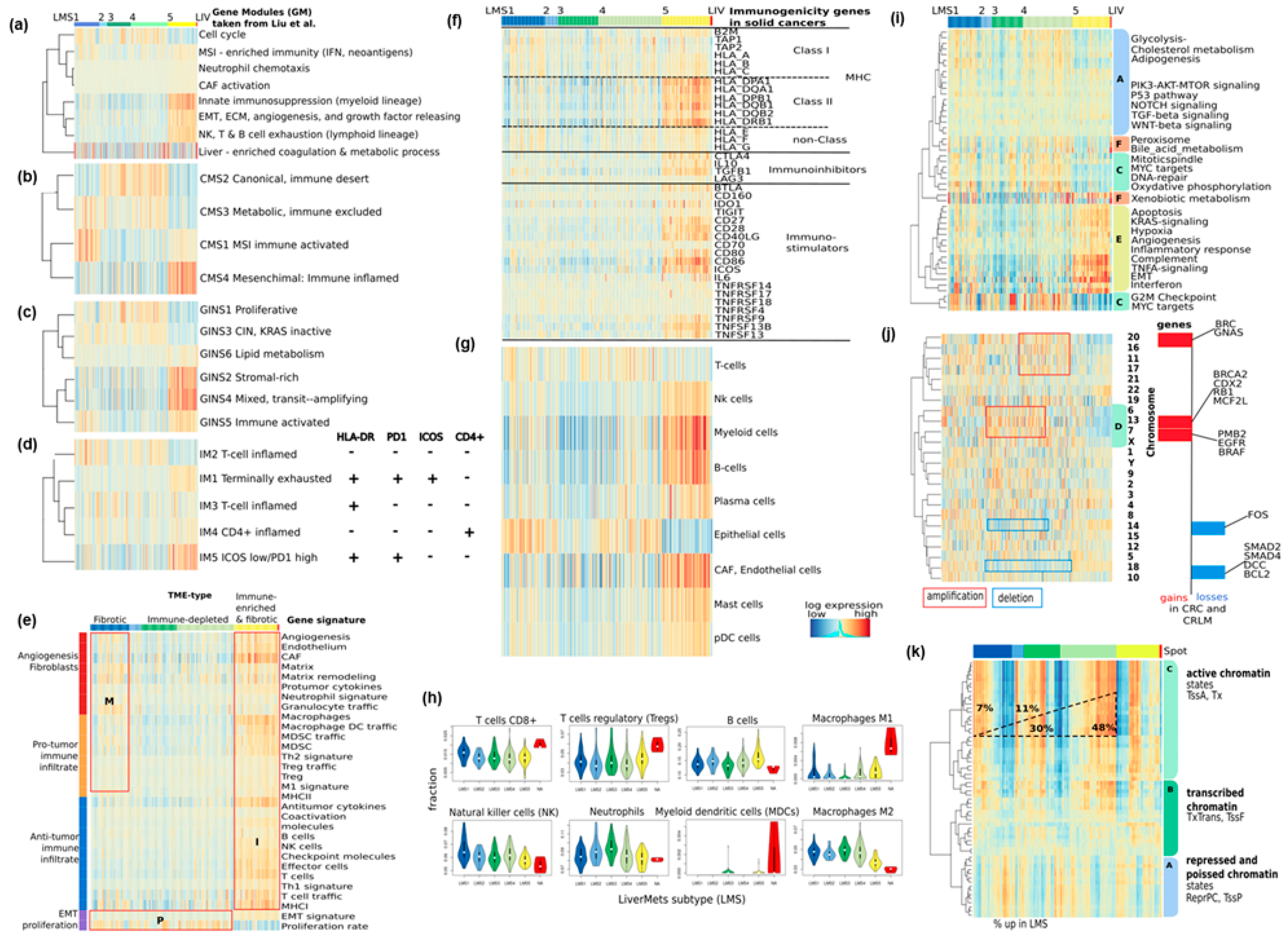
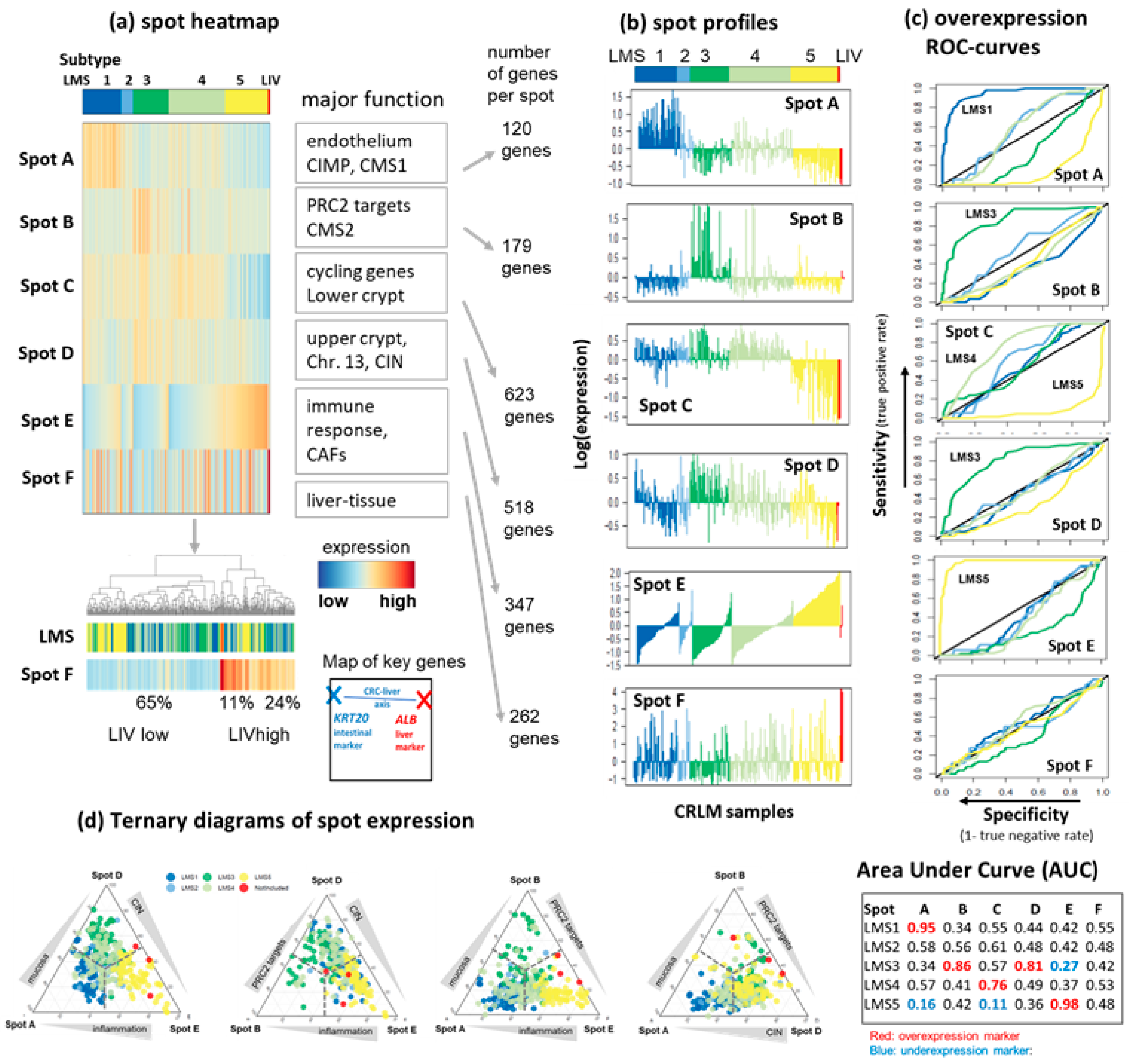
3.2. LMS Are Governed by Six Major Modules of Co-Regulated Genes
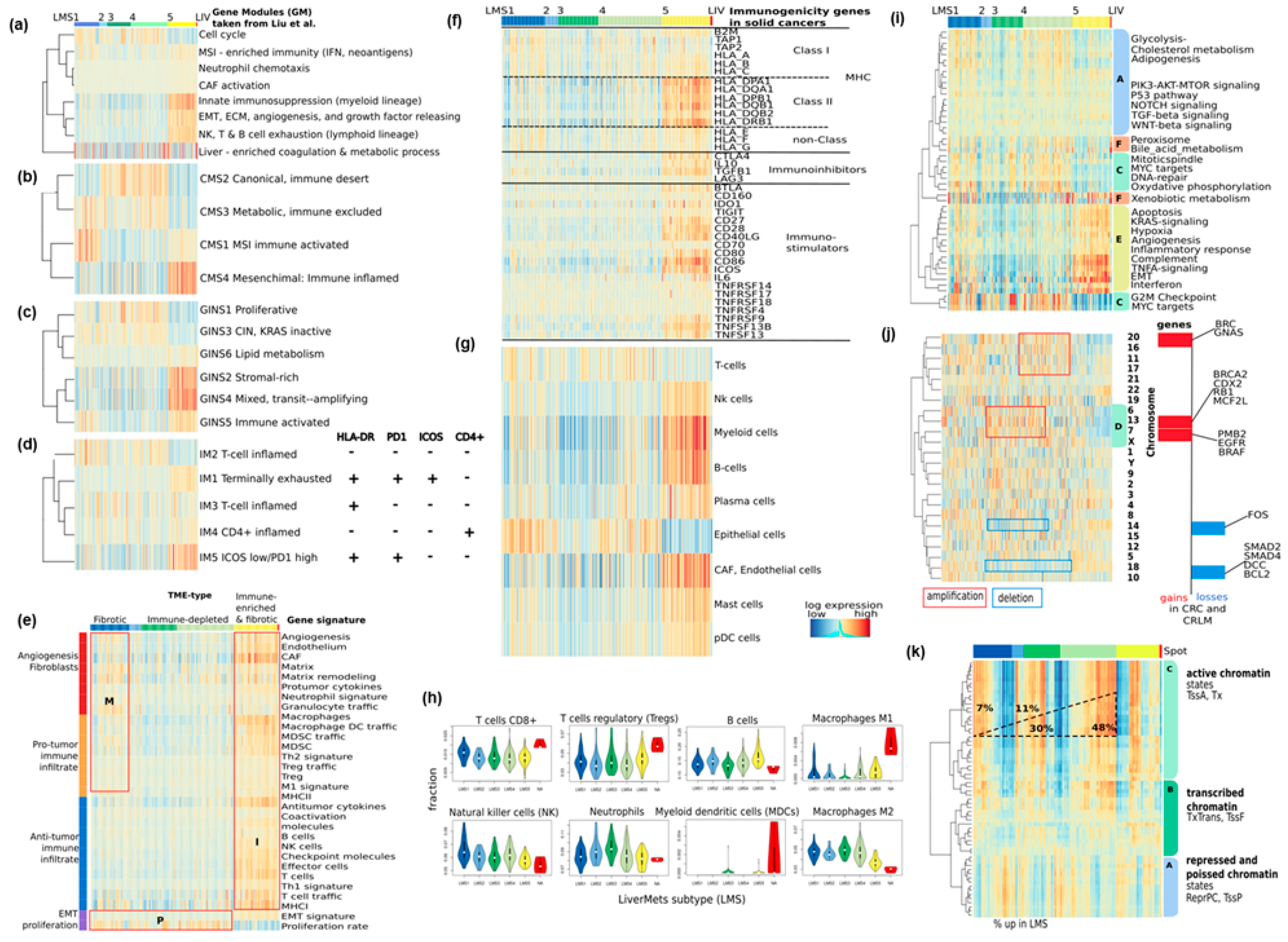
3.3. Functional and TME Context and Relation to Primary CRC Characteristics
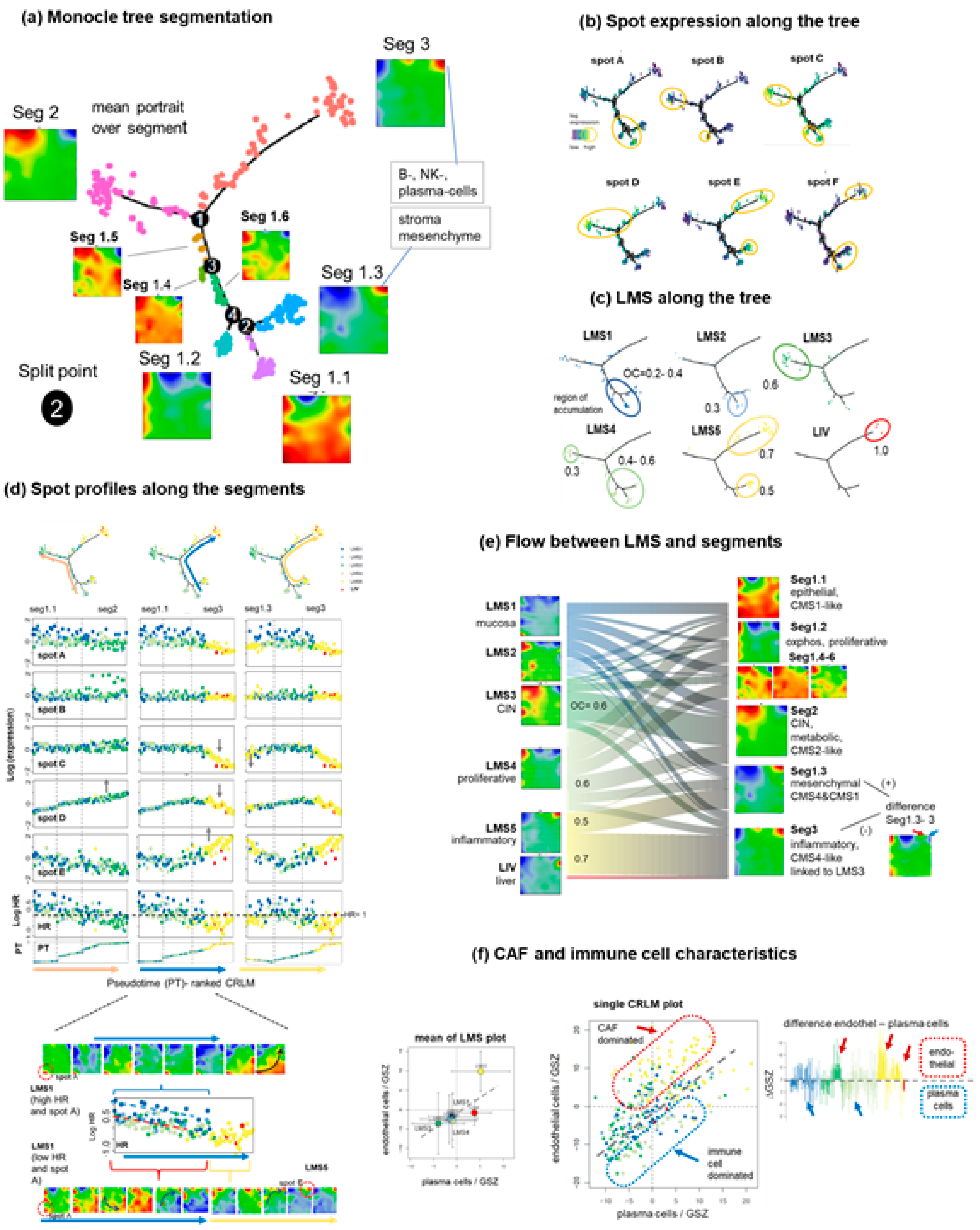
3.4. Trajectory Inference Indicates Continuous Alterations of Cellular States along Developmental Paths and Gradual Changes of the TME
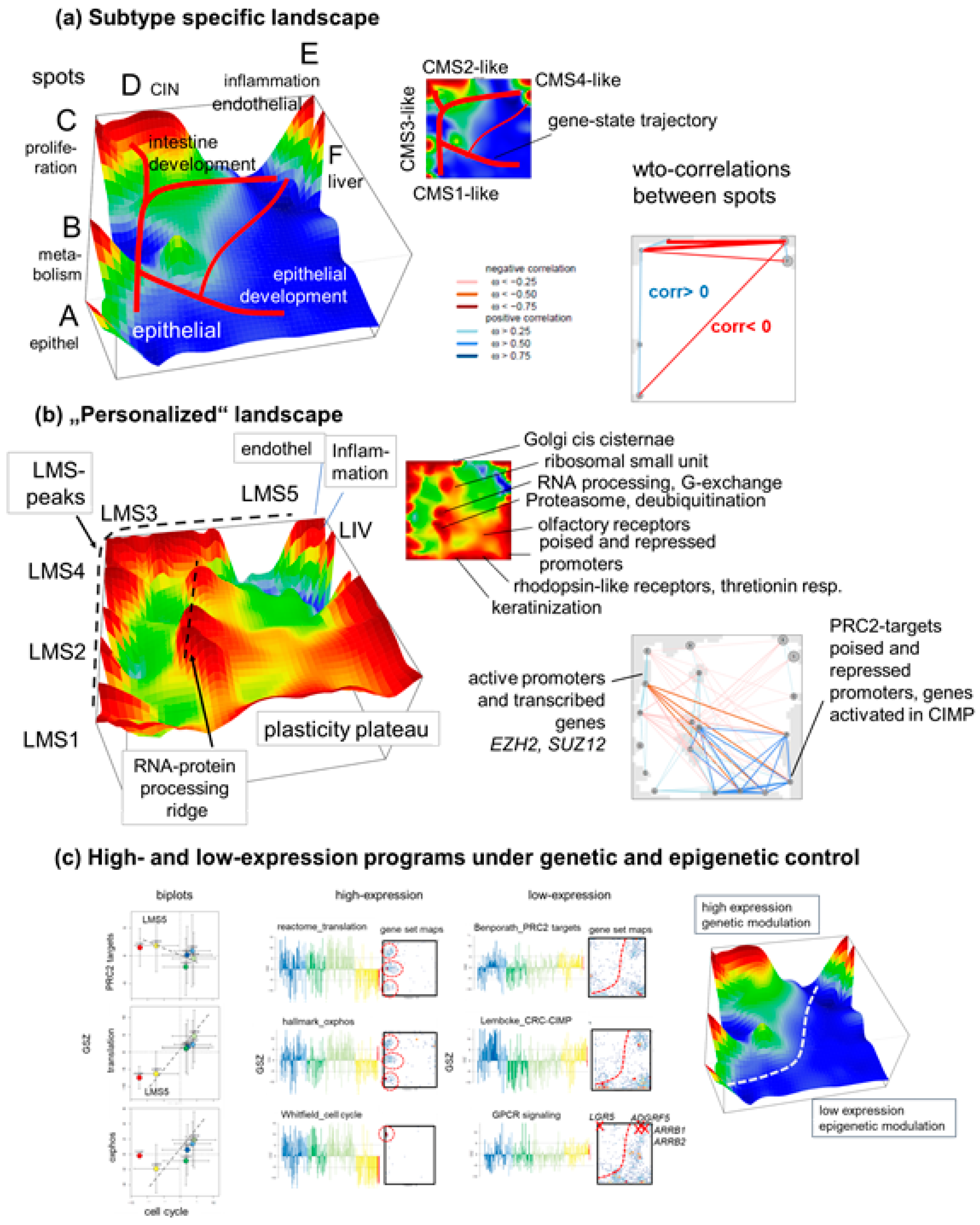
3.5. High-Resolution Expression Cartography Deciphers an Interplay between Genetic and Epigenetic Regulation of Metastasis
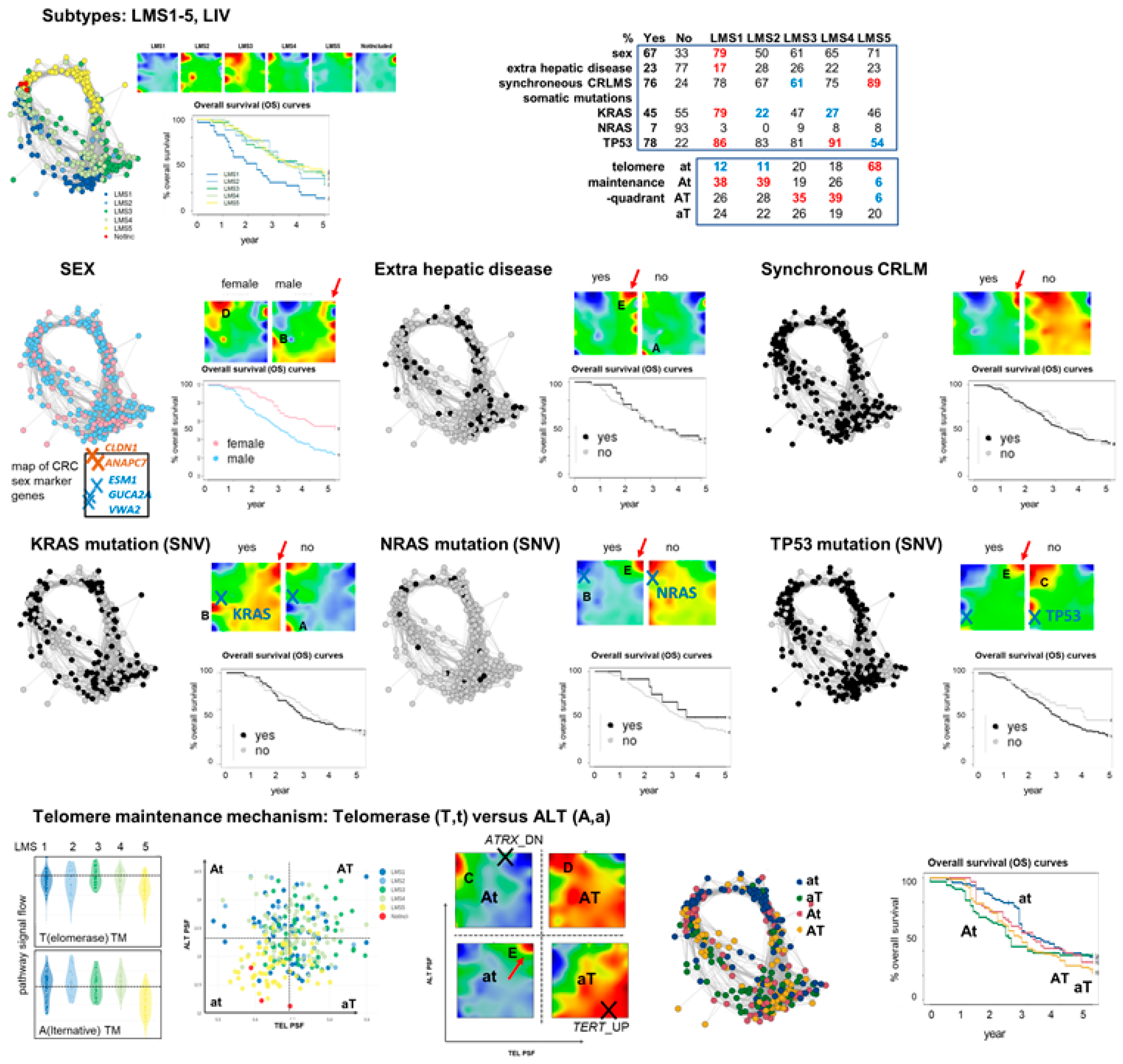
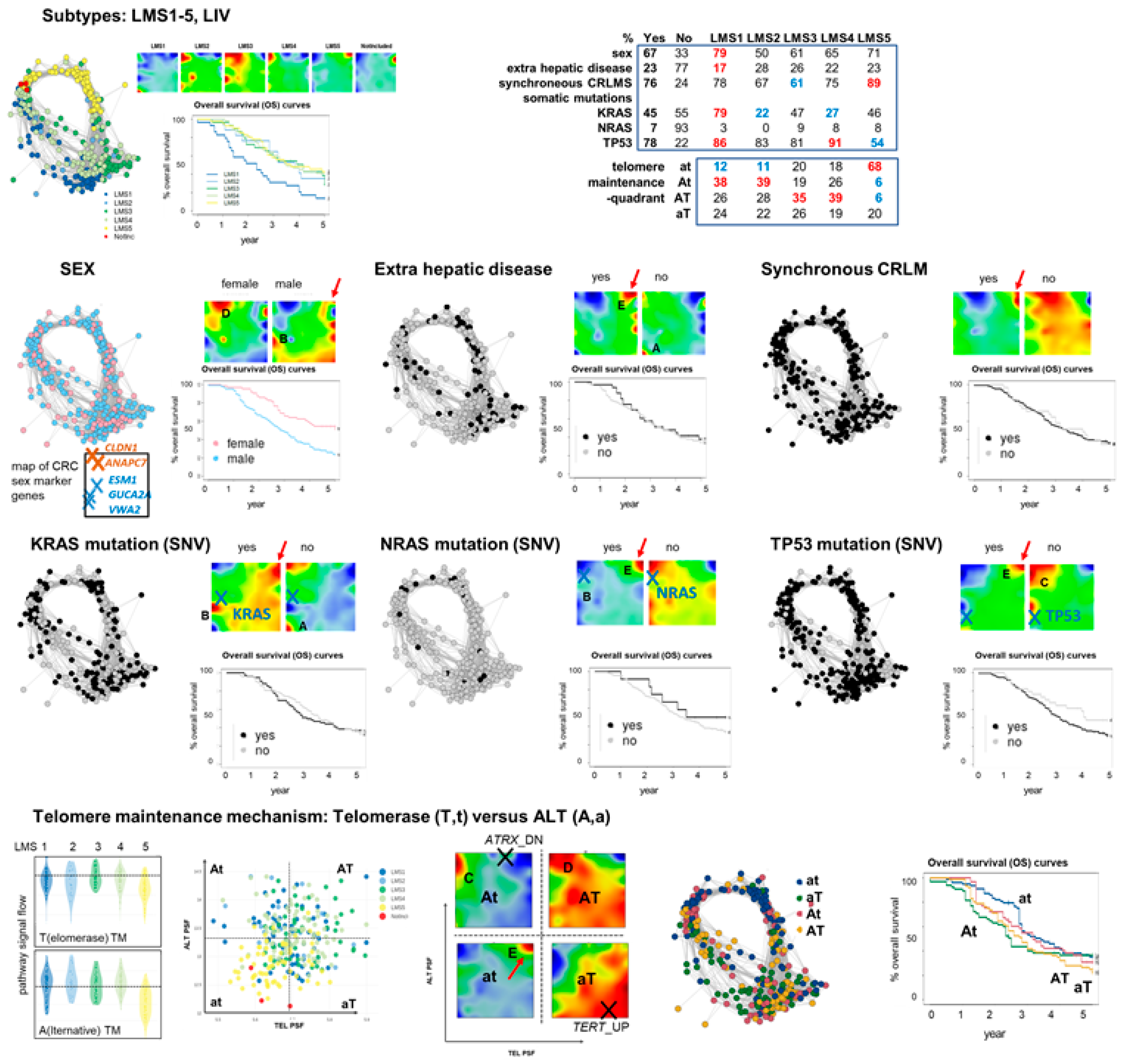
3.6. Portrayal of Clinical, Mutational, and Telomere Maintenance Characteristics
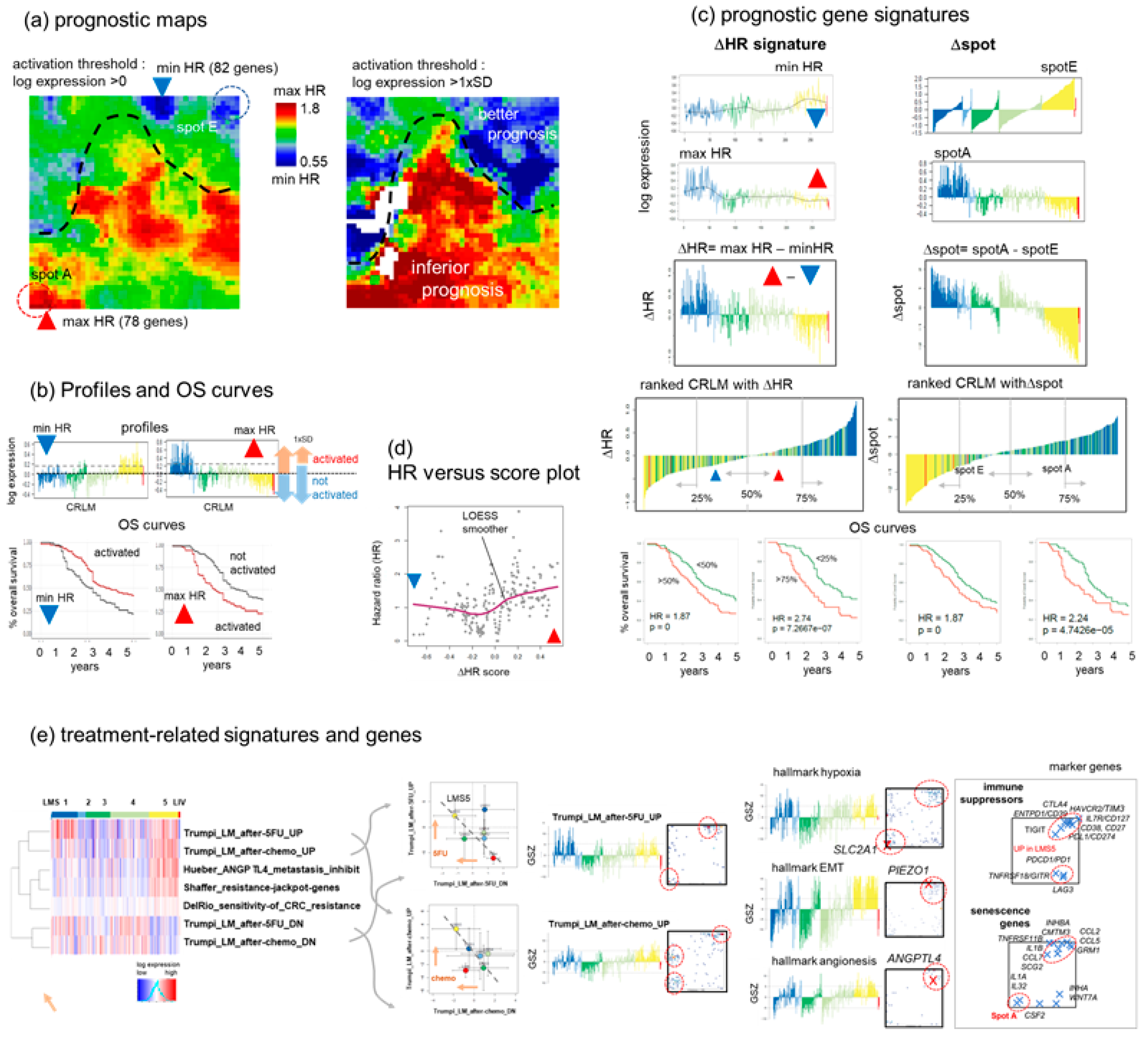
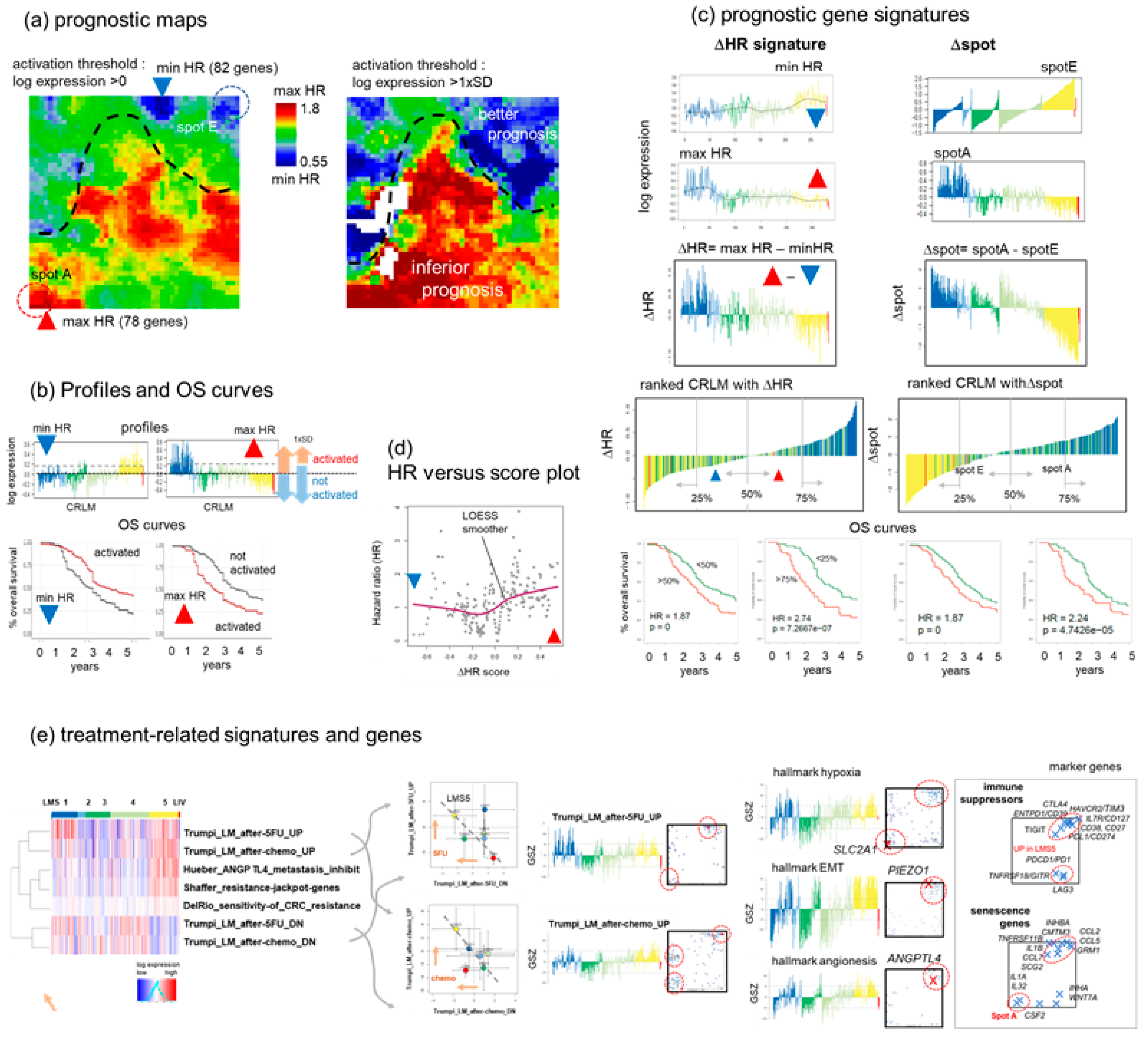
3.7. Prognostic Maps Provide an HR-Score along the EMT-Axis Related to Treatment
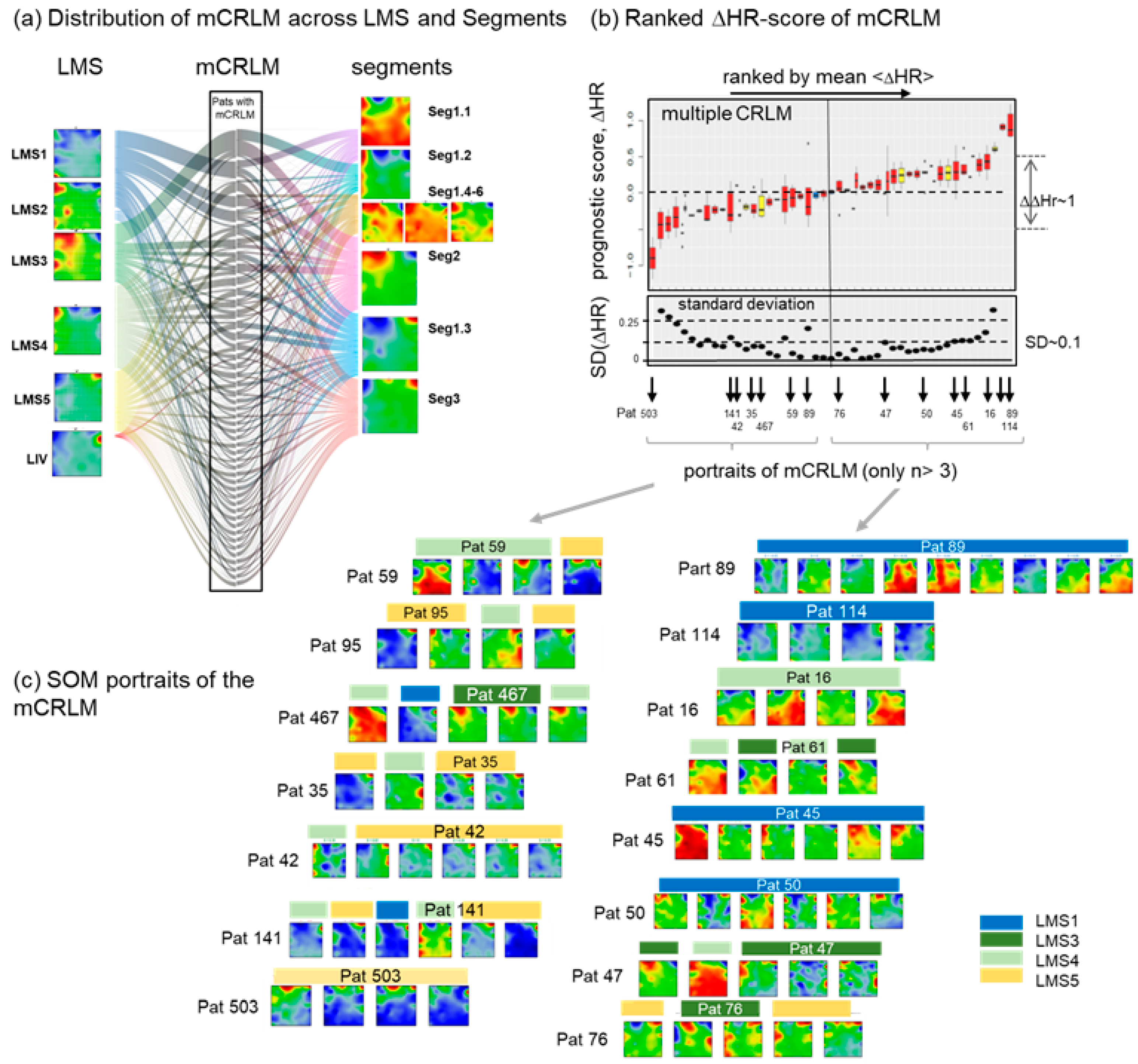
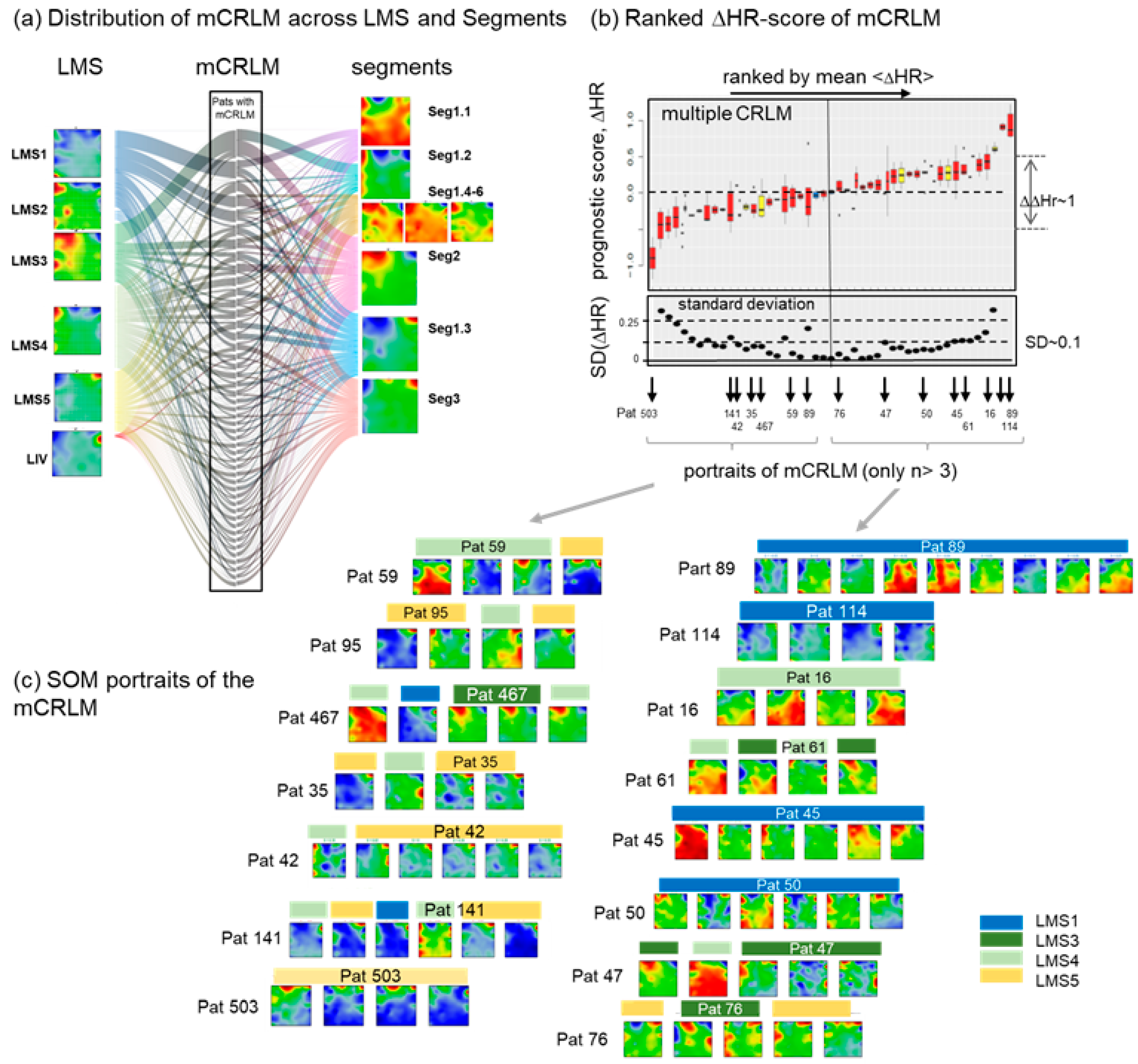
3.8. Intra-Patient Heterogeneity of Metastasis
4. Discussion
4.1. Subtyping Stratifies CRLM along Cancer Hallmarks and TME Education
4.2. Trajectory Inference and Personalized Analysis Discover a Hidden Universe of Continuous Epigenetic States Shaping Metastasis
4.3. Towards Precision Diagnostics and Treatment Decisions of CRLM
4.4. Limitations and Open Points
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | Alternative lengthening of telomeres |
| AUC | Area Under the ROC Curve |
| CAF | Cancer-associated fibroblast |
| CIN | Chromosomal instability |
| CRC | Colorectal cancer |
| CRLM | Colorectal liver metastases |
| CMS | Consensus molecular subtyping |
| CNV | Copy number variations |
| CIMP | CPG island methylation phenotype |
| EPC | Endothelial progenitor cell |
| ECM | Extracellular matrix |
| FU | Fluorouracil |
| GM | Gene module |
| GO | Gene ontology |
| GINS | Gene-interaction-perturbation-network-based subtyping |
| GPCR | G-protein coupled receptor |
| HR | Hazard ratio |
| HepSC | Hepatic stellate cell |
| HGP | Histopathological growth patterns |
| ICI | Immune checkpoint inhibitor |
| ICA | Independent component analysis |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LIV | Liver |
| LM | Liver metastases |
| LMS | Liver metastasis subtype |
| LSEC | Liver sinusoidal endothelial cell |
| MSI | Microsatellite instable |
| NK | Natural killer |
| OS | Overall survival |
| OC | lOverlap coefficient |
| PSF | Pathway signal flow |
| PC | Plasma cells |
| pDC | Plasmacytoid dendritic cells |
| PRC2 | Polycomb repressive complex 2 |
| PCA | Principal component analysis |
| PT | Pseudotime |
| ROC | Receiver operator characteristic |
| SOM | Self-organizing map |
| scRNAseq | Single cell RNA sequencing |
| TAF | Tumor associated macrophages |
| TEL | Telomerase |
| TM | Telomere maintenance |
| TCGA | The cancer genome atlas |
| TF | Transcription factor |
| TME | Tumor microenvironment |
| UMAP | Uniform manifold approximation and projection for dimension reduction |
| UMI | Unique molecular identifier |
| WTO | Weighted topological overlap |
References
- Tsilimigras, D.I.; Brodt, P.; Clavien, P.A.; Muschel, R.J.; D’Angelica, M.I.; Endo, I.; Parks, R.W.; Doyle, M.; de Santibañes, E.; Pawlik, T.M. Liver metastases. Nat. Rev. Dis. Primers 2021, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Vanaclocha, F. The liver prometastatic reaction of cancer patients: Implications for microenvironment-dependent colon cancer gene regulation. Cancer Microenviron. 2011, 4, 163–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Qian, L.; Li, Z.; Li, Y.; Bai, J.; Zheng, B.; Chen, K.; Qiu, X.; Cai, G.; Wang, S.; et al. Integrated Multi-Omics Landscape of Liver Metastases. Gastroenterology 2023, 164, 407–423.e17. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cho, Y.B.; Hong, H.K.; Wu, S.; Ebert, P.J.; Bray, S.M.; Wong, S.S.; Ting, J.C.; Calley, J.N.; Whittington, C.F.; et al. Molecular dissection of CRC primary tumors and their matched liver metastases reveals critical role of immune microenvironment, EMT and angiogenesis in cancer metastasis. Sci. Rep. 2020, 10, 10725. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, S.H.; Eide, P.W.; Eilertsen, I.A.; Brunsell, T.H.; Berg, K.C.G.; Røsok, B.I.; Brudvik, K.W.; Bjørnbeth, B.A.; Guren, M.G.; Nesbakken, A.; et al. De novo transcriptomic subtyping of colorectal cancer liver metastases in the context of tumor heterogeneity. Genome Med. 2021, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Trumpi, K.; Ubink, I.; Trinh, A.; Djafarihamedani, M.; Jongen, J.M.; Govaert, K.M.; Elias, S.G.; van Hooff, S.R.; Medema, J.P.; Lacle, M.M.; et al. Neoadjuvant chemotherapy affects molecular classification of colorectal tumors. Oncogenesis 2017, 6, e357. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2022, 12, 134–153. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Levchenko, A. Epigenetics as a mediator of plasticity in cancer. Science 2023, 379, eaaw3835. [Google Scholar] [CrossRef]
- Waddington, C.H. The Strategy of the Genes; Routledge: Abingdon, UK, 2014. [Google Scholar]
- Zhang, Y.; Chen, F.; Creighton, C.J. Pan-cancer molecular subtypes of metastasis reveal distinct and evolving transcriptional programs. Cell Rep. Med. 2023, 4, 100932. [Google Scholar] [CrossRef]
- Willscher, E.; Hopp, L.; Kreuz, M.; Schmidt, M.; Hakobyan, S.; Arakelyan, A.; Hentschel, B.; Jones, D.T.W.; Pfister, S.M.; Loeffler, M.; et al. High-Resolution Cartography of the Transcriptome and Methylome Landscapes of Diffuse Gliomas. Cancers 2021, 13, 3198. [Google Scholar] [CrossRef]
- Loeffler-Wirth, H.; Kreuz, M.; Hopp, L.; Arakelyan, A.; Haake, A.; Cogliatti, S.B.; Feller, A.C.; Hansmann, M.-L.; Lenze, D.; Möller, P.; et al. A modular transcriptome map of mature B cell lymphomas. Genome Med. 2019, 11, 27. [Google Scholar] [CrossRef] [Green Version]
- Kunz, M.; Löffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 2018, 37, 6136–6151. [Google Scholar] [CrossRef]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Jühling, F.; Kerick, M.; Timmermann, B.; Siebert, S.; Grimm, C.; Nersisyan, L.; et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Mortensen, L.S.; Loeffler-Wirth, H.; Kosnopfel, C.; Krohn, K.; Binder, H.; Kunz, M. Single-cell trajectories of melanoma cell resistance to targeted treatment. Cancer Biol. Med. 2022, 19, 56–73. [Google Scholar] [CrossRef]
- Hopp, L.; Willscher, E.; Wirth-Loeffler, H.; Binder, H. Function Shapes Content: DNA-Methylation Marker Genes and their Impact for Molecular Mechanisms of Glioma. J. Cancer Res. Updates 2015, 4, 127–148. [Google Scholar] [CrossRef] [Green Version]
- Hopp, L.; Löffler-Wirth, H.; Binder, H. Epigenetic Heterogeneity of B-Cell Lymphoma: DNA Methylation, Gene Expression and Chromatin States. Genes 2015, 6, 812–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, H.; Löffler, M.; von Bergen, M.; Binder, H. Expression cartography of human tissues using self organizing maps. BMC Bioinform. 2011, 12, 306. [Google Scholar] [CrossRef] [Green Version]
- Binder, H.; Wirth, H. Analysis of large-scale OMIC data using Self Organizing Maps. In Encyclopedia of Information Science and Technology Third Edition; Khosrow-Pour, M., Ed.; IGI Global: Hershey, PA, USA, 2014; pp. 1642–1654. [Google Scholar]
- Wirth, H.; von Bergen, M.; Binder, H. Mining SOM expression portraits: Feature selection and integrating concepts of molecular function. BioData Min. 2012, 5, 18. [Google Scholar] [PubMed] [Green Version]
- Löffler-Wirth, H.; Kalcher, M.; Binder, H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on bioconductor. Bioinformatics 2015, 31, 3225–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, L.-H.; Liu, J.-W.; Huo, J.-P.; Luo, R.; Xu, R.-M.; He, C.; Li, Y.-Q.; Zhou, A.-J.; Huang, P.; Chen, Y.-Y.; et al. A single-cell atlas of liver metastases of colorectal cancer reveals reprogramming of the tumor microenvironment in response to preoperative chemotherapy. Cell Discov. 2021, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Loeffler-Wirth, H.; Reikowski, J.; Hakobyan, S.; Wagner, J.; Binder, H. oposSOM-Browser: An interactive tool to explore omics data landscapes in health science. BMC Bioinform. 2020, 21, 465. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Mendelaar, P.A.J.; Smid, M.; van Riet, J.; Angus, L.; Labots, M.; Steeghs, N.; Hendriks, M.P.; Cirkel, G.A.; van Rooijen, J.M.; Ten Tije, A.J.; et al. Whole genome sequencing of metastatic colorectal cancer reveals prior treatment effects and specific metastasis features. Nat. Commun. 2021, 12, 574. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Scott Shaffer, J.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Weng, S.; Dang, Q.; Xu, H.; Ren, Y.; Guo, C.; Xing, Z.; Sun, Z.; Han, X. Gene interaction perturbation network deciphers a high-resolution taxonomy in colorectal cancer. eLife 2022, 11, e81114. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Lawrence, T.; Liang, Y. The Role of Plasmacytoid Dendritic Cells in Cancers. Front. Immunol. 2021, 12, 749190. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Cortese, N.; Soldani, C.; Franceschini, B.; Barbagallo, M.; Marchesi, F.; Torzilli, G.; Donadon, M. Macrophages in Colorectal Cancer Liver Metastases. Cancers 2019, 11, 633. [Google Scholar] [PubMed] [Green Version]
- Konstantinov, A.S.; Kovaleva, O.V.; Samoilova, D.V.; Shelekhova, K.V. Role of macrophages in progression of colorectal cancer: A contrast with the traditional paradigm. Int. J. Clin. Exp. Pathol. 2022, 15, 403–411. [Google Scholar] [PubMed]
- Kloosterman, D.J.; Akkari, L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell 2023, 186, 1627–1651. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [PubMed] [Green Version]
- Binder, H.; Schmidt, M.; Hopp, L.; Davitavyan, S.; Arakelyan, A.; Loeffler-Wirth, H. Integrated Multi-Omics Maps of Lower-Grade Gliomas. Cancers 2022, 14, 2797. [Google Scholar]
- Tan, E.S.; Knepper, T.C.; Wang, X.; Permuth, J.B.; Wang, L.; Fleming, J.B.; Xie, H. Copy Number Alterations as Novel Biomarkers and Therapeutic Targets in Colorectal Cancer. Cancers 2022, 14, 2223. [Google Scholar]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Gerber, T.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Schadendorf, D.; Schartl, M.; Anderegg, U.; Camp, G.; Treutlein, B.; Binder, H.; et al. Mapping heterogeneity in patient-derived melanoma cultures by single-cell RNA-seq. Oncotarget 2017, 8, 846–862. [Google Scholar] [CrossRef] [Green Version]
- Sobral, D.; Martins, M.; Kaplan, S.; Golkaram, M.; Salmans, M.; Khan, N.; Vijayaraghavan, R.; Casimiro, S.; Fernandes, A.; Borralho, P.; et al. Genetic and microenvironmental intra-tumor heterogeneity impacts colorectal cancer evolution and metastatic development. Commun. Biol. 2022, 5, 937. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef] [PubMed]
- Merotto, L.; Sturm, G. Immunedeconv: Methods for Immune Cell Deconvolution. 2022. Available online: https:/omnideconv.org/immunedeconv (accessed on 25 July 2023).
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Lorzadeh, A.; Romero-Wolf, M.; Goel, A.; Jadhav, U. Epigenetic Regulation of Intestinal Stem Cells and Disease: A Balancing Act of DNA and Histone Methylation. Gastroenterology 2021, 160, 2267–2282. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiera, F.; Rossi, A.; Jammula, S.; Zanotti, M.; Pasini, D. PRC2 preserves intestinal progenitors and restricts secretory lineage commitment. EMBO J. 2016, 35, 2301–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.A.; Li, A.X.; Wu, X.; Yang, R.; Drew, D.A.; Rosenberg, D.W.; Pfeifer, G.P. Loss of the Polycomb Mark from Bivalent Promoters Leads to Activation of Cancer-Promoting Genes in Colorectal Tumors. Cancer Res. 2014, 74, 3617–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Applegate, C.C.; Lane, M.A. Role of retinoids in the prevention and treatment of colorectal cancer. World J. Gastrointest. Oncol. 2015, 7, 184–203. [Google Scholar] [CrossRef]
- Majumdar, D.; Tiernan, J.P.; Lobo, A.J.; Evans, C.A.; Corfe, B.M. Keratins in colorectal epithelial function and disease. Int. J. Exp. Pathol. 2012, 93, 305–318. [Google Scholar] [CrossRef]
- Lähdeniemi, I.A.K.; Misiorek, J.O.; Antila, C.J.M.; Landor, S.K.J.; Stenvall, C.-G.A.; Fortelius, L.E.; Bergström, L.K.; Sahlgren, C.; Toivola, D.M. Keratins regulate colonic epithelial cell differentiation through the Notch1 signalling pathway. Cell Death Differ. 2017, 24, 984–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Wu, J.; Fu, X.; Du, W.; Zhou, L.; Meng, X.; Yu, H.; Lin, J.; Ye, W.; Liu, J.; et al. OTUB1 promotes metastasis and serves as a marker of poor prognosis in colorectal cancer. Mol. Cancer 2014, 13, 258. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Song, M.J.; Jung, Y.; Lee, W.S.; Jang, H.H. Proteomic Analysis of Primary Colon Cancer and Synchronous Solitary Liver Metastasis. Cancer Genom. Proteom. 2019, 16, 583–592. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. The ubiquitin-proteasome system in colorectal cancer. Biochim. Et Biophys. Acta BBA—Mol. Basis Dis. 2008, 1782, 800–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, H.; Mathew, J.K.K.; Visweswariah, S.S. Receptor Guanylyl Cyclase C and Cyclic GMP in Health and Disease: Perspectives and Therapeutic Opportunities. Front. Endocrinol. 2022, 13, 911459. [Google Scholar] [CrossRef]
- Nait Slimane, S.; Marcel, V.; Fenouil, T.; Catez, F.; Saurin, J.C.; Bouvet, P.; Diaz, J.J.; Mertani, H.C. Ribosome Biogenesis Alterations in Colorectal Cancer. Cells 2020, 9, 2361. [Google Scholar] [CrossRef]
- Bui, S.; Mejia, I.; Díaz, B.; Wang, Y. Adaptation of the Golgi Apparatus in Cancer Cell Invasion and Metastasis. Front. Cell Dev. Biol. 2021, 9, 806482. [Google Scholar] [CrossRef]
- Hopp, L.; Wirth, H.; Fasold, M.; Binder, H. Portraying the expression landscapes of cancer subtypes: A glioblastoma multiforme and prostate cancer case study. Syst. Biomed. 2013, 1, 99–121. [Google Scholar]
- Hebenstreit, D.; Fang, M.; Gu, M.; Charoensawan, V.; van Oudenaarden, A.; Teichmann, S.A. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 2011, 7, 497. [Google Scholar] [CrossRef] [Green Version]
- Prorok, P.; Forouzanfar, F.; Murugarren, N.; Peiffer, I.; Charton, R.; Akerman, I.; Méchali, M. Loss of Ezh2 function remodels the DNA replication initiation landscape. Cell Rep. 2023, 42, 112280. [Google Scholar] [CrossRef]
- Schmidt, M.; Loeffler-Wirth, H.; Binder, H. Developmental scRNAseq Trajectories in Gene- and Cell-State Space—The Flatworm Example. Genes 2020, 11, 1214. [Google Scholar]
- Zeng, Z.; Ma, C.; Chen, K.; Jiang, M.; Vasu, R.; Liu, R.; Zhao, Y.; Zhang, H. Roles of G Protein-Coupled Receptors (GPCRs) in Gastrointestinal Cancers: Focus on Sphingosine 1-Shosphate Receptors, Angiotensin II Receptors, and Estrogen-Related GPCRs. Cells 2021, 10, 2988. [Google Scholar]
- Kumari, N.; Reabroi, S.; North, B.J. Unraveling the Molecular Nexus between GPCRs, ERS, and EMT. Mediat. Inflamm. 2021, 2021, 6655417. [Google Scholar] [CrossRef]
- Kang, H.; Fichna, J.; Matlawska-Wasowska, K.; Jacenik, D. The Expression Pattern of Adhesion G Protein-Coupled Receptor F5 Is Related to Cell Adhesion and Metastatic Pathways in Colorectal Cancer—Comprehensive Study Based on In Silico Analysis. Cells 2022, 11, 3876. [Google Scholar]
- Song, Q.; Han, Z.; Wu, X.; Wang, Y.; Zhou, L.; Yang, L.; Liu, N.; Sui, H.; Cai, J.; Ji, Q.; et al. β-Arrestin1 Promotes Colorectal Cancer Metastasis Through GSK-3β/β-Catenin Signaling- Mediated Epithelial-to-Mesenchymal Transition. Front. Cell Dev. Biol. 2021, 9, 650067. [Google Scholar] [CrossRef]
- Hirsch, D.; Barker, N.; McNeil, N.; Hu, Y.; Camps, J.; McKinnon, K.; Clevers, H.; Ried, T.; Gaiser, T. LGR5 positivity defines stem-like cells in colorectal cancer. Carcinogenesis 2014, 35, 849–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Lin, W.; Wen, L.; Li, G. Lgr5 in cancer biology: Functional identification of Lgr5 in cancer progression and potential opportunities for novel therapy. Stem Cell Res. Ther. 2019, 10, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitfield, M.L.; Sherlock, G.; Saldanha, A.J.; Murray, J.I.; Ball, C.A.; Alexander, K.E.; Matese, J.C.; Perou, C.M.; Hurt, M.M.; Brown, P.O.; et al. Identification of Genes Periodically Expressed in the Human Cell Cycle and Their Expression in Tumors. Mol. Biol. Cell 2002, 13, 1977–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Hopp, L.; Lembcke, K.; Wirth, H. Personalized Disease Phenotypes from Massive OMICs Data. In Big Data Analytics in Bioinformatics and Healthcare; Baoying, W., Ruowang, L., William, P., Eds.; IGI Global: Hershey, PA, USA, 2015; pp. 359–378. [Google Scholar] [CrossRef] [Green Version]
- Mooi, J.K.; Wirapati, P.; Asher, R.; Lee, C.K.; Savas, P.; Price, T.J.; Townsend, A.; Hardingham, J.; Buchanan, D.; Williams, D.; et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: Molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 2018, 29, 2240–2246. [Google Scholar] [CrossRef] [PubMed]
- Majek, O.; Gondos, A.; Jansen, L.; Emrich, K.; Holleczek, B.; Katalinic, A.; Nennecke, A.; Eberle, A.; Brenner, H. Sex differences in colorectal cancer survival: Population-based analysis of 164,996 colorectal cancer patients in Germany. PLoS ONE 2013, 8, e68077. [Google Scholar] [CrossRef] [Green Version]
- Abancens, M.; Bustos, V.; Harvey, H.; McBryan, J.; Harvey, B.J. Sexual Dimorphism in Colon Cancer. Front. Oncol. 2020, 10, 607909. [Google Scholar] [CrossRef]
- White, A.; Ironmonger, L.; Steele, R.J.C.; Ormiston-Smith, N.; Crawford, C.; Seims, A. A review of sex-related differences in colorectal cancer incidence, screening uptake, routes to diagnosis, cancer stage and survival in the UK. BMC Cancer 2018, 18, 906. [Google Scholar] [CrossRef] [Green Version]
- Milette, S.; Hashimoto, M.; Perrino, S.; Qi, S.; Chen, M.; Ham, B.; Wang, N.; Istomine, R.; Lowy, A.M.; Piccirillo, C.A.; et al. Sexual dimorphism and the role of estrogen in the immune microenvironment of liver metastases. Nat. Commun. 2019, 10, 5745. [Google Scholar] [CrossRef] [Green Version]
- Hases, L.; Ibrahim, A.; Chen, X.; Liu, Y.; Hartman, J.; Williams, C. The Importance of Sex in the Discovery of Colorectal Cancer Prognostic Biomarkers. Int. J. Mol. Sci. 2021, 22, 1354. [Google Scholar] [CrossRef] [PubMed]
- Nersisyan, L.; Simonyan, A.; Binder, H.; Arakelyan, A. Telomere Maintenance Pathway Activity Analysis Enables Tissue- and Gene-Level Inferences. Front. Genet. 2021, 12, 662464. [Google Scholar] [CrossRef]
- Nersisyan, L.; Hopp, L.; Loeffler-Wirth, H.; Galle, J.; Loeffler, M.; Arakelyan, A.; Binder, H. Telomere Length Maintenance and Its Transcriptional Regulation in Lynch Syndrome and Sporadic Colorectal Carcinoma. Front. Oncol. 2019, 9, 1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012, 142, 1021–1031.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, L.E.; Sengupta, S.; Chen, I.H.; den Boon, J.A.; Cheng, Y.J.; Westra, W.; Newton, M.A.; Mittl, B.F.; McShane, L.; Chen, C.J.; et al. Genes involved in DNA repair and nitrosamine metabolism and those located on chromosome 14q32 are dysregulated in nasopharyngeal carcinoma. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2216–2225. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.R.; Szabolcs, A.; Allen, J.N.; Clark, J.W.; Wo, J.Y.; Raabe, M.; Thel, H.; Hoyos, D.; Mehta, A.; Arshad, S.; et al. Radiation therapy enhances immunotherapy response in microsatellite stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nat. Cancer 2021, 2, 1124–1135. [Google Scholar] [CrossRef]
- Matsuda, S.; Revandkar, A.; Dubash, T.D.; Ravi, A.; Wittner, B.S.; Lin, M.; Morris, R.; Burr, R.; Guo, H.; Seeger, K.; et al. TGF-β in the microenvironment induces a physiologically occurring immune-suppressive senescent state. Cell Rep. 2023, 42, 112129. [Google Scholar] [CrossRef]
- He, J.; Shan, S.; Li, Q.; Fang, B.; Xie, Y. Mechanical Stretch Triggers Epithelial-Mesenchymal Transition in Keratinocytes Through Piezo1 Channel. Front. Physiol. 2022, 13, 745572. [Google Scholar] [CrossRef]
- Dombroski, J.A.; Hope, J.M.; Sarna, N.S.; King, M.R. Channeling the Force: Piezo1 Mechanotransduction in Cancer Metastasis. Cells 2021, 10, 2815. [Google Scholar] [CrossRef]
- De Felice, D.; Alaimo, A. Mechanosensitive Piezo Channels in Cancer: Focus on altered Calcium Signaling in Cancer Cells and in Tumor Progression. Cancers 2020, 12, 1780. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Garcia, J.; Herrera-Fernández, V.; Serra, S.A.; Rubio-Moscardo, F.; Vogel-Gonzalez, M.; Doñate-Macian, P.; Hevia, C.F.; Pujades, C.; Valverde, M.A. The mechanosensitive Piezo1 channel controls endosome trafficking for an efficient cytokinetic abscission. Sci. Adv. 2021, 7, eabi7785. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, T.E.; Dyer, D.P.; Allen, J.E. The extracellular matrix and the immune system: A mutually dependent relationship. Science 2023, 379, eabp8964. [Google Scholar] [CrossRef] [PubMed]
- Hübers, C.; Abdul Pari, A.A.; Grieshober, D.; Petkov, M.; Schmidt, A.; Messmer, T.; Heyer, C.M.; Schölch, S.; Kapel, S.S.; Gengenbacher, N.; et al. Primary tumor–derived systemic nANGPTL4 inhibits metastasis. J. Exp. Med. 2022, 220, e20202595. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, M.; Molina, F.; Bascoul-Mollevi, C.; Copois, V.; Bibeau, F.; Chalbos, P.; Bareil, C.; Kramar, A.; Salvetat, N.; Fraslon, C.; et al. Gene expression signature in advanced colorectal cancer patients select drugs and response for the use of leucovorin, fluorouracil, and irinotecan. J. Clin. Oncol. 2007, 25, 773–780. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Loeffler-Wirth, H.; Kreuz, M.; Schmidt, M.; Ott, G.; Siebert, R.; Binder, H. Classifying Germinal Center Derived Lymphomas-Navigate a Complex Transcriptional Landscape. Cancers 2022, 14, 3434. [Google Scholar] [CrossRef]
- Ciner, A.T.; Jones, K.; Muschel, R.J.; Brodt, P. The unique immune microenvironment of liver metastases: Challenges and opportunities. Semin. Cancer Biol. 2021, 71, 143–156. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Wang, H.; Pan, J.; Barsky, L.; Jacob, J.C.; Zheng, Y.; Gao, C.; Wang, S.; Zhu, W.; Sun, H.; Lu, L.; et al. Characteristics of pre-metastatic niche: The landscape of molecular and cellular pathways. Mol. Biomed. 2021, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.P.; Rai, S.; Pandey, A.; Singh, N.K.; Srivastava, S. Molecular subtypes of colorectal cancer: An emerging therapeutic opportunity for personalized medicine. Genes Dis. 2021, 8, 133–145. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, S.; Jia, Y.; Zhang, X. Telomere maintenance mechanism dysregulation serves as an early predictor of adjuvant therapy response and a potential therapeutic target in human cancers. Int. J. Cancer 2022, 151, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Movasati, A.; Brunner, S.R.; Nguyen, L.; Priestley, P.; Cuppen, E.; Van Hoeck, A. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature 2023, 618, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Pich, O.; Muiños, F.; Lolkema, M.P.; Steeghs, N.; Gonzalez-Perez, A.; Lopez-Bigas, N. The mutational footprints of cancer therapies. Nat. Genet. 2019, 51, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Nallasamy, P.; Nimmakayala, R.K.; Parte, S.; Are, A.C.; Batra, S.K.; Ponnusamy, M.P. Tumor microenvironment enriches the stemness features: The architectural event of therapy resistance and metastasis. Mol. Cancer 2022, 21, 225. [Google Scholar] [CrossRef] [PubMed]
- Patras, L.; Shaashua, L.; Matei, I.; Lyden, D. Immune determinants of the pre-metastatic niche. Cancer Cell 2023, 41, 546–572. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Tien, F.-M.; Lu, H.-H.; Lin, S.-Y.; Tsai, H.-C. Epigenetic remodeling of the immune landscape in cancer: Therapeutic hurdles and opportunities. J. Biomed. Sci. 2023, 30, 3. [Google Scholar] [CrossRef]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Jiao, W.; Atwal, G.; Polak, P.; Karlic, R.; Cuppen, E.; Danyi, A.; de Ridder, J.; van Herpen, C.; Lolkema, M.P.; Steeghs, N.; et al. A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns. Nat. Commun. 2020, 11, 728. [Google Scholar] [CrossRef] [Green Version]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Kilmister, E.J.; Koh, S.P.; Weth, F.R.; Gray, C.; Tan, S.T. Cancer Metastasis and Treatment Resistance: Mechanistic Insights and Therapeutic Targeting of Cancer Stem Cells and the Tumor Microenvironment. Biomedicines 2022, 10, 2988. [Google Scholar] [CrossRef]
- Binder, H.; Steiner, L.; Wirth, H.; Rohlf, T.; Prohaska, S.; Galle, J. Transcriptional regulation by histone modifications: Towards a theory of chromatin re-organization during stem cell differentiation. Phys. Biol. 2013, 10, 026006. [Google Scholar] [CrossRef]
- Morgan, R.G.; Mortensson, E.; Williams, A.C. Targeting LGR5 in Colorectal Cancer: Therapeutic gold or too plastic? Br. J. Cancer 2018, 118, 1410–1418. [Google Scholar] [CrossRef]
- Cross, W.C.; Graham, T.A.; Wright, N.A. New paradigms in clonal evolution: Punctuated equilibrium in cancer. J. Pathol. 2016, 240, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [Green Version]
- Avagyan, S.; Binder, H. Subtyping or not subtyping—Quo vadis for precision medicine of colorectal cancer. Transl. Cancer Res. 2023, 12, 321–339. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Spurr, L.F.; Martinez, C.A.; Katipally, R.R.; Iyer, S.C.; Pugh, S.A.; Bridgewater, J.A.; Primrose, J.N.; Domingo, E.; Maughan, T.S.; D’Angelica, M.I.; et al. A proliferative subtype of colorectal liver metastases exhibits hypersensitivity to cytotoxic chemotherapy. Npj Precis. Oncol. 2022, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Pitroda, S.P.; Khodarev, N.N.; Huang, L.; Uppal, A.; Wightman, S.C.; Ganai, S.; Joseph, N.; Pitt, J.; Brown, M.; Forde, M.; et al. Integrated molecular subtyping defines a curable oligometastatic state in colorectal liver metastasis. Nat. Commun. 2018, 9, 1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katipally, R.R.; Martinez, C.A.; Pugh, S.A.; Bridgewater, J.A.; Primrose, J.N.; Domingo, E.; Maughan, T.S.; Talamonti, M.S.; Posner, M.C.; Weichselbaum, R.R.; et al. Integrated Clinical-Molecular Classification of Colorectal Liver Metastases: A Biomarker Analysis of the Phase 3 New EPOC Randomized Clinical Trial. JAMA Oncol. 2023. [Google Scholar] [CrossRef]
- Avraham-Davidi, I.; Mages, S.; Klughammer, J.; Moriel, N.; Imada, S.; Hofree, M.; Murray, E.; Chen, J.; Pelka, K.; Mehta, A.; et al. Integrative single cell and spatial transcriptomics of colorectal cancer reveals multicellular functional units that support tumor progression. bioRxiv, 2022. [Google Scholar] [CrossRef]
- Fleischer, J.R.; Schmitt, A.M.; Haas, G.; Xu, X.; Zeisberg, E.M.; Bohnenberger, H.; Küffer, S.; Teuwen, L.-A.; Karras, P.J.; Beißbarth, T.; et al. Molecular differences of angiogenic versus vessel co-opting colorectal cancer liver metastases at single-cell resolution. Mol. Cancer 2023, 22, 17. [Google Scholar] [CrossRef]
- Wood, C.S.; Pennel, K.A.F.; Leslie, H.; Legrini, A.; Cameron, A.J.; Melissourgou-Syka, L.; Quinn, J.A.; van Wyk, H.C.; Hay, J.; Roseweir, A.K.; et al. Spatially Resolved Transcriptomics Deconvolutes Prognostic Histological Subgroups in Patients with Colorectal Cancer and Synchronous Liver Metastases. Cancer Res. 2023, 83, 1329–1344. [Google Scholar] [CrossRef]
- Hu, M.; Chen, Z.; Hu, D.; Xi, S.; Wang, D.; Zhang, X.; Fong, W.P.; Wen, L.; Cai, Y.; Yuan, Y.; et al. Delineating the molecular landscape of different histopathological growth patterns in colorectal cancer liver metastases. Front. Immunol. 2022, 13, 1045329. [Google Scholar] [CrossRef]
- Nersisyan, L.; Loeffler-Wirth, H.; Arakelyan, A.; Binder, H. Gene set- and pathway- centered knowledge discovery assigns transcriptional activation patterns in brain, blood and colon cancer A bioinformatics perspective. J. Bioinform. Knowl. Min. 2016, 4, 46–70. [Google Scholar]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015, 16, 64. [Google Scholar] [CrossRef]
- Kosinski, C.; Li, V.S.; Chan, A.S.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; Yuen, S.T.; et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc. Natl. Acad. Sci. USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.; Gerds, T.A.; Seidelin, J.B.; Csillag, C.; Bjerrum, J.T.; Troelsen, J.T.; Nielsen, O.H. Diagnosis of ulcerative colitis before onset of inflammation by multivariate modeling of genome-wide gene expression data. Inflamm. Bowel Dis. 2009, 15, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.S.; Shu, W.P.; Cohen, A.M.; Guillem, J.G. Matrix metalloproteinase-7 expression in colorectal cancer liver metastases: Evidence for involvement of MMP-7 activation in human cancer metastases. Clin. Cancer Res. 2002, 8, 144–148. [Google Scholar] [PubMed]
- Bakker, E.R.; Das, A.M.; Helvensteijn, W.; Franken, P.F.; Swagemakers, S.; van der Valk, M.A.; ten Hagen, T.L.; Kuipers, E.J.; van Veelen, W.; Smits, R. Wnt5a promotes human colon cancer cell migration and invasion but does not augment intestinal tumorigenesis in Apc 1638N mice. Carcinogenesis 2013, 34, 2629–2638. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tang, Z.; Gong, H.; Zhu, L.; Liu, X. Wnt5a promotes epithelial-to-mesenchymal transition and metastasis in non-small-cell lung cancer. Biosci. Rep. 2017, 37, BSR20171092. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashekyan, O.; Shahbazyan, N.; Bareghamyan, Y.; Kudryavzeva, A.; Mandel, D.; Schmidt, M.; Loeffler-Wirth, H.; Uduman, M.; Chand, D.; Underwood, D.; et al. Transcriptomic Maps of Colorectal Liver Metastasis: Machine Learning of Gene Activation Patterns and Epigenetic Trajectories in Support of Precision Medicine. Cancers 2023, 15, 3835. https://doi.org/10.3390/cancers15153835
Ashekyan O, Shahbazyan N, Bareghamyan Y, Kudryavzeva A, Mandel D, Schmidt M, Loeffler-Wirth H, Uduman M, Chand D, Underwood D, et al. Transcriptomic Maps of Colorectal Liver Metastasis: Machine Learning of Gene Activation Patterns and Epigenetic Trajectories in Support of Precision Medicine. Cancers. 2023; 15(15):3835. https://doi.org/10.3390/cancers15153835
Chicago/Turabian StyleAshekyan, Ohanes, Nerses Shahbazyan, Yeva Bareghamyan, Anna Kudryavzeva, Daria Mandel, Maria Schmidt, Henry Loeffler-Wirth, Mohamed Uduman, Dhan Chand, Dennis Underwood, and et al. 2023. "Transcriptomic Maps of Colorectal Liver Metastasis: Machine Learning of Gene Activation Patterns and Epigenetic Trajectories in Support of Precision Medicine" Cancers 15, no. 15: 3835. https://doi.org/10.3390/cancers15153835
APA StyleAshekyan, O., Shahbazyan, N., Bareghamyan, Y., Kudryavzeva, A., Mandel, D., Schmidt, M., Loeffler-Wirth, H., Uduman, M., Chand, D., Underwood, D., Armen, G., Arakelyan, A., Nersisyan, L., & Binder, H. (2023). Transcriptomic Maps of Colorectal Liver Metastasis: Machine Learning of Gene Activation Patterns and Epigenetic Trajectories in Support of Precision Medicine. Cancers, 15(15), 3835. https://doi.org/10.3390/cancers15153835