
PTHrP Regulates Fatty Acid Metabolism via Novel lncRNA in Breast Cancer Initiation and Progression Models

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
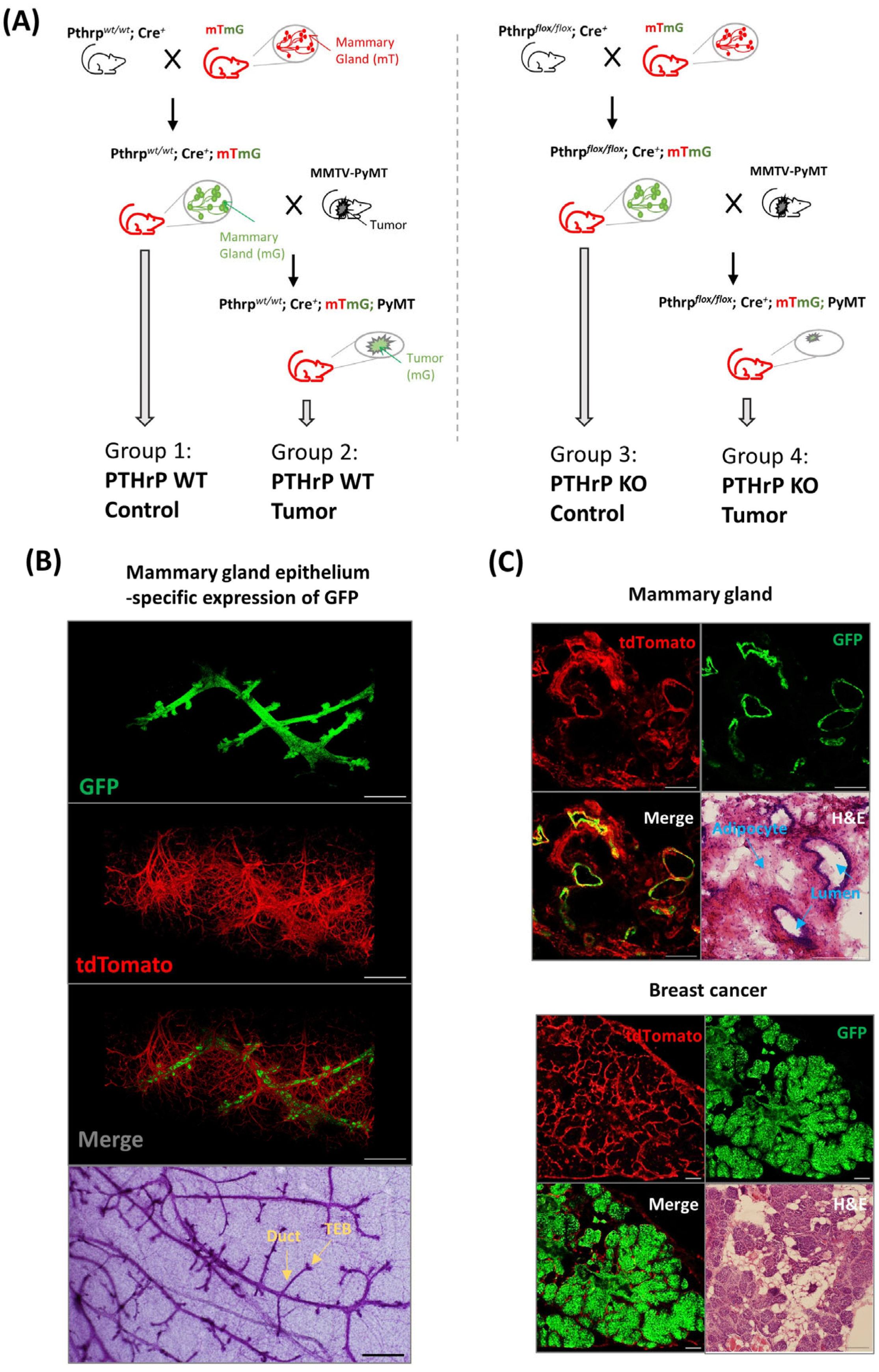
2.1. Mouse Lines and Breeding
2.2. Mice Genotyping
2.3. HE Staining
2.4. Confocal Microscopy
2.5. Isolation of Primary Mammary Gland and Breast Tumor
2.6. FACS of Single GFP+ Cells
2.7. RNA Isolation and Sequencing
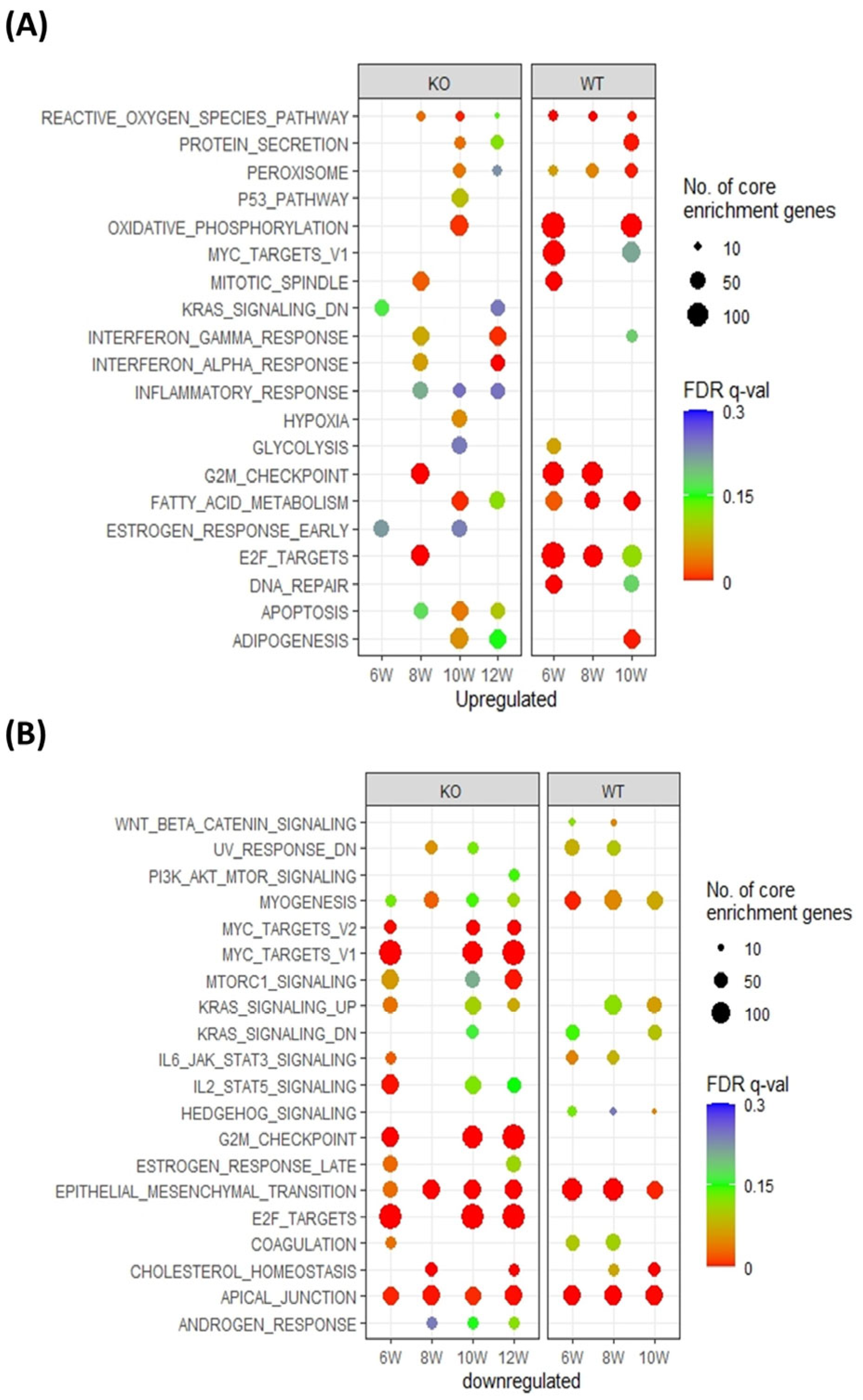
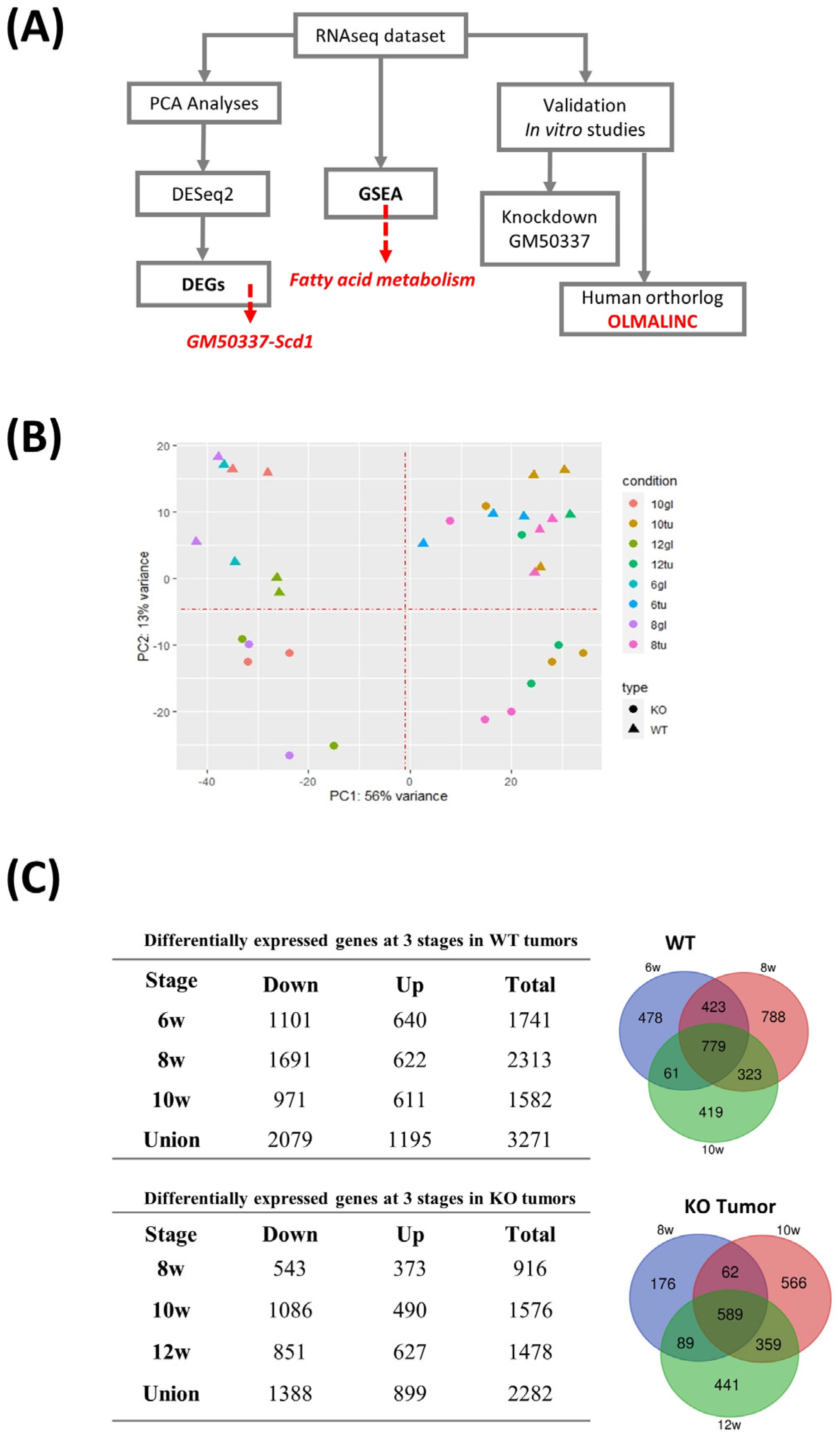
2.8. RNA-Seq Analyses
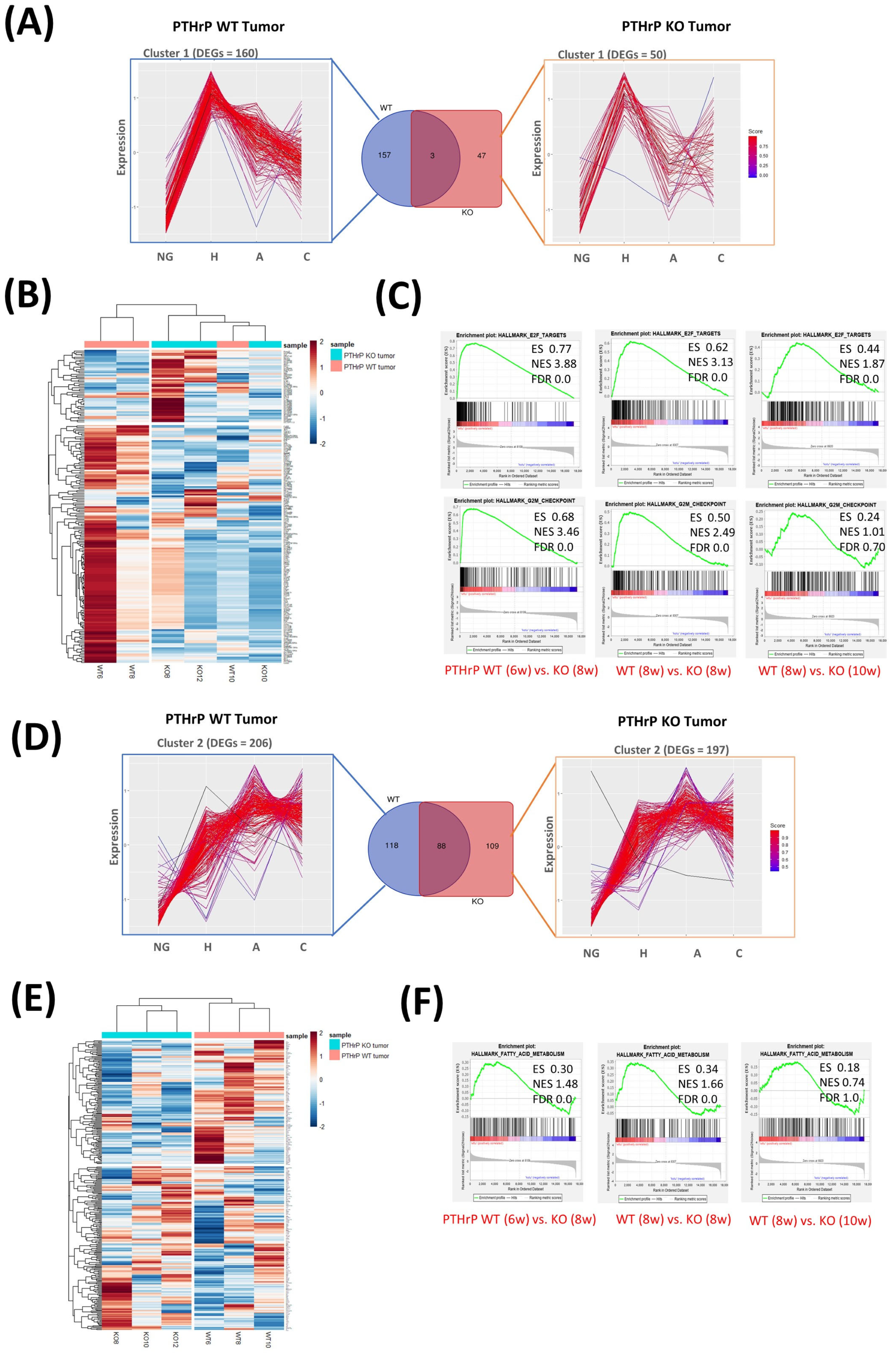
2.9. Time-Course Data Analyses
2.10. Correlation Analyses between lncRNA and Protein Coding Genes
2.11. Cell Culture
2.12. CRISPR/Cas9 Genetic Knockout
2.13. Quantitative Real-Time PCR (qRT-PCR) Assays
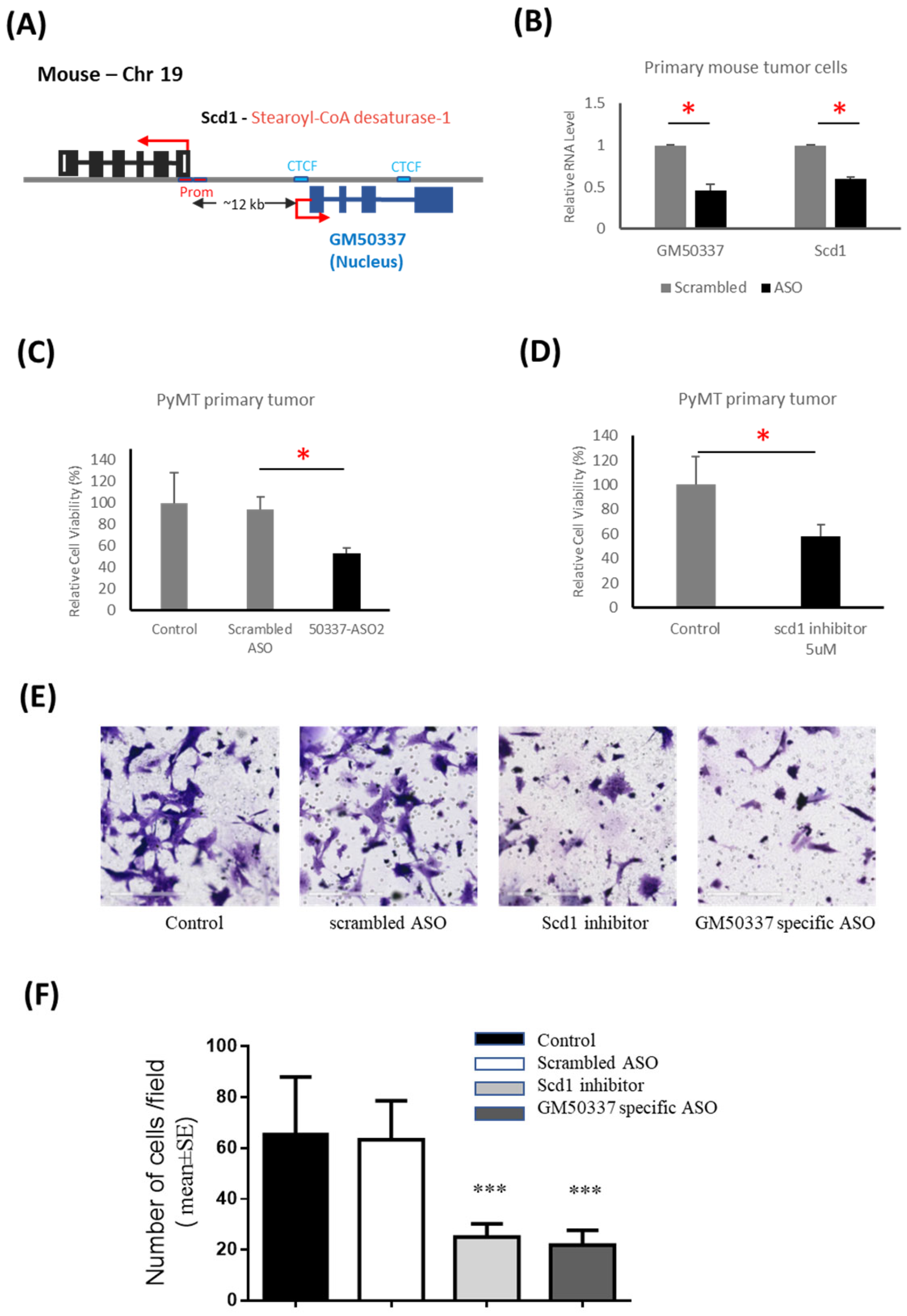
2.14. Antisense Oligonucleotide (ASO)-Mediated Knockdown (KD)
2.15. Cell Viability Assays
2.16. Cell Invasion Assay
3. Results
3.1. Mouse Model for Fluorescence-Based Mapping of Mammary Epithelium
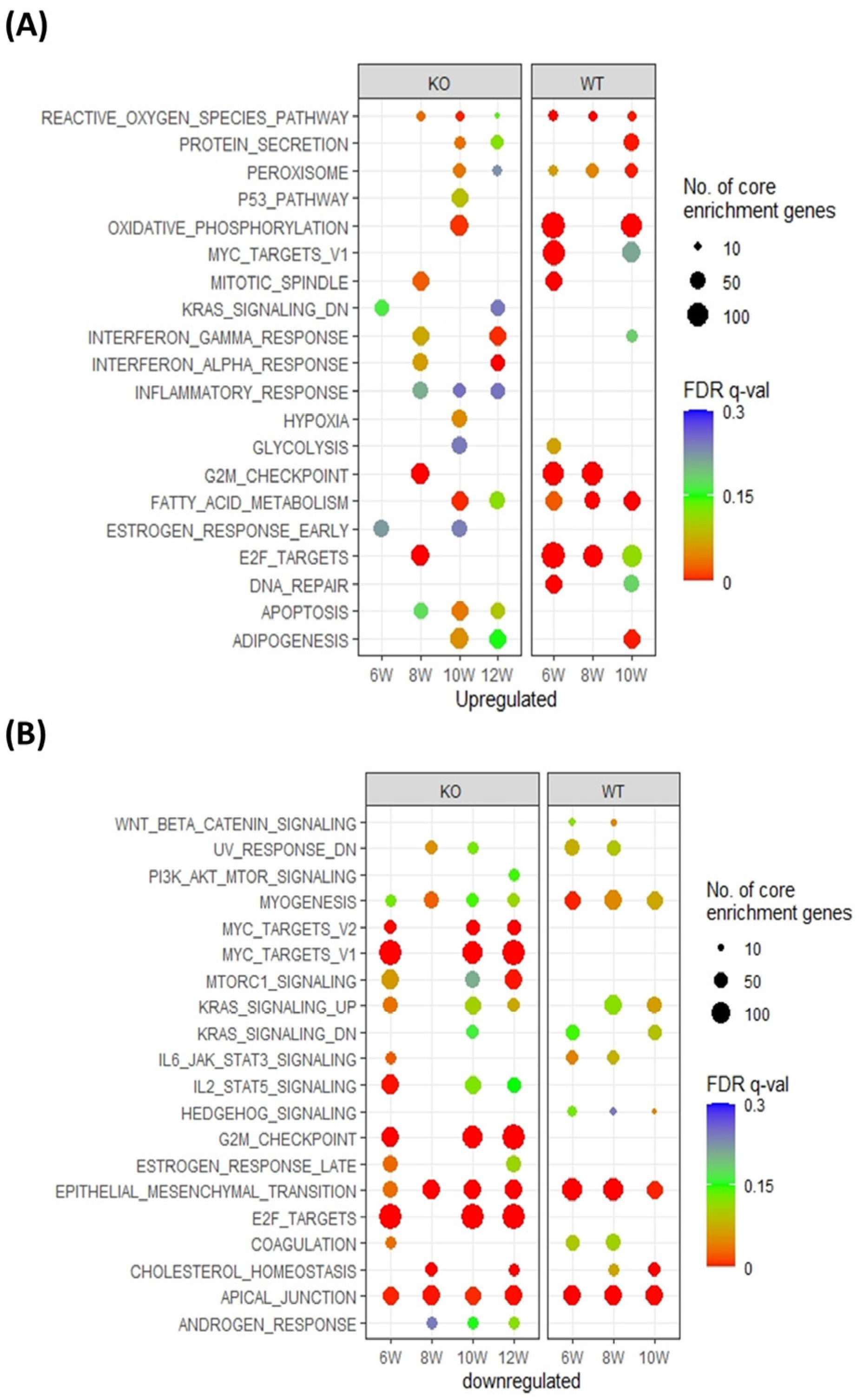
3.2. Pthrp Ablation Modifies Fatty Acid Metabolism as well as Cell Cycle Events in BC Initiation and Progression
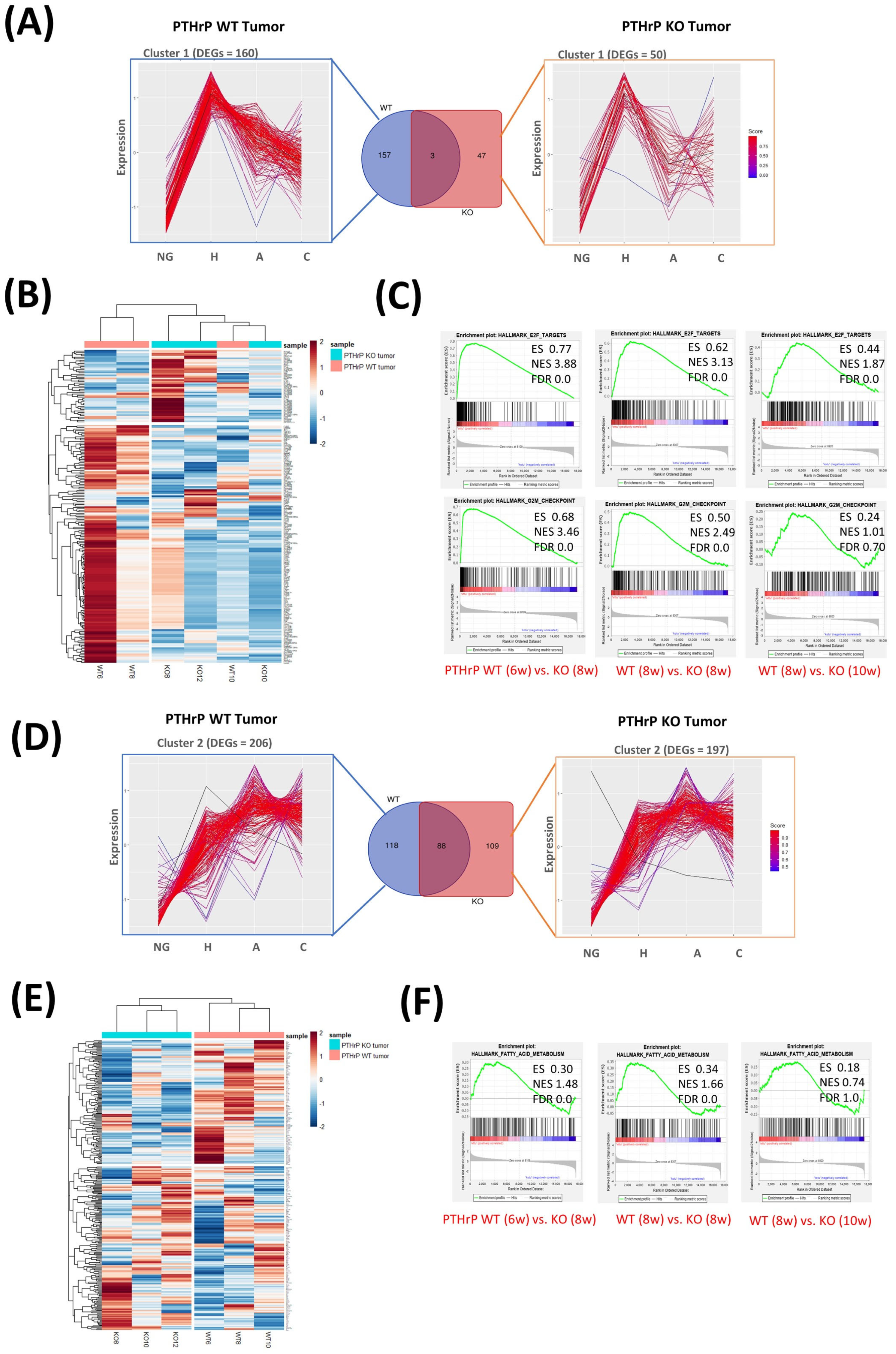
3.3. Pthrp Ablation Deregulated lncRNA Expression in BC Initiation and Progression
3.4. Interruption of GM50337-Scd1 Decreases Cell Viability
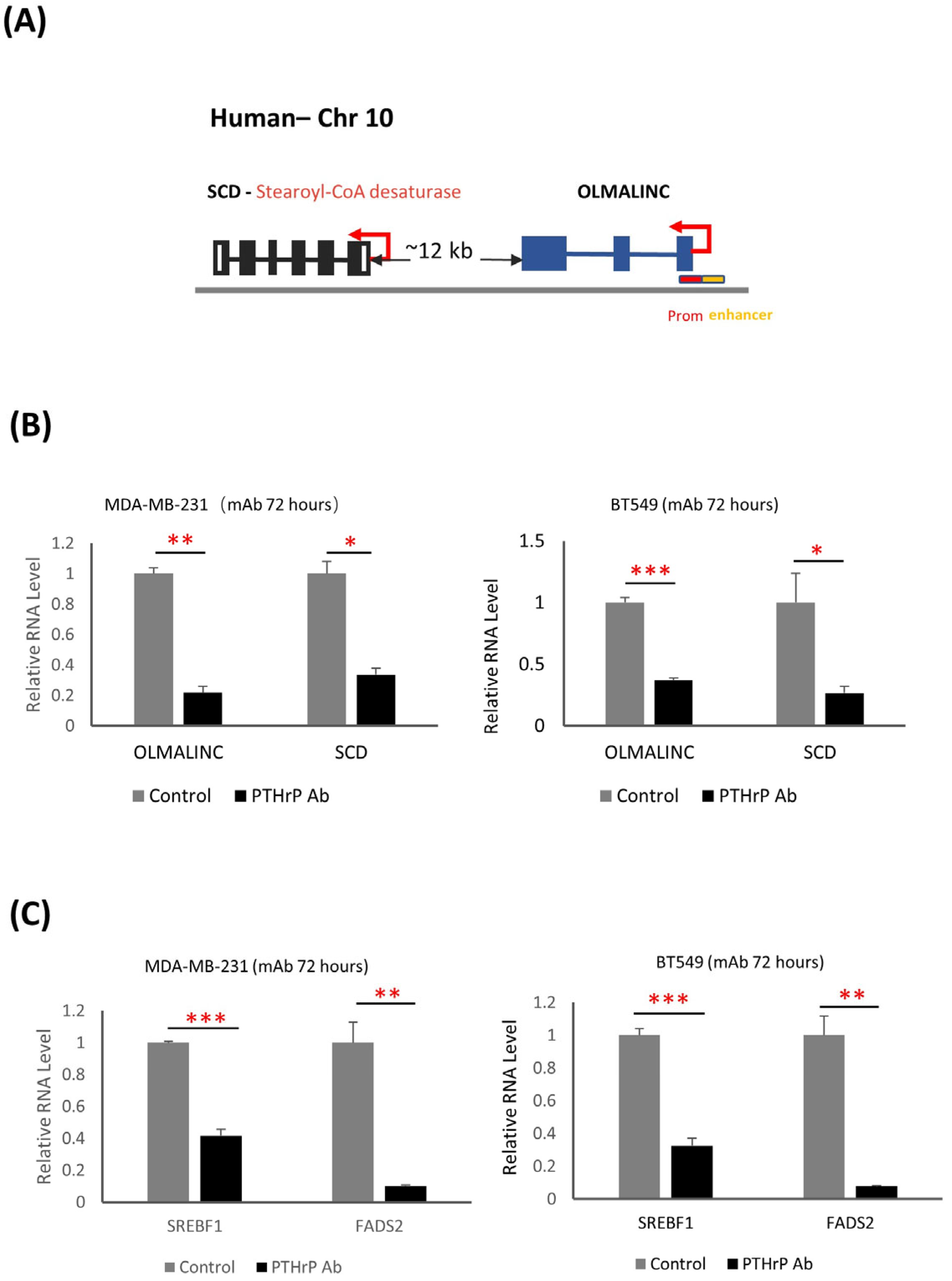
3.5. PTHrP Regulates BC initiation via Fatty Acid Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Shaughnessy, J. Extending survival with chemotherapy in metastatic breast cancer. Oncologist 2005, 10 (Suppl. 3), 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Vargo-Gogola, T.; Rosen, J.M. Modelling breast cancer: One size does not fit all. Nat. Rev. Cancer 2007, 7, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Attalla, S.; Taifour, T.; Bui, T.; Muller, W. Insights from transgenic mouse models of PyMT-induced breast cancer: Recapitulating human breast cancer progression in vivo. Oncogene 2021, 40, 475–491. [Google Scholar] [CrossRef]
- Albright, F. Case Records of the Massachusetts General Hospital. Case 27461. N. Engl. J. Med. 1941, 225, 789–791. [Google Scholar]
- Zhang, R.; Li, J.; Assaker, G.; Camirand, A.; Sabri, S.; Karaplis, A.C.; Kremer, R. Parathyroid Hormone-Related Protein (PTHrP): An Emerging Target in Cancer Progression and Metastasis. Adv. Exp. Med. Biol. 2019, 1164, 161–178. [Google Scholar] [CrossRef]
- Kremer, R.; Li, J.; Camirand, A.; Karaplis, A.C. Parathyroid hormone related protein (PTHrP) in tumor progression. Adv. Exp. Med. Biol. 2011, 720, 145–160. [Google Scholar] [CrossRef]
- Pitarresi, J.R.; Norgard, R.J.; Chiarella, A.M.; Suzuki, K.; Bakir, B.; Sahu, V.; Li, J.; Zhao, J.; Marchand, B.; Wengyn, M.D.; et al. PTHrP Drives Pancreatic Cancer Growth and Metastasis and Reveals a New Therapeutic Vulnerability. Cancer Discov. 2021, 11, 1774–1791. [Google Scholar] [CrossRef]
- Chang, W.M.; Lin, Y.F.; Su, C.Y.; Peng, H.Y.; Chang, Y.C.; Hsiao, J.R.; Chen, C.L.; Chang, J.Y.; Shieh, Y.S.; Hsiao, M.; et al. Parathyroid Hormone-Like Hormone is a Poor Prognosis Marker of Head and Neck Cancer and Promotes Cell Growth via RUNX2 Regulation. Sci. Rep. 2017, 7, 41131. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Xue, M.; Liu, C.; Xie, F.; Bai, L. Parathyroid Hormone-Like Hormone Induces Epithelial-to-Mesenchymal Transition of Intestinal Epithelial Cells by Activating the Runt-Related Transcription Factor 2. Am. J. Pathol. 2018, 188, 1374–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Liao, Y.; He, S.; Shi, J.; Peng, L.; Xu, X.; Xie, F.; Diao, N.; Huang, J.; Xie, Q.; et al. Autocrine parathyroid hormone-like hormone promotes intrahepatic cholangiocarcinoma cell proliferation via increased ERK/JNK-ATF2-cyclinD1 signaling. J. Transl. Med. 2017, 15, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walia, M.K.; Ho, P.M.; Taylor, S.; Ng, A.J.; Gupte, A.; Chalk, A.M.; Zannettino, A.C.; Martin, T.J.; Walkley, C.R. Activation of PTHrP-cAMP-CREB1 signaling following p53 loss is essential for osteosarcoma initiation and maintenance. eLife 2016, 5, 13446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Karaplis, A.C.; Huang, D.C.; Siegel, P.M.; Camirand, A.; Yang, X.F.; Muller, W.J.; Kremer, R. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J. Clin. Investig. 2011, 121, 4655–4669. [Google Scholar] [CrossRef] [Green Version]
- Kremer, R.; Shustik, C.; Tabak, T.; Papavasiliou, V.; Goltzman, D. Parathyroid-hormone-related peptide in hematologic malignancies. Am. J. Med. 1996, 100, 406–411. [Google Scholar] [CrossRef]
- Lv, Z.; Wu, X.; Cao, W.; Shen, Z.; Wang, L.; Xie, F.; Zhang, J.; Ji, T.; Yan, M.; Chen, W. Parathyroid hormone-related protein serves as a prognostic indicator in oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2014, 33, 100. [Google Scholar] [CrossRef] [Green Version]
- Urosevic, J.; Garcia-Albeniz, X.; Planet, E.; Real, S.; Cespedes, M.V.; Guiu, M.; Fernandez, E.; Bellmunt, A.; Gawrzak, S.; Pavlovic, M.; et al. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat. Cell. Biol. 2014, 16, 685–694. [Google Scholar] [CrossRef]
- Truong, N.U.; Edwardes, M.D.D.; Papavasiliou, V.; Goltzman, D.; Kremer, R. Parathyroid hormone-related peptide and survival of patients with cancer and hypercalcemia. Am. J. Med. 2003, 115, 115–121. [Google Scholar] [CrossRef]
- Assaker, G.; Camirand, A.; Abdulkarim, B.; Omeroglu, A.; Deschenes, J.; Joseph, K.; Noman, A.S.M.; Ramana Kumar, A.V.; Kremer, R.; Sabri, S. PTHrP, A Biomarker for CNS Metastasis in Triple-Negative Breast Cancer and Selection for Adjuvant Chemotherapy in Node-Negative Disease. JNCI Cancer Spectr. 2020, 4, pkz063. [Google Scholar] [CrossRef]
- Fleming, N.I.; Trivett, M.K.; George, J.; Slavin, J.L.; Murray, W.K.; Moseley, J.M.; Anderson, R.L.; Thomas, D.M. Parathyroid hormone-related protein protects against mammary tumor emergence and is associated with monocyte infiltration in ductal carcinoma in situ. Cancer Res. 2009, 69, 7473–7479. [Google Scholar] [CrossRef] [Green Version]
- Henderson, M.; Danks, J.; Moseley, J.; Slavin, J.; Harris, T.; McKinlay, M.; Hopper, J.; Martin, T. Parathyroid hormone-related protein production by breast cancers, improved survival, and reduced bone metastases. J. Natl. Cancer Inst. 2001, 93, 234–237. [Google Scholar] [CrossRef] [Green Version]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Cruz-Gil, S.; Fernandez, L.P.; Sanchez-Martinez, R.; Gomez de Cedron, M.; Ramirez de Molina, A. Non-Coding and Regulatory RNAs as Epigenetic Remodelers of Fatty Acid Homeostasis in Cancer. Cancers 2020, 12, 2890. [Google Scholar] [CrossRef]
- Cui, M.; Xiao, Z.; Wang, Y.; Zheng, M.; Song, T.; Cai, X.; Sun, B.; Ye, L.; Zhang, X. Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer Res. 2015, 75, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Taniue, K.; Kurimoto, A.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Iwasaki, K.; Nagashima, T.; Okada-Hatakeyama, M.; Oyama, M.; Kozuka-Hata, H.; et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc. Natl. Acad. Sci. USA 2016, 113, 1273–1278. [Google Scholar] [CrossRef]
- Li, J.; Camirand, A.; Zakikhani, M.; Sellin, K.; Guo, Y.; Luan, X.R.; Mihalcioiu, C.; Kremer, R. Parathyroid Hormone-Related Protein Inhibition Blocks Triple-Negative Breast Cancer Expansion in Bone Through Epithelial to Mesenchymal Transition Reversal. J. Bone Miner. Res. 2021, 6, e10587. [Google Scholar] [CrossRef]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef]
- Wagner, K.U.; McAllister, K.; Ward, T.; Davis, B.; Wiseman, R.; Hennighausen, L. Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res. 2001, 10, 545–553. [Google Scholar] [CrossRef]
- He, B.; Deckelbaum, R.A.; Miao, D.; Lipman, M.L.; Pollak, M.; Goltzman, D.; Karaplis, A.C. Tissue-specific targeting of the pthrp gene: The generation of mice with floxed alleles. Endocrinology 2001, 142, 2070–2077. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aken, B.L.; Achuthan, P.; Akanni, W.; Amode, M.R.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; et al. Ensembl 2017. Nucleic Acids Res. 2017, 45, D635–D642. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Tabix: Fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics 2011, 27, 718–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graubert, A.; Aguet, F.; Ravi, A.; Ardlie, K.G.; Getz, G. RNA-SeQC 2: Efficient RNA-seq quality control and quantification for large cohorts. Bioinformatics 2021, 37, 3048–3050. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Team, R. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com/ (accessed on 1 January 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Diermeier, S.D.; Chang, K.C.; Freier, S.M.; Song, J.; El Demerdash, O.; Krasnitz, A.; Rigo, F.; Bennett, C.F.; Spector, D.L. Mammary Tumor-Associated RNAs Impact Tumor Cell Proliferation, Invasion, and Migration. Cell Rep. 2016, 17, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Diederichs, S. The four dimensions of noncoding RNA conservation. Trends Genet. 2014, 30, 121–123. [Google Scholar] [CrossRef]
- Benhammou, J.N.; Ko, A.; Alvarez, M.; Kaikkonen, M.U.; Rankin, C.; Garske, K.M.; Padua, D.; Bhagat, Y.; Kaminska, D.; Karja, V.; et al. Novel Lipid Long Intervening Noncoding RNA, Oligodendrocyte Maturation-Associated Long Intergenic Noncoding RNA, Regulates the Liver Steatosis Gene Stearoyl-Coenzyme A Desaturase As an Enhancer RNA. Hepatol. Commun. 2019, 3, 1356–1372. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Lin, X.; Wang, G. Targeting SREBP-1-Mediated Lipogenesis as Potential Strategies for Cancer. Front. Oncol. 2022, 12, 952371. [Google Scholar] [CrossRef]
- Hollern, D.P.; Swiatnicki, M.R.; Andrechek, E.R. Histological subtypes of mouse mammary tumors reveal conserved relationships to human cancers. PLoS Genet. 2018, 14, e1007135. [Google Scholar] [CrossRef] [Green Version]
- Oshi, M.; Takahashi, H.; Tokumaru, Y.; Yan, L.; Rashid, O.M.; Nagahashi, M.; Matsuyama, R.; Endo, I.; Takabe, K. The E2F Pathway Score as a Predictive Biomarker of Response to Neoadjuvant Therapy in ER+/HER2- Breast Cancer. Cells 2020, 9, 1643. [Google Scholar] [CrossRef]
- Weigelt, B.; Peterse, J.L.; van ‘t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [Green Version]
- Holder, A.M.; Gonzalez-Angulo, A.M.; Chen, H.; Akcakanat, A.; Do, K.A.; Fraser Symmans, W.; Pusztai, L.; Hortobagyi, G.N.; Mills, G.B.; Meric-Bernstam, F. High stearoyl-CoA desaturase 1 expression is associated with shorter survival in breast cancer patients. Breast Cancer Res. Treat. 2013, 137, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Mason, P.; Liang, B.; Li, L.; Fremgen, T.; Murphy, E.; Quinn, A.; Madden, S.L.; Biemann, H.P.; Wang, B.; Cohen, A.; et al. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS ONE 2012, 7, e33823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roongta, U.V.; Pabalan, J.G.; Wang, X.; Ryseck, R.P.; Fargnoli, J.; Henley, B.J.; Yang, W.P.; Zhu, J.; Madireddi, M.T.; Lawrence, R.M.; et al. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res. 2011, 9, 1551–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Ren, J.; Yang, L.; Li, Y.; Fu, J.; Li, Y.; Tian, Y.; Qiu, F.; Liu, Z.; Qiu, Y. Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Sci. Rep. 2016, 6, 19665. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.; Chisholm, J.W.; Igal, R.A. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS ONE 2010, 5, e11394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef] [Green Version]
- Piao, C.; Cui, X.; Zhan, B.; Li, J.; Li, Z.; Li, Z.; Liu, X.; Bi, J.; Zhang, Z.; Kong, C. Inhibition of stearoyl CoA desaturase-1 activity suppresses tumour progression and improves prognosis in human bladder cancer. J. Cell. Mol. Med. 2019, 23, 2064–2076. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Bai, H.; Liu, S.; Cao, D.; Wu, H.; Shen, K.; Tai, Y.; Yang, J. Targeting stearoyl-CoA desaturase 1 to repress endometrial cancer progression. Oncotarget 2018, 9, 12064–12078. [Google Scholar] [CrossRef] [Green Version]
- Pinkham, K.; Park, D.J.; Hashemiaghdam, A.; Kirov, A.B.; Adam, I.; Rosiak, K.; da Hora, C.C.; Teng, J.; Cheah, P.S.; Carvalho, L.; et al. Stearoyl CoA Desaturase Is Essential for Regulation of Endoplasmic Reticulum Homeostasis and Tumor Growth in Glioblastoma Cancer Stem Cells. Stem Cell Rep. 2019, 12, 712–727. [Google Scholar] [CrossRef] [Green Version]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avancès, C.; Allory, Y.; de la Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Zhu, L.; Zhu, Q.; Su, J.; Liu, M.; Huang, W. SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol. Lett. 2016, 12, 2409–2416. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Cheng, M.; Niu, Y.; Chi, X.; Liu, X.; Fan, J.; Fan, H.; Chang, Y.; Yang, W. Identification of a novel human long non-coding RNA that regulates hepatic lipid metabolism by inhibiting SREBP-1c. Int. J. Biol. Sci. 2017, 13, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liang, C.; Zhang, Y.; Wang, Z.; Li, R.; Wei, Z.; Guo, J. The role of FOXD2-AS1 in cancer: A comprehensive study based on data mining and published articles. Biosci. Rep. 2020, 40, BSR20190372. [Google Scholar] [CrossRef]
- Zhang, Q.; Jin, X.; Shi, W.; Chen, X.; Pang, W.; Yu, X.; Yang, L. A long non-coding RNA LINC00461-dependent mechanism underlying breast cancer invasion and migration via the miR-144-3p/KPNA2 axis. Cancer Cell Int. 2020, 20, 137. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Esmaeili, M.; Taheri, M. H19 lncRNA: Roles in tumorigenesis. Biomed. Pharmacother. 2020, 123, 109774. [Google Scholar] [CrossRef]
- Cantile, M.; Di Bonito, M.; Cerrone, M.; Collina, F.; De Laurentiis, M.; Botti, G. Long Non-Coding RNA HOTAIR in Breast Cancer Therapy. Cancers 2020, 12, 1197. [Google Scholar] [CrossRef]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LncRNAs | Protein Coding Genes | PCC | p Value | Description | Functions |
|---|---|---|---|---|---|
| Up only in PTHrP WT tumors | |||||
| A730020E08Rik | Ccser1 | 0.74 | 7.05 × 10−7 | Coiled-Coil Serine Rich Protein 1 | Modulate cell division |
| BC016548 | Elf5 | 0.97 | 3.14 × 10−20 | E74 Like ETS Transcription Factor 5 | Transcription factor |
| Gm14133 | Syndig1 | 0.79 | 4.27 × 10−8 | Synapse Differentiation Inducing 1 | Regulator of excitatory synapse development |
| Gm26902 | Ch25h | 0.75 | 5.07 × 10−7 | Cholesterol 25-hydroxylase | Enzyme that converts cholesterol to 25-hydroxycholesterol |
| Gm50337 | Scd1 | 0.91 | 1.34 × 10−13 | Stearoyl-CoA desaturase 1 | Synthesis of monounsaturated fatty acids |
| Rps18-ps4 | Rps18-ps6 | 0.71 | 3.46 × 10−6 | Ribosomal Protein S18, Pseudogene 6 | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Li, J.; Badescu, D.; Karaplis, A.C.; Ragoussis, J.; Kremer, R. PTHrP Regulates Fatty Acid Metabolism via Novel lncRNA in Breast Cancer Initiation and Progression Models. Cancers 2023, 15, 3763. https://doi.org/10.3390/cancers15153763
Zhang R, Li J, Badescu D, Karaplis AC, Ragoussis J, Kremer R. PTHrP Regulates Fatty Acid Metabolism via Novel lncRNA in Breast Cancer Initiation and Progression Models. Cancers. 2023; 15(15):3763. https://doi.org/10.3390/cancers15153763
Chicago/Turabian StyleZhang, Rui, Jiarong Li, Dunarel Badescu, Andrew C. Karaplis, Jiannis Ragoussis, and Richard Kremer. 2023. "PTHrP Regulates Fatty Acid Metabolism via Novel lncRNA in Breast Cancer Initiation and Progression Models" Cancers 15, no. 15: 3763. https://doi.org/10.3390/cancers15153763
APA StyleZhang, R., Li, J., Badescu, D., Karaplis, A. C., Ragoussis, J., & Kremer, R. (2023). PTHrP Regulates Fatty Acid Metabolism via Novel lncRNA in Breast Cancer Initiation and Progression Models. Cancers, 15(15), 3763. https://doi.org/10.3390/cancers15153763