
Microtubule Dynamics Deregulation Induces Apoptosis in Human Urothelial Bladder Cancer Cells via a p53-Independent Pathway

 , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Cell Viability MTT Assays
2.3. Flow Cytometry for Cell Cycle Analysis
2.4. Western Blotting
2.5. RT-PCR
2.6. DepMap Data Analysis
2.7. ATAC-Seq. of Primary Human Tumors
2.8. Statistical Analysis
3. Results
3.1. Urothelial Bladder Cancer Cells Are Significantly Sensitive to Microtubule Assembly Inhibition
3.2. Paclitaxel Treatment Induces Cell Cycle Arrest and Growth Inhibition in Bladder Cancer Cells
3.3. Paclitaxel Treatment Induces Apoptosis in Bladder Cancer Cells
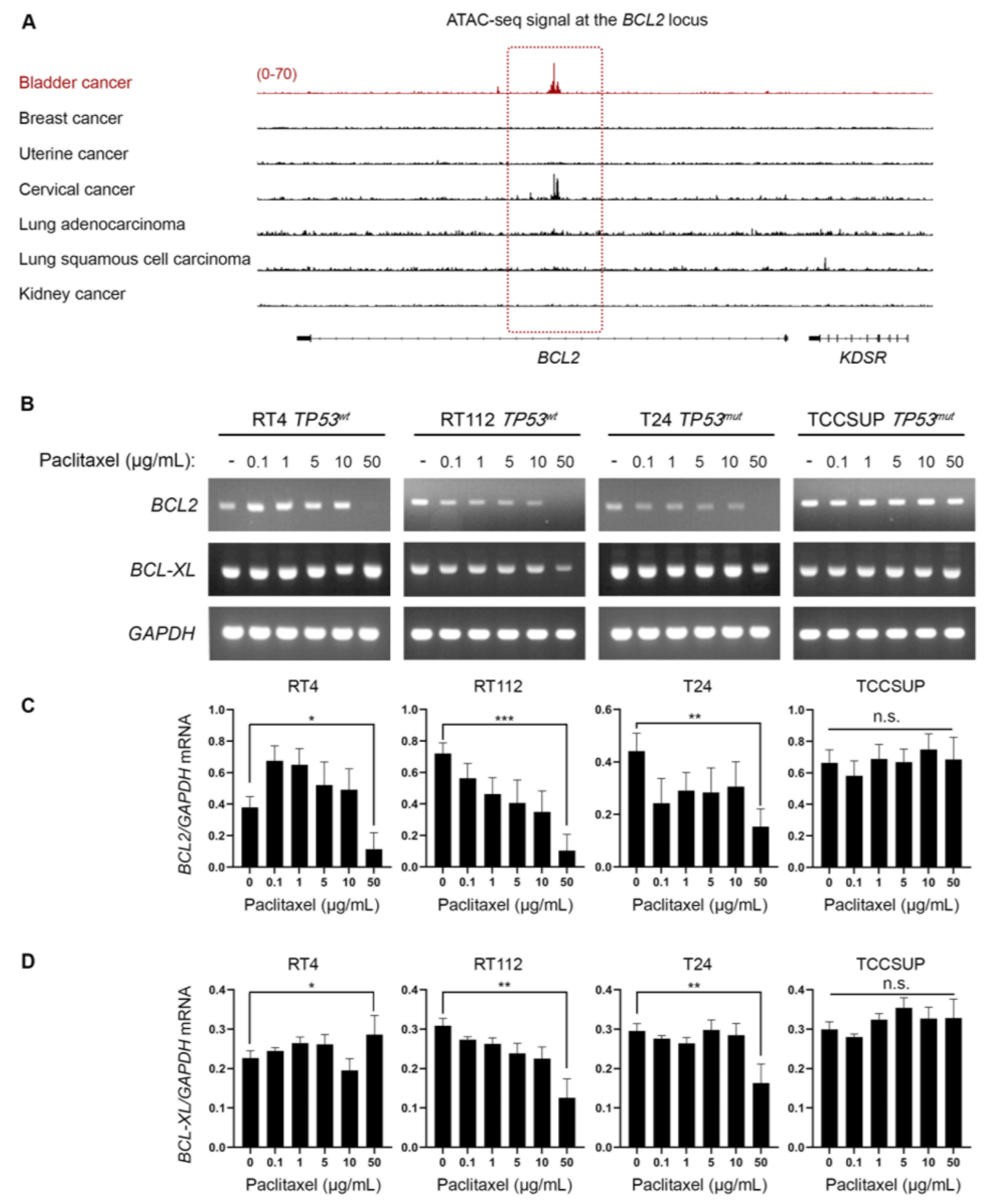
3.4. Bladder Cancer Cells Have Selectively Open Chromatin at the BCL2 Locus and Undergo BLC2 Downregulation upon Paclitaxel Treatment
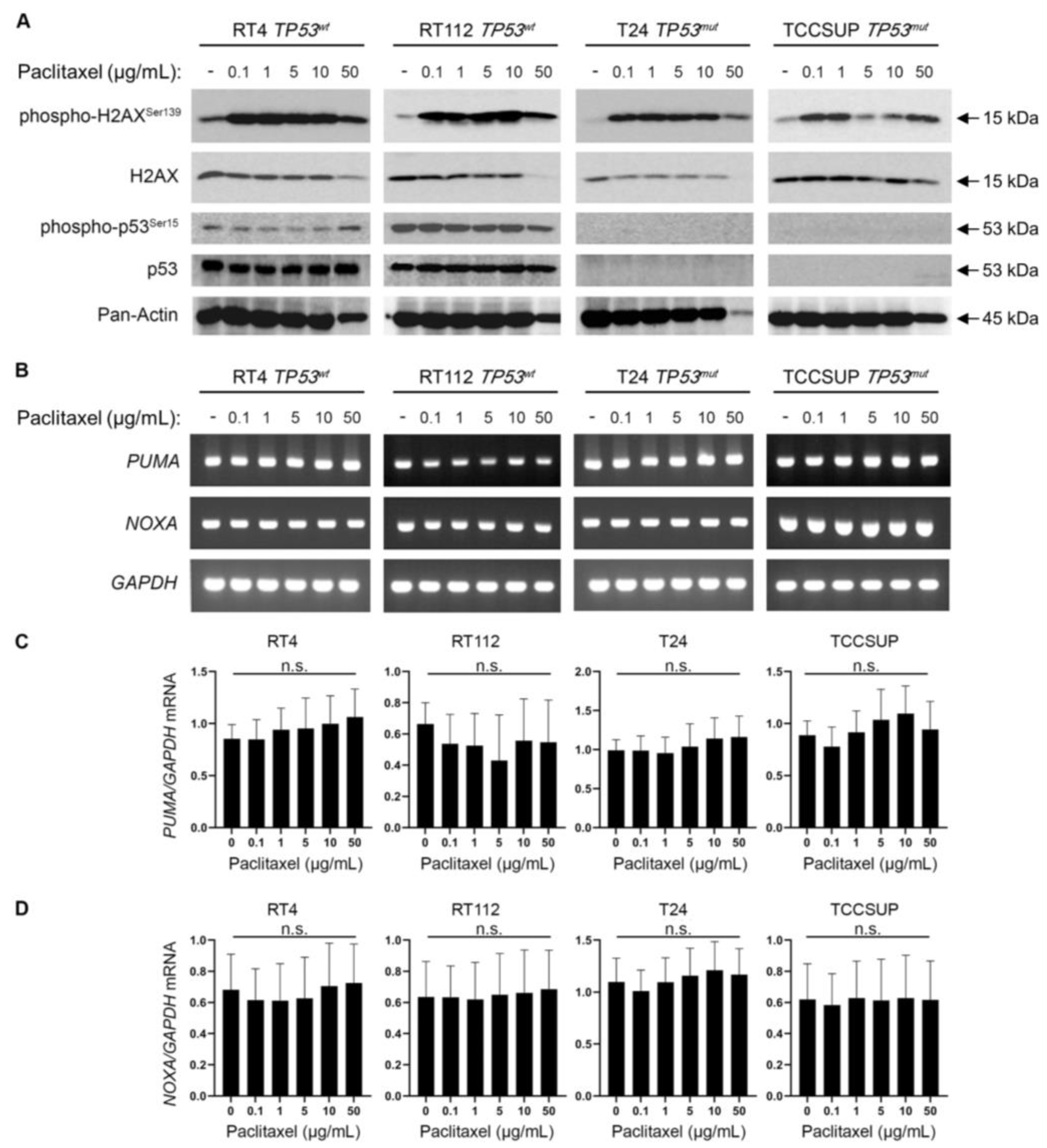
3.5. Paclitaxel Treatment Induces DNA Damage without p53 Activation
3.6. Bladder Cancer Cell Sensitivity to Paclitaxel Is Independent of TP53 Activity but Highly Dependent on the Expression of DNA Repair Genes
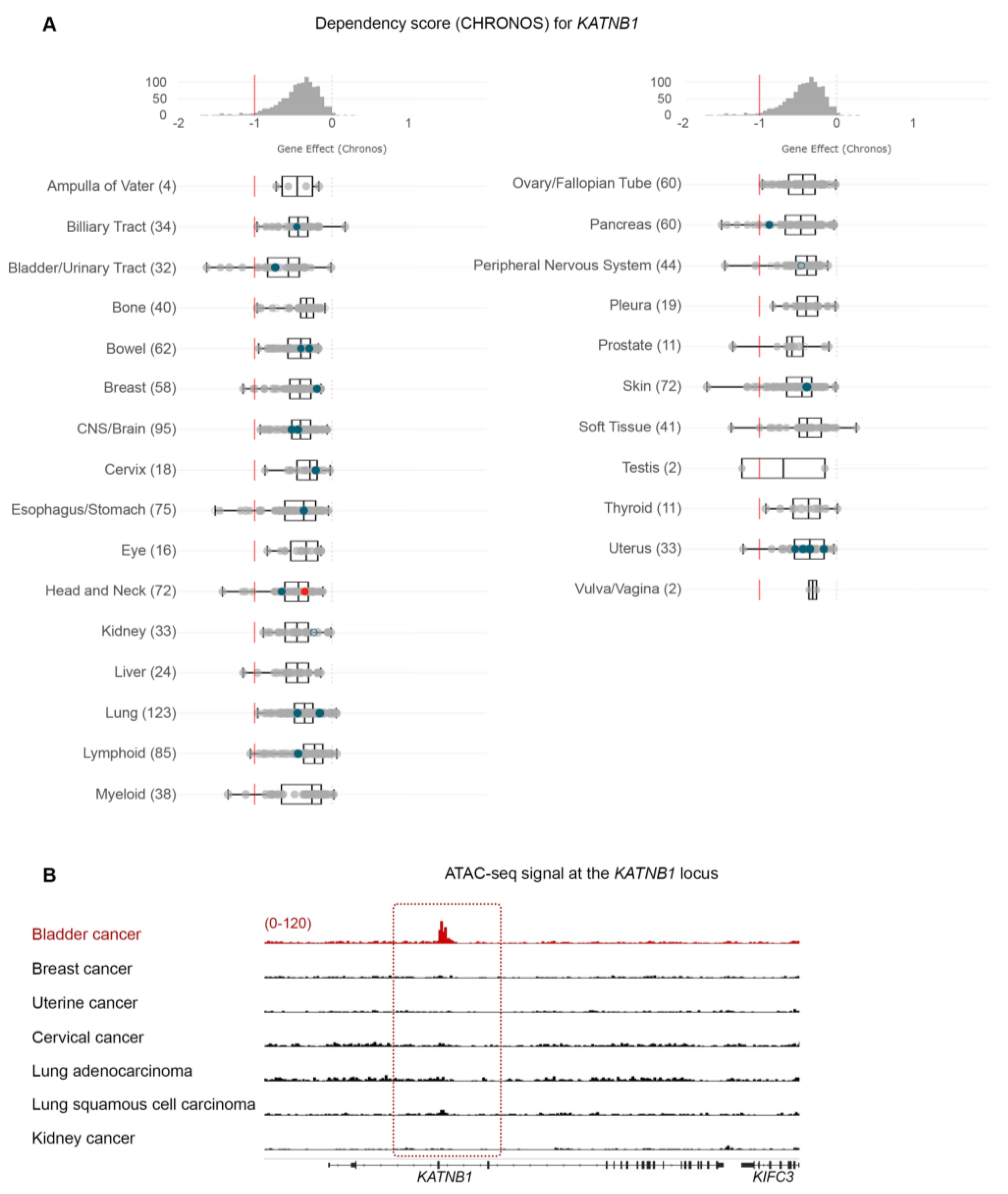
3.7. Bladder Cancer Cells Are Highly Dependent on PPARG Expression for Survival and Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Comperat, E.M.; Cowan, N.C.; Gakis, G.; Hernandez, V.; Linares Espinos, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2021, 79, 82–104. [Google Scholar] [CrossRef]
- Netto, G.J. Molecular biomarkers in urothelial carcinoma of the bladder: Are we there yet? Nat. Rev. Urol. 2012, 9, 41–51. [Google Scholar] [CrossRef]
- Wu, X.-R. Urothelial tumorigenesis: A tale of divergent pathways. Nat. Rev. Cancer 2005, 5, 713–725. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Kim, J.; Akbani, R.; Creighton, C.J.; Lerner, S.P.; Weinstein, J.N.; Getz, G.; Kwiatkowski, D.J. Invasive Bladder Cancer: Genomic Insights and Therapeutic Promise. Clin. Cancer Res. 2015, 21, 4514–4524. [Google Scholar] [CrossRef]
- Galsky, M.D.; Hahn, N.M.; Rosenberg, J.; Sonpavde, G.; Hutson, T.; Oh, W.K.; Dreicer, R.; Vogelzang, N.; Sternberg, C.N.; Bajorin, D.F.; et al. Treatment of patients with metastatic urothelial cancer “unfit” for Cisplatin-based chemotherapy. J. Clin. Oncol. 2011, 29, 2432–2438. [Google Scholar] [CrossRef] [PubMed]
- Boehm, J.S.; Garnett, M.J.; Adams, D.J.; Francies, H.E.; Golub, T.R.; Hahn, W.C.; Iorio, F.; McFarland, J.M.; Parts, L.; Vazquez, F. Cancer research needs a better map. Nature 2021, 589, 514–516. [Google Scholar] [CrossRef]
- Hahn, W.C.; Bader, J.S.; Braun, T.P.; Califano, A.; Clemons, P.A.; Druker, B.J.; Ewald, A.J.; Fu, H.; Jagu, S.; Kemp, C.J.; et al. An expanded universe of cancer targets. Cell 2021, 184, 1142–1155. [Google Scholar] [CrossRef]
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.-L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P.; et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 2016, 34, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Konstantakou, E.G.; Voutsinas, G.E.; Velentzas, A.D.; Basogianni, A.S.; Paronis, E.; Balafas, E.; Kostomitsopoulos, N.; Syrigos, K.N.; Anastasiadou, E.; Stravopodis, D.J. 3-BrPA eliminates human bladder cancer cells with highly oncogenic signatures via engagement of specific death programs and perturbation of multiple signaling and metabolic determinants. Mol. Cancer 2015, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019, 720243. [Google Scholar] [CrossRef]
- Pacini, C.; Dempster, J.M.; Boyle, I.; Goncalves, E.; Najgebauer, H.; Karakoc, E.; van der Meer, D.; Barthorpe, A.; Lightfoot, H.; Jaaks, P.; et al. Integrated cross-study datasets of genetic dependencies in cancer. Nat. Commun. 2021, 12, 1661. [Google Scholar] [CrossRef]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A CRISPR cell population dynamics model. bioRxiv 2021, 432728. [Google Scholar] [CrossRef]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E.; et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 2018, 50, 515–523. [Google Scholar] [CrossRef]
- Zimmerman, M.W.; Liu, Y.; He, S.; Durbin, A.D.; Abraham, B.J.; Easton, J.; Shao, Y.; Xu, B.; Zhu, S.; Zhang, X.; et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov. 2018, 8, 320–335. [Google Scholar] [CrossRef]
- Ferlini, C.; Cicchillitti, L.; Raspaglio, G.; Bartollino, S.; Cimitan, S.; Bertucci, C.; Mozzetti, S.; Gallo, D.; Persico, M.; Fattorusso, C.; et al. Paclitaxel Directly Binds to Bcl-2 and Functionally Mimics Activity of Nur77. Cancer Res. 2009, 69, 6906–6914. [Google Scholar] [CrossRef] [PubMed]
- Rodi, D.J.; Janes, R.W.; Sanganee, H.J.; Holton, R.A.; Wallace, B.A.; Makowski, L. Screening of a library of phage-displayed peptides identifies human bcl-2 as a taxol-binding protein. J. Mol. Biol. 1999, 285, 197–203. [Google Scholar] [CrossRef]
- Rodi, D.J.; Makowski, L. Similarity between the sequences of taxol-selected peptides and the disordered loop of the anti-apoptotic protein, Bcl-2. Pac. Symp. Biocomput. 1999, 4, 532–541. [Google Scholar] [CrossRef]
- Whitaker, R.H.; Placzek, W.J. Regulating the BCL2 Family to Improve Sensitivity to Microtubule Targeting Agents. Cells 2019, 8, 346. [Google Scholar] [CrossRef] [PubMed]
- Bal, E.; Kumar, R.; Hadigol, M.; Holmes, A.B.; Hilton, L.K.; Loh, J.W.; Dreval, K.; Wong, J.C.H.; Vlasevska, S.; Corinaldesi, C.; et al. Super-enhancer hypermutation alters oncogene expression in B cell lymphoma. Nature 2022, 607, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.; Kornblau, S.; Safe, S.; Watt, J.C.; Ruvolo, V.; Chen, W.; Qiu, Y.; Coombes, K.R.; Ju, Z.; Abdelrahim, M.; et al. Role of peroxisome proliferator-activated receptor-gamma and its coactivator DRIP205 in cellular responses to CDDO (RTA-401) in acute myelogenous leukemia. Cancer Res. 2010, 70, 4949–4960. [Google Scholar] [CrossRef]
- Vazquez, F.; Sellers, W.R. Are CRISPR Screens Providing the Next Generation of Therapeutic Targets? Cancer Res. 2021, 81, 5806–5809. [Google Scholar] [CrossRef] [PubMed]
- Chetverina, D.; Vorobyeva, N.E.; Gyorffy, B.; Shtil, A.A.; Erokhin, M. Analyses of Genes Critical to Tumor Survival Reveal Potential ‘Supertargets’: Focus on Transcription. Cancers 2023, 15, 3042. [Google Scholar] [CrossRef]
- Wittmann, T.; Hyman, A.; Desai, A. The spindle: A dynamic assembly of microtubules and motors. Nat. Cell Biol. 2001, 3, E28–E34. [Google Scholar] [CrossRef]
- Valdez, V.A.; Neahring, L.; Petry, S.; Dumont, S. Mechanisms underlying spindle assembly and robustness. Nat. Rev. Mol. Cell Biol. 2023, 24, 523–542. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Yuan, Z.M.; Huang, Y.; Ishiko, T.; Nakada, S.; Utsugisawa, T.; Kharbanda, S.; Wang, R.; Sung, P.; Shinohara, A.; Weichselbaum, R.; et al. Regulation of Rad51 function by c-Abl in response to DNA damage. J. Biol. Chem. 1998, 273, 3799–3802. [Google Scholar] [CrossRef]
- Niida, H.; Katsuno, Y.; Sengoku, M.; Shimada, M.; Yukawa, M.; Ikura, M.; Ikura, T.; Kohno, K.; Shima, H.; Suzuki, H.; et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev. 2010, 24, 333–338. [Google Scholar] [CrossRef]
- Peng, T.; Wang, G.; Cheng, S.; Xiong, Y.; Cao, R.; Qian, K.; Ju, L.; Wang, X.; Xiao, Y. The role and function of PPARγ in bladder cancer. J. Cancer 2020, 11, 3965–3975. [Google Scholar] [CrossRef] [PubMed]
- Chi, T.; Wang, M.; Wang, X.; Yang, K.; Xie, F.; Liao, Z.; Wei, P. PPAR-γ Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, 737776. [Google Scholar] [CrossRef]
- Smallridge, R.C.; Copland, J.A.; Brose, M.S.; Wadsworth, J.T.; Houvras, Y.; Menefee, M.E.; Bible, K.C.; Shah, M.H.; Gramza, A.W.; Klopper, J.P.; et al. Efatutazone, an Oral PPAR-γ Agonist, in Combination With Paclitaxel in Anaplastic Thyroid Cancer: Results of a Multicenter Phase 1 Trial. J. Clin. Endocrinol. Metab. 2013, 98, 2392–2400. [Google Scholar] [CrossRef]
- Mierzejewski, K.; Paukszto, Ł.; Kurzyńska, A.; Kunicka, Z.; Jastrzębski, J.P.; Makowczenko, K.G.; Golubska, M.; Bogacka, I. PPARγ regulates the expression of genes involved in the DNA damage response in an inflamed endometrium. Sci. Rep. 2022, 12, 4026. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Nag, A.; Gangenahalli, G.; Sharma, K. Peroxisome Proliferator Activated Receptor Gamma Sensitizes Non-small Cell Lung Carcinoma to Gamma Irradiation Induced Apoptosis. Front. Genet. 2019, 10, 554. [Google Scholar] [CrossRef]
- Khandekar, M.J.; Banks, A.S.; Laznik-Bogoslavski, D.; White, J.P.; Choi, J.H.; Kazak, L.; Lo, J.C.; Cohen, P.; Wong, K.K.; Kamenecka, T.M.; et al. Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2018, 115, 561–566. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drosos, Y.; Konstantakou, E.G.; Bassogianni, A.-S.; Nikolakopoulos, K.-S.; Koumoundourou, D.G.; Markaki, S.P.; Tsitsilonis, O.E.; Voutsinas, G.E.; Valakos, D.; Anastasiadou, E.; et al. Microtubule Dynamics Deregulation Induces Apoptosis in Human Urothelial Bladder Cancer Cells via a p53-Independent Pathway. Cancers 2023, 15, 3730. https://doi.org/10.3390/cancers15143730
Drosos Y, Konstantakou EG, Bassogianni A-S, Nikolakopoulos K-S, Koumoundourou DG, Markaki SP, Tsitsilonis OE, Voutsinas GE, Valakos D, Anastasiadou E, et al. Microtubule Dynamics Deregulation Induces Apoptosis in Human Urothelial Bladder Cancer Cells via a p53-Independent Pathway. Cancers. 2023; 15(14):3730. https://doi.org/10.3390/cancers15143730
Chicago/Turabian StyleDrosos, Yiannis, Eumorphia G. Konstantakou, Aggeliki-Stefania Bassogianni, Konstantinos-Stylianos Nikolakopoulos, Dimitra G. Koumoundourou, Sophia P. Markaki, Ourania E. Tsitsilonis, Gerassimos E. Voutsinas, Dimitrios Valakos, Ema Anastasiadou, and et al. 2023. "Microtubule Dynamics Deregulation Induces Apoptosis in Human Urothelial Bladder Cancer Cells via a p53-Independent Pathway" Cancers 15, no. 14: 3730. https://doi.org/10.3390/cancers15143730
APA StyleDrosos, Y., Konstantakou, E. G., Bassogianni, A.-S., Nikolakopoulos, K.-S., Koumoundourou, D. G., Markaki, S. P., Tsitsilonis, O. E., Voutsinas, G. E., Valakos, D., Anastasiadou, E., Thanos, D., Velentzas, A. D., & Stravopodis, D. J. (2023). Microtubule Dynamics Deregulation Induces Apoptosis in Human Urothelial Bladder Cancer Cells via a p53-Independent Pathway. Cancers, 15(14), 3730. https://doi.org/10.3390/cancers15143730